
Region-Specific Effects of Metformin on Gut Microbiome and Metabolome in High-Fat Diet-Induced Type 2 Diabetes Mouse Model

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
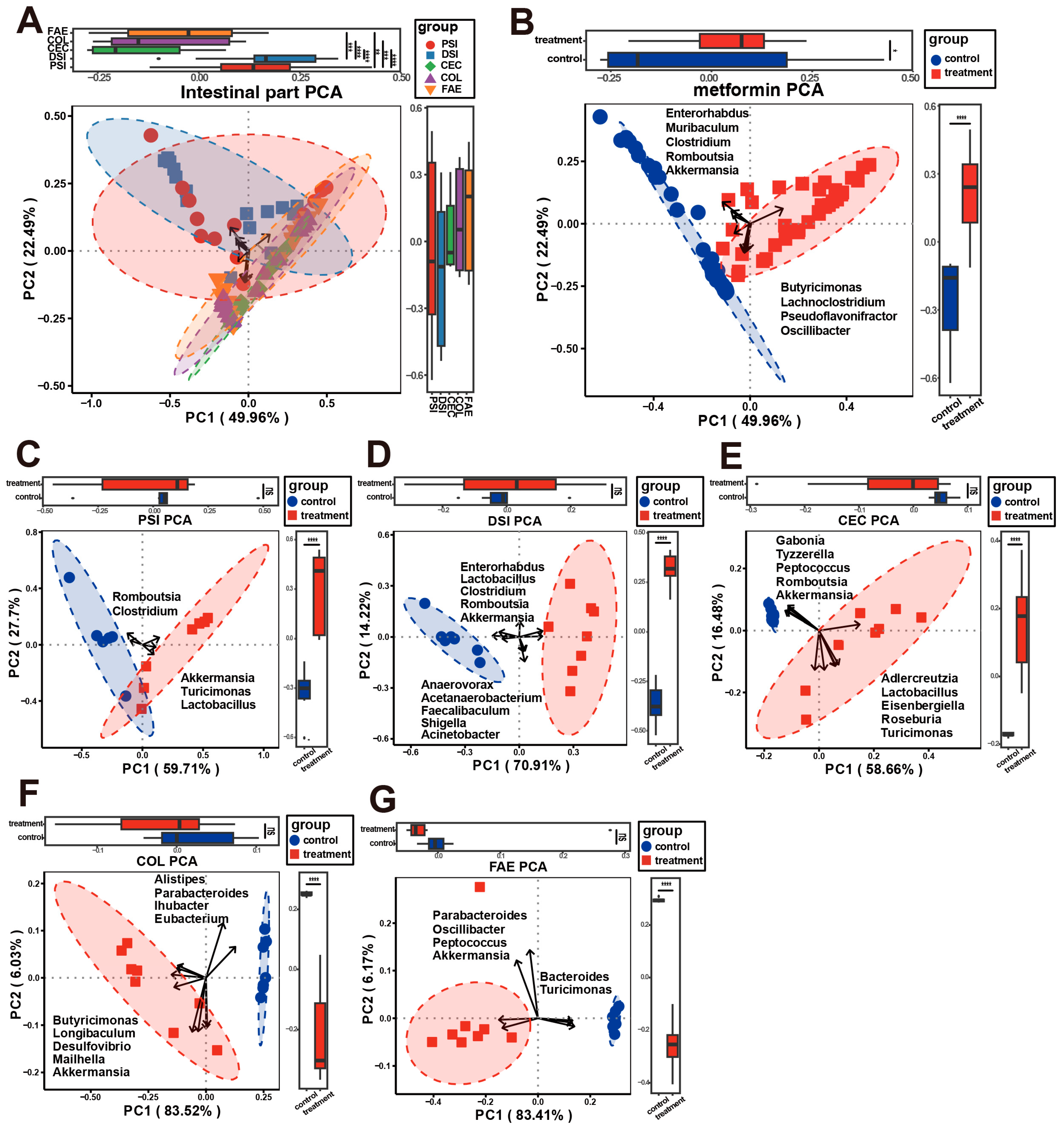
2.1. Microbial Composition of Metformin-Treated and Control Groups
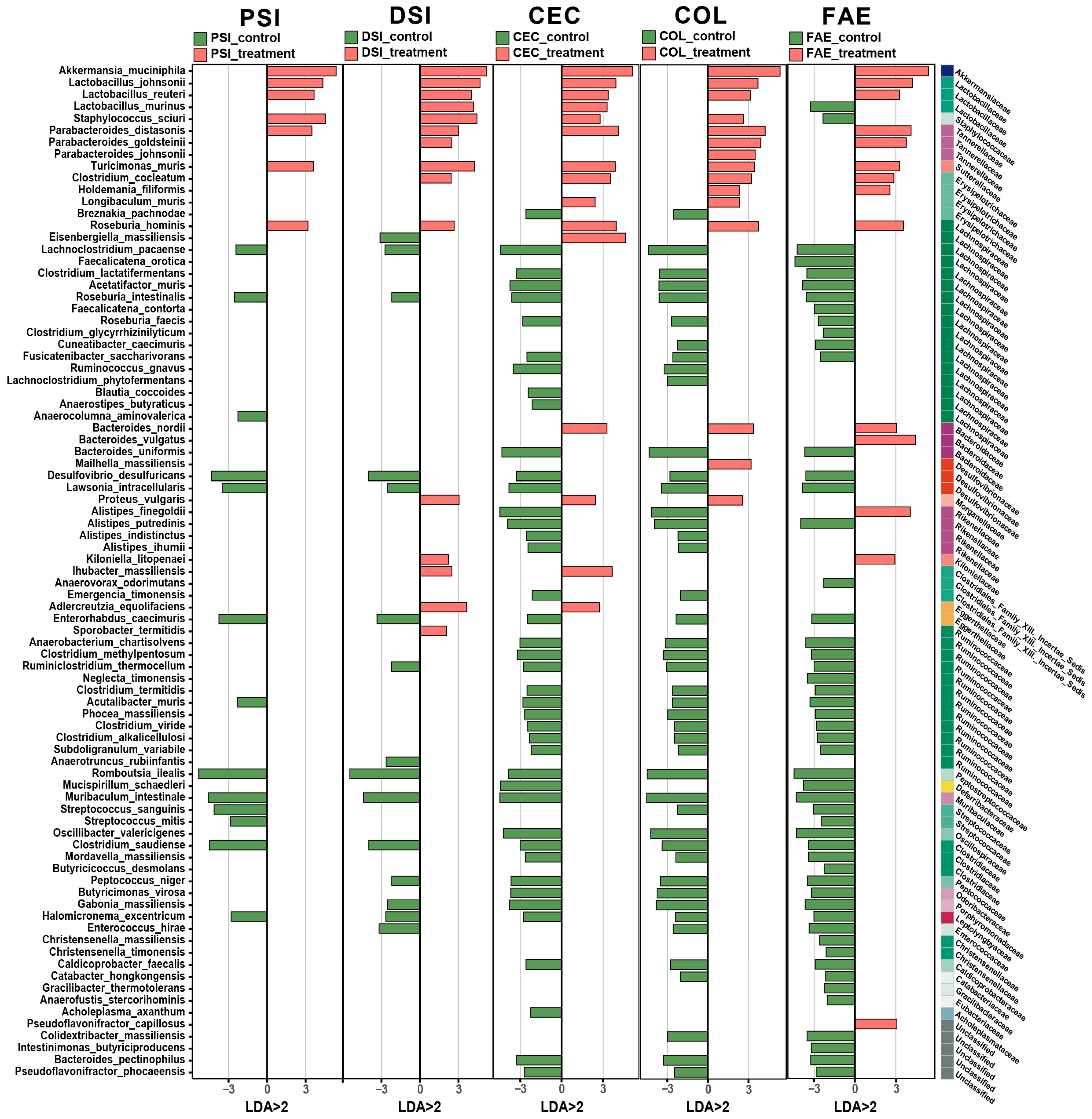
2.2. Alterations in Microbial Composition in Metformin-Treated Mice
2.3. Alterations in Metabolomic Composition in Metformin-Treated Mice
2.4. Correlation Analysis of Microbiome and Metabolome Data
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Sample Collection
4.3. DNA Isolation and Full-Length 16S rRNA Gene Amplification
4.4. Data Analysis of Full-Length 16S rRNA Gene Sequences
4.5. Targeted Metabolomics Profiling
4.6. Correlation Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sanchez-Rangel, E.; Inzucchi, S.E. Metformin: Clinical Use in Type 2 Diabetes. Diabetologia 2017, 60, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- McCreight, L.J.; Bailey, C.J.; Pearson, E.R. Metformin and the Gastrointestinal Tract. Diabetologia 2016, 59, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Horowitz, M.; Rayner, C.K. New Insights into the Anti-Diabetic Actions of Metformin: From the Liver to the Gut. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Ren, L.; Jia, X.; Wang, J.; Cong, B. Understanding the Action Mechanisms of Metformin in the Gastrointestinal Tract. Front. Pharmacol. 2024, 15, 1347047. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.V.; Duca, F.A.; Waise, T.M.Z.; Rasmussen, B.A.; Abraham, M.A.; Dranse, H.J.; Puri, A.; O’Brien, C.A.; Lam, T.K.T. Metformin Alters Upper Small Intestinal Microbiota That Impact a Glucose-SGLT1-Sensing Glucoregulatory Pathway. Cell Metab. 2018, 27, 101–117.e5. [Google Scholar] [CrossRef] [PubMed]
- MetaHIT Consortium; Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; et al. Disentangling Type 2 Diabetes and Metformin Treatment Signatures in the Human Gut Microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Silamiķele, L.; Saksis, R.; Silamiķelis, I.; Kotoviča, P.P.; Brīvība, M.; Kalniņa, I.; Kalniņa, Z.; Fridmanis, D.; Kloviņš, J. Spatial Variation of the Gut Microbiome in Response to Long-Term Metformin Treatment in High-Fat Diet-Induced Type 2 Diabetes Mouse Model of Both Sexes. Gut Microbes 2023, 15, 2188663. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W. Metabolomic Approaches to Investigate the Effect of Metformin: An Overview. Int. J. Mol. Sci. 2021, 22, 10275. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut Microbiota and Intestinal FXR Mediate the Clinical Benefits of Metformin. Nat. Med. 2018, 24, 1919–1929. [Google Scholar] [CrossRef]
- Palau-Rodriguez, M.; Tulipani, S.; Isabel Queipo-Ortuño, M.; Urpi-Sarda, M.; Tinahones, F.J.; Andres-Lacueva, C. Metabolomic Insights into the Intricate Gut Microbial–Host Interaction in the Development of Obesity and Type 2 Diabetes. Front. Microbiol. 2015, 6, 1151. [Google Scholar] [CrossRef]
- Belizário, J.E.; Napolitano, M. Human Microbiomes and Their Roles in Dysbiosis, Common Diseases, and Novel Therapeutic Approaches. Front. Microbiol. 2015, 6, 1050. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lang, T.; Shen, J.; Dai, J.; Tian, L.; Wang, X. Core Gut Bacteria Analysis of Healthy Mice. Front. Microbiol. 2019, 10, 887. [Google Scholar] [CrossRef] [PubMed]
- Beura, S.; Kundu, P.; Das, A.K.; Ghosh, A. Metagenome-Scale Community Metabolic Modelling for Understanding the Role of Gut Microbiota in Human Health. Comput. Biol. Med. 2022, 149, 105997. [Google Scholar] [CrossRef] [PubMed]
- Fujisaka, S.; Watanabe, Y.; Tobe, K. The Gut Microbiome: A Core Regulator of Metabolism. J. Endocrinol. 2023, 256, e220111. [Google Scholar] [CrossRef] [PubMed]
- de Vos, W.M.; Tilg, H.; Van Hul, M.; Cani, P.D. Gut Microbiome and Health: Mechanistic Insights. Gut 2022, 71, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, S.; Nie, Q.; He, H.; Tan, H.; Geng, F.; Ji, H.; Hu, J.; Nie, S. Gut Firmicutes: Relationship with Dietary Fiber and Role in Host Homeostasis. Crit. Rev. Food Sci. Nutr. 2023, 63, 12073–12088. [Google Scholar] [CrossRef]
- Blaak, E.E.; Canfora, E.E.; Theis, S.; Frost, G.; Groen, A.K.; Mithieux, G.; Nauta, A.; Scott, K.; Stahl, B.; van Harsselaar, J.; et al. Short Chain Fatty Acids in Human Gut and Metabolic Health. Benef. Microbes 2020, 11, 411–455. [Google Scholar] [CrossRef]
- Zafar, H.; Saier, M.H. Gut Bacteroides Species in Health and Disease. Gut Microbes 2021, 13, 1848158. [Google Scholar] [CrossRef]
- Wu, T.-R.; Lin, C.-S.; Chang, C.-J.; Lin, T.-L.; Martel, J.; Ko, Y.-F.; Ojcius, D.M.; Lu, C.-C.; Young, J.D.; Lai, H.-C. Gut Commensal Parabacteroides Goldsteinii Plays a Predominant Role in the Anti-Obesity Effects of Polysaccharides Isolated from Hirsutella Sinensis. Gut 2019, 68, 248–262. [Google Scholar] [CrossRef]
- Jiang, T.; Savaiano, D.A. Modification of Colonic Fermentation by Bifidobacteria and pH in Vitro (Impact on Lactose Metabolism, Short-Chain Fatty Acid, and Lactate Production). Dig. Dis. Sci. 1997, 42, 2370–2377. [Google Scholar] [CrossRef]
- Ricci, L.; Mackie, J.; Donachie, G.E.; Chapuis, A.; Mezerová, K.; Lenardon, M.D.; Brown, A.J.P.; Duncan, S.H.; Walker, A.W. Human Gut Bifidobacteria Inhibit the Growth of the Opportunistic Fungal Pathogen Candida Albicans. FEMS Microbiol. Ecol. 2022, 98, fiac095. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. The Gut Microbiota Shapes Intestinal Immune Responses during Health and Disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial Signature of Dysbiosis in Gut Microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut Biogeography of the Bacterial Microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef]
- McCallum, G.; Tropini, C. The Gut Microbiota and Its Biogeography. Nat. Rev. Microbiol. 2024, 22, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.A.; Ahmed, T.; Brüssow, H. Hunger and Microbiology: Is a Low Gastric Acid-Induced Bacterial Overgrowth in the Small Intestine a Contributor to Malnutrition in Developing Countries? Microb. Biotechnol. 2017, 10, 1025–1030. [Google Scholar] [CrossRef]
- Martinez-Guryn, K.; Leone, V.; Chang, E.B. Regional Diversity of the Gastrointestinal Microbiome. Cell Host Microbe 2019, 26, 314–324. [Google Scholar] [CrossRef]
- Brown, K.; Abbott, D.W.; Uwiera, R.R.E.; Inglis, G.D. Removal of the Cecum Affects Intestinal Fermentation, Enteric Bacterial Community Structure, and Acute Colitis in Mice. Gut Microbes 2018, 9, 218–235. [Google Scholar] [CrossRef]
- Milla, P.J. Advances in Understanding Colonic Function. J. Pediatr. Gastroenterol. Nutr. 2009, 48 (Suppl. S2), S43–S45. [Google Scholar] [CrossRef]
- Song, N.-N.; Li, Q.-S.; Liu, C.-X. Intestinal Permeability of Metformin Using Single-Pass Intestinal Perfusion in Rats. World J. Gastroenterol. 2006, 12, 4064–4070. [Google Scholar] [CrossRef]
- Boccard, J.; Rutledge, D.N. A Consensus Orthogonal Partial Least Squares Discriminant Analysis (OPLS-DA) Strategy for Multiblock Omics Data Fusion. Anal. Chim. Acta 2013, 769, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Shen, J.; Feng, S.; Huang, C.; Wang, H.; Huo, F.; Liu, H. Akkermansia Muciniphila, Which Is Enriched in the Gut Microbiota by Metformin, Improves Cognitive Function in Aged Mice by Reducing the Proinflammatory Cytokine Interleukin-6. Microbiome 2023, 11, 120. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Li, F.; Deng, W.; Li, Z.; Wang, S.; Lv, P.; Chen, Y. Metformin Exerts Anti-Inflammatory and Mucus Barrier Protective Effects by Enriching Akkermansia Muciniphila in Mice With Ulcerative Colitis. Front. Pharmacol. 2021, 12, 726707. [Google Scholar] [CrossRef] [PubMed]
- de la Cuesta-Zuluaga, J.; Mueller, N.T.; Corrales-Agudelo, V.; Velásquez-Mejía, E.P.; Carmona, J.A.; Abad, J.M.; Escobar, J.S. Metformin Is Associated With Higher Relative Abundance of Mucin-Degrading Akkermansia Muciniphila and Several Short-Chain Fatty Acid-Producing Microbiota in the Gut. Diabetes Care 2017, 40, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.-R.; Lee, J.-C.; Lee, H.-Y.; Kim, M.-S.; Whon, T.W.; Lee, M.-S.; Bae, J.-W. An Increase in the Akkermansia Spp. Population Induced by Metformin Treatment Improves Glucose Homeostasis in Diet-Induced Obese Mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef]
- Hu, N.; Zhang, Q.; Wang, H.; Yang, X.; Jiang, Y.; Chen, R.; Wang, L. Comparative Evaluation of the Effect of Metformin and Insulin on Gut Microbiota and Metabolome Profiles of Type 2 Diabetic Rats Induced by the Combination of Streptozotocin and High-Fat Diet. Front. Pharmacol. 2021, 12, 794103. [Google Scholar] [CrossRef] [PubMed]
- Lkhagva, E.; Chung, H.-J.; Hong, J.; Tang, W.H.W.; Lee, S.-I.; Hong, S.-T.; Lee, S. The Regional Diversity of Gut Microbiome along the GI Tract of Male C57BL/6 Mice. BMC Microbiol. 2021, 21, 44. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Depommier, C.; Derrien, M.; Everard, A.; de Vos, W.M. Akkermansia Muciniphila: Paradigm for next-Generation Beneficial Microorganisms. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 625–637. [Google Scholar] [CrossRef] [PubMed]
- He, K.-Y.; Lei, X.-Y.; Wu, D.-H.; Zhang, L.; Li, J.-Q.; Li, Q.-T.; Yin, W.-T.; Zhao, Z.-L.; Liu, H.; Xiang, X.-Y.; et al. Akkermansia Muciniphila Protects the Intestine from Irradiation-Induced Injury by Secretion of Propionic Acid. Gut Microbes 2023, 15, 2293312. [Google Scholar] [CrossRef]
- Ruigrok, R.A.A.A.; Weersma, R.K.; Vich Vila, A. The Emerging Role of the Small Intestinal Microbiota in Human Health and Disease. Gut Microbes 2023, 15, 2201155. [Google Scholar] [CrossRef]
- Kastl, A.J.; Terry, N.A.; Wu, G.D.; Albenberg, L.G. The Structure and Function of the Human Small Intestinal Microbiota: Current Understanding and Future Directions. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 33–45. [Google Scholar] [CrossRef]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef] [PubMed]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef]
- Kameyama, K.; Itoh, K. Intestinal Colonization by a Lachnospiraceae Bacterium Contributes to the Development of Diabetes in Obese Mice. Microbes Environ. 2014, 29, 427–430. [Google Scholar] [CrossRef]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.-Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA Gene Sequencing for Species and Strain-Level Microbiome Analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liao, M.; Zhou, N.; Bao, L.; Ma, K.; Zheng, Z.; Wang, Y.; Liu, C.; Wang, W.; Wang, J.; et al. Parabacteroides Distasonis Alleviates Obesity and Metabolic Dysfunctions via Production of Succinate and Secondary Bile Acids. Cell Rep. 2019, 26, 222–235.e5. [Google Scholar] [CrossRef]
- Ashraf, R.; Shah, N.P. Immune System Stimulation by Probiotic Microorganisms. Crit. Rev. Food Sci. Nutr. 2014, 54, 938–956. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhao, L.; Wu, J.; Pan, Y.; Zhao, G.; Li, Z.; Zhang, L. The Effects of Lactobacillus Johnsonii on Diseases and Its Potential Applications. Microorganisms 2023, 11, 2580. [Google Scholar] [CrossRef]
- Mu, Q.; Tavella, V.J.; Luo, X.M. Role of Lactobacillus Reuteri in Human Health and Diseases. Front. Microbiol. 2018, 9, 757. [Google Scholar] [CrossRef]
- Al-Hadidi, A.; Navarro, J.; Goodman, S.D.; Bailey, M.T.; Besner, G.E. Lactobacillus Reuteri in Its Biofilm State Improves Protection from Experimental Necrotizing Enterocolitis. Nutrients 2021, 13, 918. [Google Scholar] [CrossRef]
- Mooranian, A.; Zamani, N.; Takechi, R.; Luna, G.; Mikov, M.; Goločorbin-Kon, S.; Kovacevic, B.; Arfuso, F.; Al-Salami, H. Modulatory Nano/Micro Effects of Diabetes Development on Pharmacology of Primary and Secondary Bile Acids Concentrations. CDR 2020, 16, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.Y.; Kelkar, Y.; Hadjipanayis, A.; Shipstone, A.; Wynn, T.A.; Hall, J.P. Metformin and 2-Deoxyglucose Collaboratively Suppress Human CD4+ T Cell Effector Functions and Activation-Induced Metabolic Reprogramming. J. Immunol. 2020, 205, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Gundpatil, D.B.; Somani, B.L.; Saha, T.K.; Bandyopadhyay, S.; Misra, P. Anticancer Effect of Dl-Glyceraldehyde and 2-Deoxyglucose in Ehrlich Ascites Carcinoma Bearing Mice and Their Effect on Liver, Kidney and Haematological Parameters. Indian J. Clin. Biochem. 2014, 29, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Lee, S.; Shiuchi, T.; Toda, C.; Kamijo, M.; Inagaki-Ohara, K.; Okamoto, S.; Minokoshi, Y. An Enzymatic Photometric Assay for 2-Deoxyglucose Uptake in Insulin-Responsive Tissues and 3T3-L1 Adipocytes. Anal. Biochem. 2011, 412, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Han, S.; Shi, M.; Ge, J.; Yu, S.; Adam, J.; Adamski, J.; Scheerer, M.F.; Neschen, S.; de Angelis, M.H.; et al. Metabolic Effects of SGLT2i and Metformin on 3-Hydroxybutyric Acid and Lactate in Db/Db Mice. Int. J. Biol. Macromol. 2024, 265, 130962. [Google Scholar] [CrossRef] [PubMed]
- Hasanpour, M.; Iranshahy, M.; Iranshahi, M. The Application of Metabolomics in Investigating Anti-Diabetic Activity of Medicinal Plants. Biomed. Pharmacother. 2020, 128, 110263. [Google Scholar] [CrossRef] [PubMed]
- Wilmanns, J.C.; Pandey, R.; Hon, O.; Chandran, A.; Schilling, J.M.; Forte, E.; Wu, Q.; Cagnone, G.; Bais, P.; Philip, V.; et al. Metformin Intervention Prevents Cardiac Dysfunction in a Murine Model of Adult Congenital Heart Disease. Mol. Metab. 2019, 20, 102–114. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Zhao, S.; Liu, X.; Dong, S.; Lv, J.; Liu, D.; Wang, J.; Meng, Z. ReSeqTools: An Integrated Toolkit for Large-Scale next-Generation Sequencing Based Resequencing Analysis. Genet. Mol. Res. 2013, 12, 6275–6283. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly Accurate OTU Sequences from Microbial Amplicon Reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Gao, X.; Lin, H.; Revanna, K.; Dong, Q. A Bayesian Taxonomic Classification Method for 16S rRNA Gene Sequences with Improved Species-Level Accuracy. BMC Bioinform. 2017, 18, 247. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, K.I.; Pratt, B.S.; Shulman, N.; Tamura, K.; MacCoss, M.J.; MacLean, B.X.; Baker, E.S. Utilizing Skyline to Analyze Lipidomics Data Containing Liquid Chromatography, Ion Mobility Spectrometry and Mass Spectrometry Dimensions. Nat. Protoc. 2022, 17, 2415–2430. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, M.; Jia, X.; Ren, L.; Chen, S.; Wang, W.; Wang, J.; Cong, B. Region-Specific Effects of Metformin on Gut Microbiome and Metabolome in High-Fat Diet-Induced Type 2 Diabetes Mouse Model. Int. J. Mol. Sci. 2024, 25, 7250. https://doi.org/10.3390/ijms25137250
Cheng M, Jia X, Ren L, Chen S, Wang W, Wang J, Cong B. Region-Specific Effects of Metformin on Gut Microbiome and Metabolome in High-Fat Diet-Induced Type 2 Diabetes Mouse Model. International Journal of Molecular Sciences. 2024; 25(13):7250. https://doi.org/10.3390/ijms25137250
Chicago/Turabian StyleCheng, Meihui, Xianxian Jia, Lili Ren, Siqian Chen, Wei Wang, Jianwei Wang, and Bin Cong. 2024. "Region-Specific Effects of Metformin on Gut Microbiome and Metabolome in High-Fat Diet-Induced Type 2 Diabetes Mouse Model" International Journal of Molecular Sciences 25, no. 13: 7250. https://doi.org/10.3390/ijms25137250