
Genomic and Epigenomic Biomarkers of Immune Checkpoint Immunotherapy Response in Melanoma: Current and Future Perspectives

Abstract
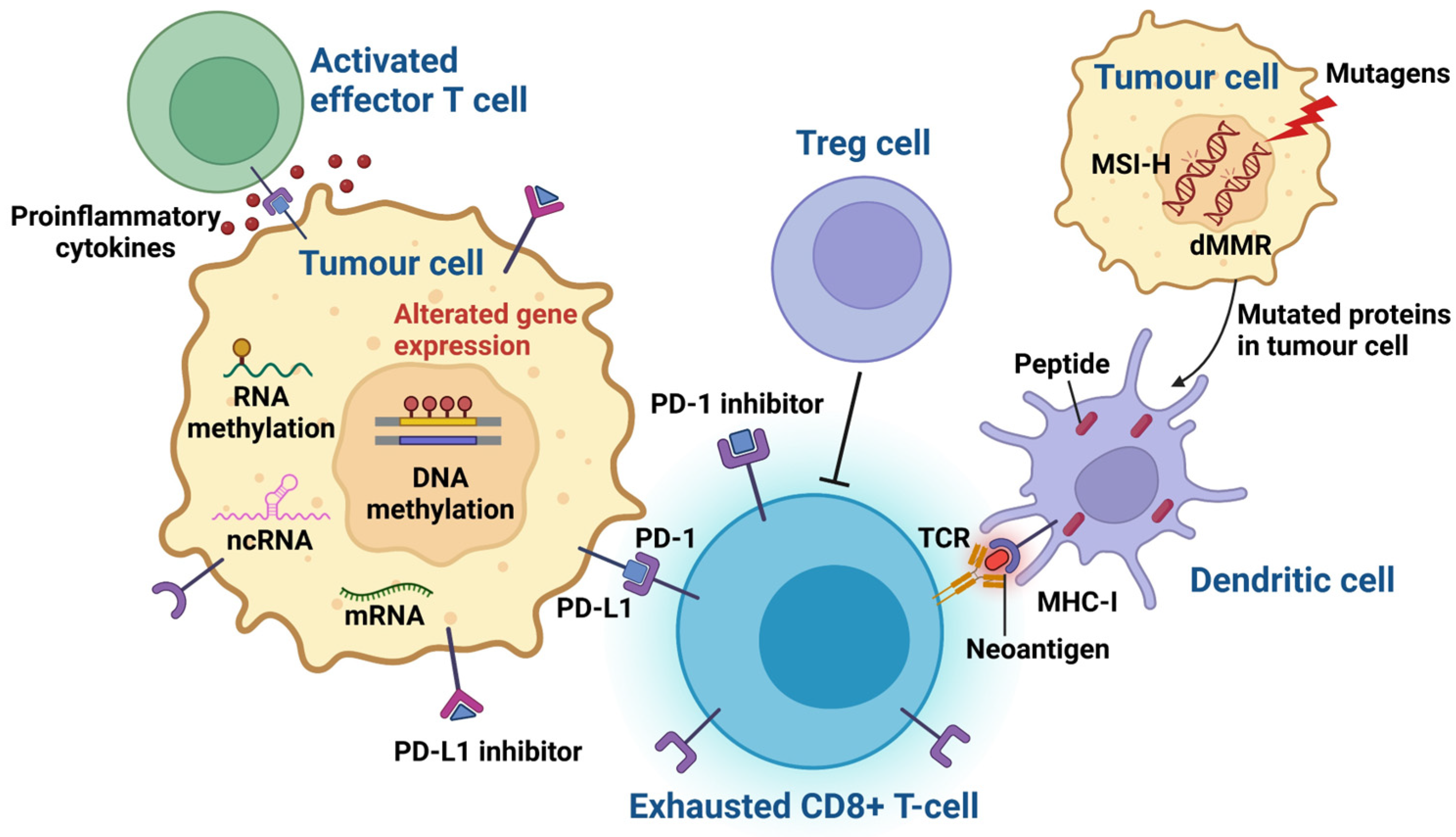
:1. Introduction
2. Genomic Biomarkers of ICI Treatment Response in Melanoma Patients
2.1. Tumour Mutational Burden (TMB)
2.2. Neoantigen Expression
2.3. Mismatch Repair Deficiency (dMMR) and High Microsatellite Instability (MSI-H)
3. Epigenomic Biomarkers with the Potential to Predict ICI Therapy Response
4. Understanding the Tumour Microenvironment from an Epigenetics Perspective
5. Currently Studied Epigenomic Biomarkers of ICI Response
5.1. DNA Methylation and Epigenomic Signatures
5.2. Non-Coding RNAs
5.3. RNA Methylation
6. Limitations and Future Directions
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Passarelli, A.; Mannavola, F.; Stucci, L.S.; Tucci, M.; Silvestris, F. Immune system and melanoma biology: A balance between immunosurveillance and immune escape. Oncotarget 2017, 8, 106132–106142. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [PubMed]
- Grigg, C.; Rizvi, N.A. PD-L1 biomarker testing for non-small cell lung cancer: Truth or fiction? J. Immunother. Cancer 2016, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Marconcini, R.; Spagnolo, F.; Stucci, L.S.; Ribero, S.; Marra, E.; Rosa, F.; Picasso, V.; Di Guardo, L.; Cimminiello, C.; Cavalieri, S.; et al. Current status and perspectives in immunotherapy for metastatic melanoma. Oncotarget 2018, 9, 12452–12470. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Rodger, E.J.; Ahn, A.; Stockwell, P.A.; Parry, M.; Motwani, J.; Gallagher, S.J.; Shklovskaya, E.; Tiffen, J.; Eccles, M.R.; et al. Marked Global DNA Hypomethylation Is Associated with Constitutive PD-L1 Expression in Melanoma. iScience 2018, 4, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Hossain, S.M.; Lynch-Sutherland, C.F.; Chatterjee, A.; Macaulay, E.C.; Eccles, M.R. Can Immune Suppression and Epigenome Regulation in Placenta Offer Novel Insights into Cancer Immune Evasion and Immunotherapy Resistance? Epigenomes 2021, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Koppolu, V.; Rekha Vasigala, V.K. Checkpoint immunotherapy by nivolumab for treatment of metastatic melanoma. J. Cancer Res. Ther. 2018, 14, 1167–1175. [Google Scholar] [CrossRef]
- Ventola, C.L. Cancer Immunotherapy, Part 3: Challenges and Future Trends. P T 2017, 42, 514–521. [Google Scholar]
- Jessurun, C.A.C.; Vos, J.A.M.; Limpens, J.; Luiten, R.M. Biomarkers for Response of Melanoma Patients to Immune Checkpoint Inhibitors: A Systematic Review. Front. Oncol. 2017, 7, 233. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef] [PubMed]
- Ankeny, J.S.; Labadie, B.; Luke, J.; Hsueh, E.; Messina, J.; Zager, J.S. Review of diagnostic, prognostic, and predictive biomarkers in melanoma. Clin. Exp. Metastasis 2018, 35, 487–493. [Google Scholar] [CrossRef]
- Maher, N.G.; Vergara, I.A.; Long, G.V.; Scolyer, R.A. Prognostic and predictive biomarkers in melanoma. Pathology 2024, 56, 259–273. [Google Scholar] [CrossRef]
- Paver, E.C.; Cooper, W.A.; Colebatch, A.J.; Ferguson, P.M.; Hill, S.K.; Lum, T.; Shin, J.S.; O’Toole, S.; Anderson, L.; Scolyer, R.A.; et al. Programmed death ligand-1 (PD-L1) as a predictive marker for immunotherapy in solid tumours: A guide to immunohistochemistry implementation and interpretation. Pathology 2021, 53, 141–156. [Google Scholar] [CrossRef]
- Lantuejoul, S.; Sound-Tsao, M.; Cooper, W.A.; Girard, N.; Hirsch, F.R.; Roden, A.C.; Lopez-Rios, F.; Jain, D.; Chou, T.Y.; Motoi, N.; et al. PD-L1 Testing for Lung Cancer in 2019: Perspective From the IASLC Pathology Committee. J. Thorac. Oncol. 2020, 15, 499–519. [Google Scholar] [CrossRef]
- Cha, J.H.; Chan, L.C.; Li, C.W.; Hsu, J.L.; Hung, M.C. Mechanisms Controlling PD-L1 Expression in Cancer. Mol. Cell 2019, 76, 359–370. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in cancer immunotherapy: Clinical implications and future considerations. Hum. Vaccines Immunother. 2019, 15, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]
- Maleki Vareki, S.; Garrigos, C.; Duran, I. Biomarkers of response to PD-1/PD-L1 inhibition. Crit. Rev. Oncol./Hematol. 2017, 116, 116–124. [Google Scholar] [CrossRef]
- Madore, J.; Vilain, R.E.; Menzies, A.M.; Kakavand, H.; Wilmott, J.S.; Hyman, J.; Yearley, J.H.; Kefford, R.F.; Thompson, J.F.; Long, G.V.; et al. PD-L1 expression in melanoma shows marked heterogeneity within and between patients: Implications for anti-PD-1/PD-L1 clinical trials. Pigment. Cell Melanoma Res. 2015, 28, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Kamel, H.F.M.; Al-Amodi, H. Exploitation of Gene Expression and Cancer Biomarkers in Paving the Path to Era of Personalized Medicine. Genom. Proteom. Bioinform. 2017, 15, 220–235. [Google Scholar] [CrossRef] [PubMed]
- Goossens, N.; Nakagawa, S.; Sun, X.; Hoshida, Y. Cancer biomarker discovery and validation. Transl. Cancer Res. 2015, 4, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Roccuzzo, G.; Bongiovanni, E.; Tonella, L.; Pala, V.; Marchisio, S.; Ricci, A.; Senetta, R.; Bertero, L.; Ribero, S.; Berrino, E.; et al. Emerging prognostic biomarkers in advanced cutaneous melanoma: A literature update. Expert. Rev. Mol. Diagn. 2024, 24, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Fumet, J.D.; Truntzer, C.; Yarchoan, M.; Ghiringhelli, F. Tumour mutational burden as a biomarker for immunotherapy: Current data and emerging concepts. Eur. J. Cancer 2020, 131, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Maleki Vareki, S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer 2018, 6, 157. [Google Scholar] [CrossRef]
- Gong, L.; He, R.; Xu, Y.; Luo, T.; Jin, K.; Yuan, W.; Zheng, Z.; Liu, L.; Liang, Z.; Li, A.; et al. Neoantigen load as a prognostic and predictive marker for stage II/III non-small cell lung cancer in Chinese patients. Thorac. Cancer 2021, 12, 2170–2181. [Google Scholar] [CrossRef]
- Zou, X.L.; Li, X.B.; Ke, H.; Zhang, G.Y.; Tang, Q.; Yuan, J.; Zhou, C.J.; Zhang, J.L.; Zhang, R.; Chen, W.Y. Prognostic Value of Neoantigen Load in Immune Checkpoint Inhibitor Therapy for Cancer. Front. Immunol. 2021, 12, 689076. [Google Scholar] [CrossRef]
- Maio, M.; Ascierto, P.A.; Manzyuk, L.; Motola-Kuba, D.; Penel, N.; Cassier, P.A.; Bariani, G.M.; De Jesus Acosta, A.; Doi, T.; Longo, F.; et al. Pembrolizumab in microsatellite instability high or mismatch repair deficient cancers: Updated analysis from the phase II KEYNOTE-158 study. Ann. Oncol. 2022, 33, 929–938. [Google Scholar] [CrossRef]
- Xue, G.; Cui, Z.J.; Zhou, X.H.; Zhu, Y.X.; Chen, Y.; Liang, F.J.; Tang, D.N.; Huang, B.Y.; Zhang, H.Y.; Hu, Z.H.; et al. DNA Methylation Biomarkers Predict Objective Responses to PD-1/PD-L1 Inhibition Blockade. Front. Genet. 2019, 10, 724. [Google Scholar] [CrossRef] [PubMed]
- Kugel, J.F.; Goodrich, J.A. Non-coding RNAs: Key regulators of mammalian transcription. Trends Biochem. Sci. 2012, 37, 144–151. [Google Scholar] [CrossRef]
- Yang, X.; Liu, M.; Li, M.; Zhang, S.; Hiju, H.; Sun, J.; Mao, Z.; Zheng, M.; Feng, B. Epigenetic modulations of noncoding RNA: A novel dimension of Cancer biology. Mol. Cancer 2020, 19, 64. [Google Scholar] [CrossRef]
- García-Giménez, J.L.; Ushijima, T.; Tollefsbol, T.O. Chapter 1—Epigenetic Biomarkers: New Findings, Perspectives, and Future Directions in Diagnostics. In Epigenetic Biomarkers and Diagnostics; Academic Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Chen, X.Y.; Zhang, J.; Zhu, J.S. The role of m(6)A RNA methylation in human cancer. Mol. Cancer 2019, 18, 103. [Google Scholar] [CrossRef] [PubMed]
- Schwitalle, Y.; Kloor, M.; Eiermann, S.; Linnebacher, M.; Kienle, P.; Knaebel, H.P.; Tariverdian, M.; Benner, A.; von Knebel Doeberitz, M. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology 2008, 134, 988–997. [Google Scholar] [CrossRef]
- Yamashita, H.; Nakayama, K.; Ishikawa, M.; Nakamura, K.; Ishibashi, T.; Sanuki, K.; Ono, R.; Sasamori, H.; Minamoto, T.; Iida, K.; et al. Microsatellite instability is a biomarker for immune checkpoint inhibitors in endometrial cancer. Oncotarget 2018, 9, 5652–5664. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Miao, D.; Margolis, C.A.; Gao, W.; Voss, M.H.; Li, W.; Martini, D.J.; Norton, C.; Bosse, D.; Wankowicz, S.M.; Cullen, D.; et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018, 359, 801–806. [Google Scholar] [CrossRef]
- Miao, D.; Margolis, C.A.; Vokes, N.I.; Liu, D.; Taylor-Weiner, A.; Wankowicz, S.M.; Adeegbe, D.; Keliher, D.; Schilling, B.; Tracy, A.; et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat. Genet. 2018, 50, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e399. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Chowell, D.; Krishna, C.; Pierini, F.; Makarov, V.; Rizvi, N.A.; Kuo, F.; Morris, L.G.T.; Riaz, N.; Lenz, T.L.; Chan, T.A. Evolutionary divergence of HLA class I genotype impacts efficacy of cancer immunotherapy. Nat. Med. 2019, 25, 1715–1720. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.D.; Warren, R.L.; Gibb, E.A.; Martin, S.D.; Spinelli, J.J.; Nelson, B.H.; Holt, R.A. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014, 24, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Mauriello, A.; Zeuli, R.; Cavalluzzo, B.; Petrizzo, A.; Tornesello, M.L.; Buonaguro, F.M.; Ceccarelli, M.; Tagliamonte, M.; Buonaguro, L. High Somatic Mutation and Neoantigen Burden Do Not Correlate with Decreased Progression-Free Survival in HCC Patients not Undergoing Immunotherapy. Cancers 2019, 11, 1824. [Google Scholar] [CrossRef] [PubMed]
- Karpanen, T.; Olweus, J. The Potential of Donor T-Cell Repertoires in Neoantigen-Targeted Cancer Immunotherapy. Front. Immunol. 2017, 8, 1718. [Google Scholar] [CrossRef]
- Koster, B.D.; de Gruijl, T.D.; van den Eertwegh, A.J. Recent developments and future challenges in immune checkpoint inhibitory cancer treatment. Curr. Opin. Oncol. 2015, 27, 482–488. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Maeurer, M.J.; Gollin, S.M.; Storkus, W.J.; Swaney, W.; Karbach, J.; Martin, D.; Castelli, C.; Salter, R.; Knuth, A.; Lotze, M.T. Tumor escape from immune recognition: Loss of HLA-A2 melanoma cell surface expression is associated with a complex rearrangement of the short arm of chromosome 6. Clin. Cancer Res. 1996, 2, 641–652. [Google Scholar]
- Gettinger, S.N.; Horn, L.; Gandhi, L.; Spigel, D.R.; Antonia, S.J.; Rizvi, N.A.; Powderly, J.D.; Heist, R.S.; Carvajal, R.D.; Jackman, D.M.; et al. Overall Survival and Long-Term Safety of Nivolumab (Anti-Programmed Death 1 Antibody, BMS-936558, ONO-4538) in Patients With Previously Treated Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2015, 33, 2004–2012. [Google Scholar] [CrossRef]
- Trabucco, S.E.; Gowen, K.; Maund, S.L.; Sanford, E.; Fabrizio, D.A.; Hall, M.J.; Yakirevich, E.; Gregg, J.P.; Stephens, P.J.; Frampton, G.M.; et al. A Novel Next-Generation Sequencing Approach to Detecting Microsatellite Instability and Pan-Tumor Characterization of 1000 Microsatellite Instability-High Cases in 67,000 Patient Samples. J. Mol. Diagn. 2019, 21, 1053–1066. [Google Scholar] [CrossRef]
- Modrich, P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006, 281, 30305–30309. [Google Scholar] [CrossRef]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef]
- Iyer, R.R.; Pluciennik, A.; Burdett, V.; Modrich, P.L. DNA mismatch repair: Functions and mechanisms. Chem. Rev. 2006, 106, 302–323. [Google Scholar] [CrossRef] [PubMed]
- Negureanu, L.; Salsbury, F.R., Jr. The molecular origin of the MMR-dependent apoptosis pathway from dynamics analysis of MutSalpha-DNA complexes. J. Biomol. Struct. Dyn. 2012, 30, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Jascur, T.; Lanspa, S.; Boland, C.R. Making sense of missense in Lynch syndrome: The clinical perspective. Cancer Prev. Res. 2010, 3, 1371–1374. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef]
- Beggs, A.D.; Domingo, E.; Abulafi, M.; Hodgson, S.V.; Tomlinson, I.P. A study of genomic instability in early preneoplastic colonic lesions. Oncogene 2013, 32, 5333–5337. [Google Scholar] [CrossRef]
- Funkhouser, W.K., Jr.; Lubin, I.M.; Monzon, F.A.; Zehnbauer, B.A.; Evans, J.P.; Ogino, S.; Nowak, J.A. Relevance, pathogenesis, and testing algorithm for mismatch repair-defective colorectal carcinomas: A report of the association for molecular pathology. J. Mol. Diagn. 2012, 14, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Timmermann, B.; Kerick, M.; Roehr, C.; Fischer, A.; Isau, M.; Boerno, S.T.; Wunderlich, A.; Barmeyer, C.; Seemann, P.; Koenig, J.; et al. Somatic mutation profiles of MSI and MSS colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PLoS ONE 2010, 5, e15661. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.; Yamane, K. DNA mismatch repair: Molecular mechanism, cancer, and ageing. Mech. Ageing Dev. 2008, 129, 391–407. [Google Scholar] [CrossRef] [PubMed]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Saeterdal, I.; Bjørheim, J.; Lislerud, K.; Gjertsen, M.K.; Bukholm, I.K.; Olsen, O.C.; Nesland, J.M.; Eriksen, J.A.; Møller, M.; Lindblom, A.; et al. Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 13255–13260. [Google Scholar] [CrossRef] [PubMed]
- Boissière-Michot, F.; Lazennec, G.; Frugier, H.; Jarlier, M.; Roca, L.; Duffour, J.; Du Paty, E.; Laune, D.; Blanchard, F.; Le Pessot, F.; et al. Characterization of an adaptive immune response in microsatellite-instable colorectal cancer. Oncoimmunology 2014, 3, e29256. [Google Scholar] [CrossRef]
- Cicek, M.S.; Lindor, N.M.; Gallinger, S.; Bapat, B.; Hopper, J.L.; Jenkins, M.A.; Young, J.; Buchanan, D.; Walsh, M.D.; Le Marchand, L.; et al. Quality assessment and correlation of microsatellite instability and immunohistochemical markers among population- and clinic-based colorectal tumors results from the Colon Cancer Family Registry. J. Mol. Diagn. 2011, 13, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Huang, B.; Cao, J.; Yan, Q.; Yin, M. Editorial: Epigenetic Regulation and Tumor Immunotherapy. Front. Oncol. 2022, 12, 893157. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Villanueva, L.; Álvarez-Errico, D.; Esteller, M. The Contribution of Epigenetics to Cancer Immunotherapy. Trends Immunol. 2020, 41, 676–691. [Google Scholar] [CrossRef]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157.e119. [Google Scholar] [CrossRef]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef]
- Crompton, J.G.; Narayanan, M.; Cuddapah, S.; Roychoudhuri, R.; Ji, Y.; Yang, W.; Patel, S.J.; Sukumar, M.; Palmer, D.C.; Peng, W.; et al. Lineage relationship of CD8(+) T cell subsets is revealed by progressive changes in the epigenetic landscape. Cell. Mol. Immunol. 2016, 13, 502–513. [Google Scholar] [CrossRef]
- Hackl, H.; Charoentong, P.; Finotello, F.; Trajanoski, Z. Computational genomics tools for dissecting tumour-immune cell interactions. Nat. Rev. Genet. 2016, 17, 441–458. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef]
- Scharer, C.D.; Barwick, B.G.; Youngblood, B.A.; Ahmed, R.; Boss, J.M. Global DNA methylation remodeling accompanies CD8 T cell effector function. J. Immunol. 2013, 191, 3419–3429. [Google Scholar] [CrossRef]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef]
- Lee, H.; Ferguson, A.L.; Quek, C.; Vergara, I.A.; Pires daSilva, I.; Allen, R.; Gide, T.N.; Conway, J.W.; Koufariotis, L.T.; Hayward, N.K.; et al. Intratumoral CD16+ Macrophages Are Associated with Clinical Outcomes of Patients with Metastatic Melanoma Treated with Combination Anti-PD-1 and Anti-CTLA-4 Therapy. Clin. Cancer Res. 2023, 29, 2513–2524. [Google Scholar] [CrossRef]
- House, I.G.; Savas, P.; Lai, J.; Chen, A.X.Y.; Oliver, A.J.; Teo, Z.L.; Todd, K.L.; Henderson, M.A.; Giuffrida, L.; Petley, E.V.; et al. Macrophage-Derived CXCL9 and CXCL10 Are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade. Clin. Cancer Res. 2020, 26, 487–504. [Google Scholar] [CrossRef]
- Antoranz, A.; Van Herck, Y.; Bolognesi, M.M.; Lynch, S.M.; Rahman, A.; Gallagher, W.M.; Boecxstaens, V.; Marine, J.C.; Cattoretti, G.; van den Oord, J.J.; et al. Mapping the Immune Landscape in Metastatic Melanoma Reveals Localized Cell-Cell Interactions That Predict Immunotherapy Response. Cancer Res. 2022, 82, 3275–3290. [Google Scholar] [CrossRef]
- Hossain, S.M.; Gimenez, G.; Stockwell, P.A.; Tsai, P.; Print, C.G.; Rys, J.; Cybulska-Stopa, B.; Ratajska, M.; Harazin-Lechowska, A.; Almomani, S.; et al. Innate immune checkpoint inhibitor resistance is associated with melanoma sub-types exhibiting invasive and de-differentiated gene expression signatures. Front. Immunol. 2022, 13, 955063. [Google Scholar] [CrossRef]
- Yang, X.; Wang, X.; Liu, D.; Yu, L.; Xue, B.; Shi, H. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol. Endocrinol. 2014, 28, 565–574. [Google Scholar] [CrossRef]
- Ishii, M.; Wen, H.; Corsa, C.A.; Liu, T.; Coelho, A.L.; Allen, R.M.; Carson, W.F.t.; Cavassani, K.A.; Li, X.; Lukacs, N.W.; et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 2009, 114, 3244–3254. [Google Scholar] [CrossRef]
- Villagra, A.; Cheng, F.; Wang, H.W.; Suarez, I.; Glozak, M.; Maurin, M.; Nguyen, D.; Wright, K.L.; Atadja, P.W.; Bhalla, K.; et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 2009, 10, 92–100. [Google Scholar] [CrossRef]
- Sahakian, E.; Powers, J.J.; Chen, J.; Deng, S.L.; Cheng, F.; Distler, A.; Woods, D.M.; Rock-Klotz, J.; Sodre, A.L.; Youn, J.I.; et al. Histone deacetylase 11: A novel epigenetic regulator of myeloid derived suppressor cell expansion and function. Mol. Immunol. 2015, 63, 579–585. [Google Scholar] [CrossRef]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef]
- Li, B.; Wang, Z.; Wu, H.; Xue, M.; Lin, P.; Wang, S.; Lin, N.; Huang, X.; Pan, W.; Liu, M.; et al. Epigenetic Regulation of CXCL12 Plays a Critical Role in Mediating Tumor Progression and the Immune Response In Osteosarcoma. Cancer Res. 2018, 78, 3938–3953. [Google Scholar] [CrossRef]
- Papaiz, D.D.; Rius, F.E.; Ayub, A.L.P.; Origassa, C.S.; Gujar, H.; Pessoa, D.O.; Reis, E.M.; Nsengimana, J.; Newton-Bishop, J.; Mason, C.E.; et al. Genes regulated by DNA methylation are involved in distinct phenotypes during melanoma progression and are prognostic factors for patients. Mol. Oncol. 2022, 16, 1913–1930. [Google Scholar] [CrossRef]
- Micevic, G.; Theodosakis, N.; Bosenberg, M. Aberrant DNA methylation in melanoma: Biomarker and therapeutic opportunities. Clin. Epigenetics 2017, 9, 34. [Google Scholar] [CrossRef]
- Huan, T.; Joehanes, R.; Song, C.; Peng, F.; Guo, Y.; Mendelson, M.; Yao, C.; Liu, C.; Ma, J.; Richard, M.; et al. Genome-wide identification of DNA methylation QTLs in whole blood highlights pathways for cardiovascular disease. Nat. Commun. 2019, 10, 4267. [Google Scholar] [CrossRef]
- Schinke, C.; Mo, Y.; Yu, Y.; Amiri, K.; Sosman, J.; Greally, J.; Verma, A. Aberrant DNA methylation in malignant melanoma. Melanoma Res. 2010, 20, 253–265. [Google Scholar] [CrossRef]
- Wajed, S.A.; Laird, P.W.; DeMeester, T.R. DNA methylation: An alternative pathway to cancer. Ann. Surg. 2001, 234, 10–20. [Google Scholar] [CrossRef]
- Zhao, R.; Choi, B.Y.; Lee, M.H.; Bode, A.M.; Dong, Z. Implications of Genetic and Epigenetic Alterations of CDKN2A (p16(INK4a)) in Cancer. EBioMedicine 2016, 8, 30–39. [Google Scholar] [CrossRef]
- Torano, E.G.; Petrus, S.; Fernandez, A.F.; Fraga, M.F. Global DNA hypomethylation in cancer: Review of validated methods and clinical significance. Clin. Chem. Lab. Med. 2012, 50, 1733–1742. [Google Scholar] [CrossRef]
- Filipski, K.; Scherer, M.; Zeiner, K.N.; Bucher, A.; Kleemann, J.; Jurmeister, P.; Hartung, T.I.; Meissner, M.; Plate, K.H.; Fenton, T.R.; et al. DNA methylation-based prediction of response to immune checkpoint inhibition in metastatic melanoma. J. Immunother. Cancer 2021, 9, e002226. [Google Scholar] [CrossRef]
- Ressler, J.M.; Tomasich, E.; Hatziioannou, T.; Ringl, H.; Heller, G.; Silmbrod, R.; Gottmann, L.; Starzer, A.M.; Zila, N.; Tschandl, P.; et al. DNA Methylation Signatures Correlate with Response to Immune Checkpoint Inhibitors in Metastatic Melanoma. Target. Oncol. 2024, 19, 263–275. [Google Scholar] [CrossRef]
- Goltz, D.; Gevensleben, H.; Vogt, T.J.; Dietrich, J.; Golletz, C.; Bootz, F.; Kristiansen, G.; Landsberg, J.; Dietrich, D. CTLA4 methylation predicts response to anti-PD-1 and anti-CTLA-4 immunotherapy in melanoma patients. JCI Insight 2018, 3, e96793. [Google Scholar] [CrossRef]
- Huang, D.; Chen, J.; Yang, L.; Ouyang, Q.; Li, J.; Lao, L.; Zhao, J.; Liu, J.; Lu, Y.; Xing, Y.; et al. NKILA lncRNA promotes tumor immune evasion by sensitizing T cells to activation-induced cell death. Nat. Immunol. 2018, 19, 1112–1125. [Google Scholar] [CrossRef]
- Chen, F.; Chen, J.; Yang, L.; Liu, J.; Zhang, X.; Zhang, Y.; Tu, Q.; Yin, D.; Lin, D.; Wong, P.P.; et al. Extracellular vesicle-packaged HIF-1alpha-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat. Cell Biol. 2019, 21, 498–510. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, W.; Li, A.; Chen, Y.; Ou, Q.; He, Z.; Zhang, Y.; Liu, R.; Yao, H.; Song, E. Association of Long Noncoding RNA Biomarkers With Clinical Immune Subtype and Prediction of Immunotherapy Response in Patients With Cancer. JAMA Netw. Open 2020, 3, e202149. [Google Scholar] [CrossRef]
- Zhang, M.; Song, J.; Yuan, W.; Zhang, W.; Sun, Z. Roles of RNA Methylation on Tumor Immunity and Clinical Implications. Front. Immunol. 2021, 12, 641507. [Google Scholar] [CrossRef]
- Tong, J.; Cao, G.; Zhang, T.; Sefik, E.; Amezcua Vesely, M.C.; Broughton, J.P.; Zhu, S.; Li, H.; Li, B.; Chen, L.; et al. m6A mRNA methylation sustains Treg suppressive functions. Cell Res. 2018, 28, 253–256. [Google Scholar] [CrossRef]
- Han, D.; Liu, J.; Chen, C.; Dong, L.; Liu, Y.; Chang, R.; Huang, X.; Liu, Y.; Wang, J.; Dougherty, U.; et al. Anti-tumour immunity controlled through mRNA m6A methylation and YTHDF1 in dendritic cells. Nature 2019, 566, 270–274. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, X.; Ma, C.; Hu, J. m6A Methylation Regulators Are Predictive Biomarkers for Tumour Metastasis in Prostate Cancer. Cancers 2022, 14, 4035. [Google Scholar] [CrossRef]
- Zhang, C.; Samanta, D.; Lu, H.; Bullen, J.W.; Zhang, H.; Chen, I.; He, X.; Semenza, G.L. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m⁶A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. USA 2016, 113, E2047–E2056. [Google Scholar] [CrossRef]
- Giancotti, F.G. Mechanisms governing metastatic dormancy and reactivation. Cell 2013, 155, 750–764. [Google Scholar] [CrossRef]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef]
- Milotti, E.; Fredrich, T.; Chignola, R.; Rieger, H. Oxygen in the Tumor Microenvironment: Mathematical and Numerical Modeling. Adv. Exp. Med. Biol. 2020, 1259, 53–76. [Google Scholar] [CrossRef]
- Reinfeld, B.I.; Madden, M.Z.; Wolf, M.M.; Chytil, A.; Bader, J.E.; Patterson, A.R.; Sugiura, A.; Cohen, A.S.; Ali, A.; Do, B.T.; et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 2021, 593, 282–288. [Google Scholar] [CrossRef]
- Slominski, A.; Zmijewski, M.A.; Pawelek, J. L-tyrosine and L-dihydroxyphenylalanine as hormone-like regulators of melanocyte functions. Pigment. Cell Melanoma Res. 2012, 25, 14–27. [Google Scholar] [CrossRef]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar] [CrossRef]
- Cabaço, L.C.; Tomás, A.; Pojo, M.; Barralm, D.C. The Dark Side of Melanin Secretion in Cutaneous Melanoma Aggressiveness. Front. Oncol. 2022, 12, 887366. [Google Scholar] [CrossRef]
- Slominski, R.M.; Raman, C.; Chen, J.Y.; Slominski, A.T. How cancer hijacks the body’s homeostasis through the neuroendocrine system. Trends Neurosci. 2023, 46, 263–275. [Google Scholar] [CrossRef]
- Dall’Olmo, L.; Papa, N.; Surdo, N.C.; Marigo, I.; Mocellin, S. Alpha-melanocyte stimulating hormone (α-MSH): Biology, clinical relevance and implication in melanoma. J. Transl. Med. 2023, 21, 562. [Google Scholar] [CrossRef]
- Slominski, R.M.; Chen, J.Y.; Raman, C.; Slominski, A.T. Photo-neuro-immuno-endocrinology: How the ultraviolet radiation regulates the body, brain, and immune system. Proc. Natl. Acad. Sci. USA 2024, 121, e2308374121. [Google Scholar] [CrossRef]
- Niebel, D.; Fröhlich, A.; Zarbl, R.; Fietz, S.; de Vos, L.; Vogt, T.J.; Dietrich, J.; Sirokay, J.; Kuster, P.; Saavedra, G.; et al. DNA methylation regulates TIGIT expression within the melanoma microenvironment, is prognostic for overall survival, and predicts progression-free survival in patients treated with anti-PD-1 immunotherapy. Clin. Epigenetics 2022, 14, 50. [Google Scholar] [CrossRef]
- Xu, Y.; Li, P.; Liu, Y.; Xin, D.; Lei, W.; Liang, A.; Han, W.; Qian, W. Epi-immunotherapy for cancers: Rationales of epi-drugs in combination with immunotherapy and advances in clinical trials. Cancer Commun. 2022, 42, 493–516. [Google Scholar] [CrossRef]
- Hogg, S.J.; Beavis, P.A.; Dawson, M.A.; Johnstone, R.W. Targeting the epigenetic regulation of antitumour immunity. Nat. Rev. Drug Discov. 2020, 19, 776–800. [Google Scholar] [CrossRef]

{kind=link}
| Biomarkers | Mechanistic Insights | Ref. |
|---|---|---|
| Genomic Biomarkers | ||
| PD-L1 expression in IHC | Epithelial cells can be induced to express PD-L1 in response to inflammatory cytokines, such as interferon-gamma, thus protecting these cells at sites of immune activation. PD-L1 may be expressed on tumour cells as well as inflammatory cells. The binding of PD-L1 with PD-1 or CD80 downregulates the response of activated T cells by inhibiting T-cell proliferation, cytokine production, and cytolytic activity, leading to the functional inactivation or exhaustion of T cells. Higher PD-L1 expression is often linked to better responses to ICI therapy. However, a lack of PD-L1 expression does not necessarily exclude the possibility of a response. Thus, the effectiveness of IHC detection of PD-L1, as a predictive biomarker in melanoma, is limited. | [15,16] |
| TMB | The evaluation of TMB is based on the hypothesis that a high number of mutations in exonic regions will lead to an increase in neoantigen production, which could then be recognized by CD8+ T cells, resulting in improved immune responses. Several variables may affect TMB determination: the depth of sequencing, length of sequencing reads, type of fixative agent, and fixation time, the latter of which influences the degree of formaldehyde-fixed, paraffin-embedded (FFPE), deamination-induced artifacts, all of which impact the analysis of TMB. In addition, a low tumour purity resulting from sampling errors may lead to reduced TMB assay sensitivity. | [26,27] |
| Neoantigen | Tumour types with a high TMB are theoretically often associated with a high neoantigen load. This is because a high TMB enhances the formation and presentation of immune neoantigens, leading to effective anti-tumour immune responses. It is speculated that tumours with a higher mutation burden possess more tumour-specific neoantigens, which in turn stimulates an increase in TILs due to the overexpression of immune checkpoint modulators such as PD-1 or PD-L1. However, TMB is not equivalent to a neoantigen load. One study found that half of oncogenic mutations did not result in neoantigens, indicating that TMB alone is not a reliable surrogate marker of immunogenic neoantigens. | [28,29,30] |
| dMMR and MSI-H | Tumours with mismatch repair deficiency (dMMR), either due to an inherited mutation or sporadic mutation, have a defect in one of the MMR genes (MLH1, PMS2, MSH2, or MSH6), resulting in the failure to repair errors in DNA replication. These errors are particularly prevalent in regions of repetitive DNA sequences known as microsatellites, resulting in high levels of microsatellite instability (MSI-H). | [31] |
Epigenomic Biomarkers | ||
| DNA methylation | Aberrant DNA methylation can alter the chromatin structure and gene transcription without altering the DNA sequence. Recent work revealed that DNA methylation affects tumourigenesis by regulating the tumour microenvironment. | [32] |
| Non-coding RNAs | Non-coding RNAs (ncRNAs) are involved in the regulation of the transcriptional activities of single genes, transcriptional programs, as well as the cell cycle, apoptosis, and differentiation. Malfunctions of ncRNAs could be involved in cancer progression, tumour growth, metastasis, and resistance to therapy by controlling the downregulation or upregulation of numerous genes. In general, ncRNAs are frequently altered in cancer tissues and are involved in innate and adaptive immunity in cancer. | [33,34,35] |
| RNA methylation | RNA methylation is a biologically reversible process and has been found to occur frequently in the mRNAs responsible for immune regulation. Moreover, RNA methylation influences immunogenicity, innate immune components, and regulates tumour immunity, making it a potential candidate as a predictive biomarker for ICI immunotherapy response. | [36] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hossain, S.M.; Carpenter, C.; Eccles, M.R. Genomic and Epigenomic Biomarkers of Immune Checkpoint Immunotherapy Response in Melanoma: Current and Future Perspectives. Int. J. Mol. Sci. 2024, 25, 7252. https://doi.org/10.3390/ijms25137252
Hossain SM, Carpenter C, Eccles MR. Genomic and Epigenomic Biomarkers of Immune Checkpoint Immunotherapy Response in Melanoma: Current and Future Perspectives. International Journal of Molecular Sciences. 2024; 25(13):7252. https://doi.org/10.3390/ijms25137252
Chicago/Turabian StyleHossain, Sultana Mehbuba, Carien Carpenter, and Michael R. Eccles. 2024. "Genomic and Epigenomic Biomarkers of Immune Checkpoint Immunotherapy Response in Melanoma: Current and Future Perspectives" International Journal of Molecular Sciences 25, no. 13: 7252. https://doi.org/10.3390/ijms25137252