
De Novo Hybrid Assembly Unveils Multi-Chromosomal Mitochondrial Genomes in Ludwigia Species, Highlighting Genomic Recombination, Gene Transfer, and RNA Editing Events

Abstract
:1. Introduction
1.1. Origin, Biological Traits, and Distribution of the Aquatic Invasive Plants, Ludwigia Species
1.2. Genomic Resources in the Onagraceae Family
1.3. Form and Size of Plant Mitogenomes
1.4. Repeat Sequences and Exogenous DNA in Plant Mitogenomes
1.5. Limits and Objectives of This Study
2. Results
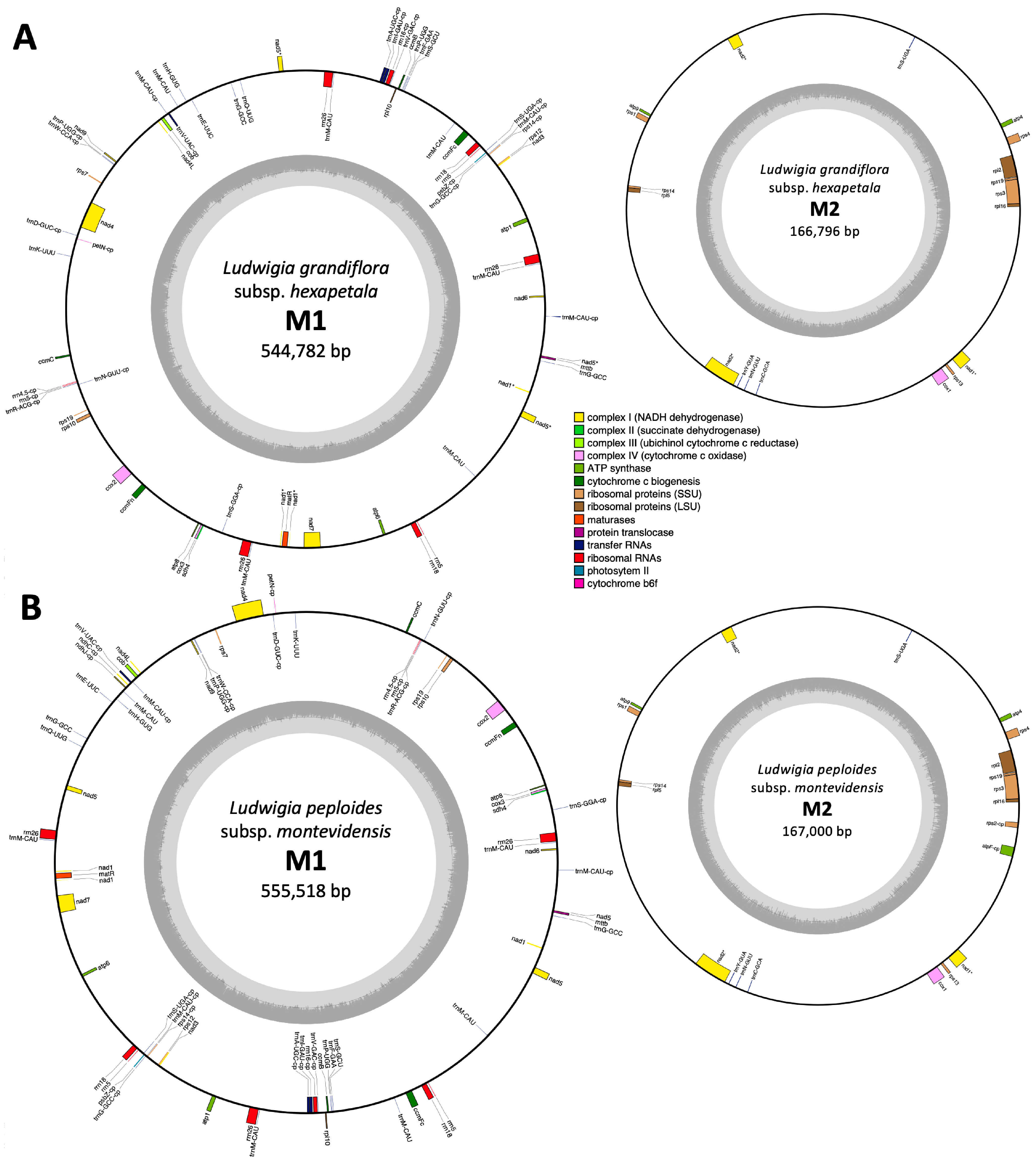
2.1. De Novo Assembly of Ludwigia Multichromosomal Mitogenomes
2.2. Annotation and Comparisons of Ludwigia Mitogenome Contents
2.2.1. Mitogenome Organization Comparisons
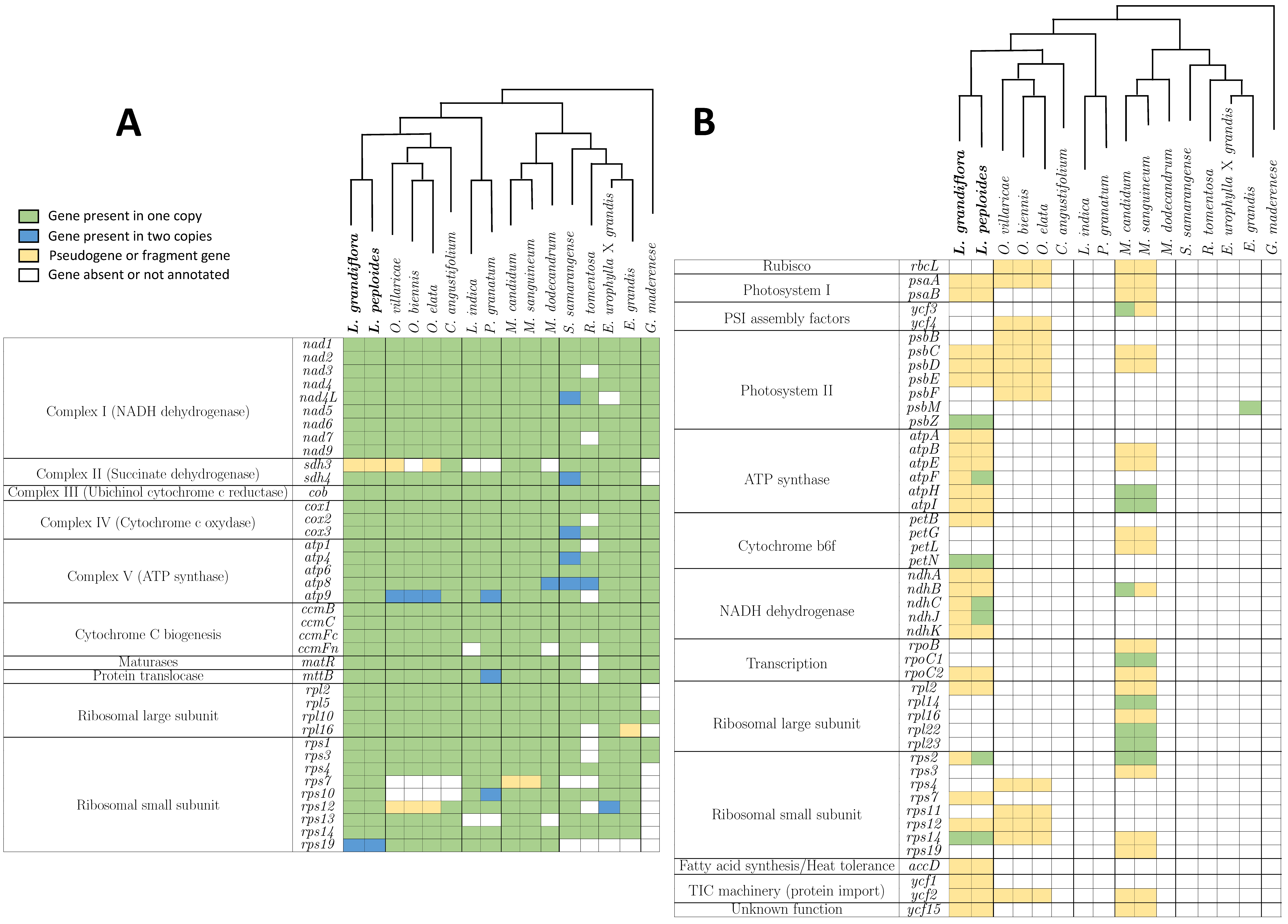
2.2.2. Protein Coding Gene Content
2.2.3. RNA Editing
2.2.4. Content of RNA Genes
2.2.5. Chloroplast-Derived Fragments
2.2.6. Fragments of Mobile Genetic Elements
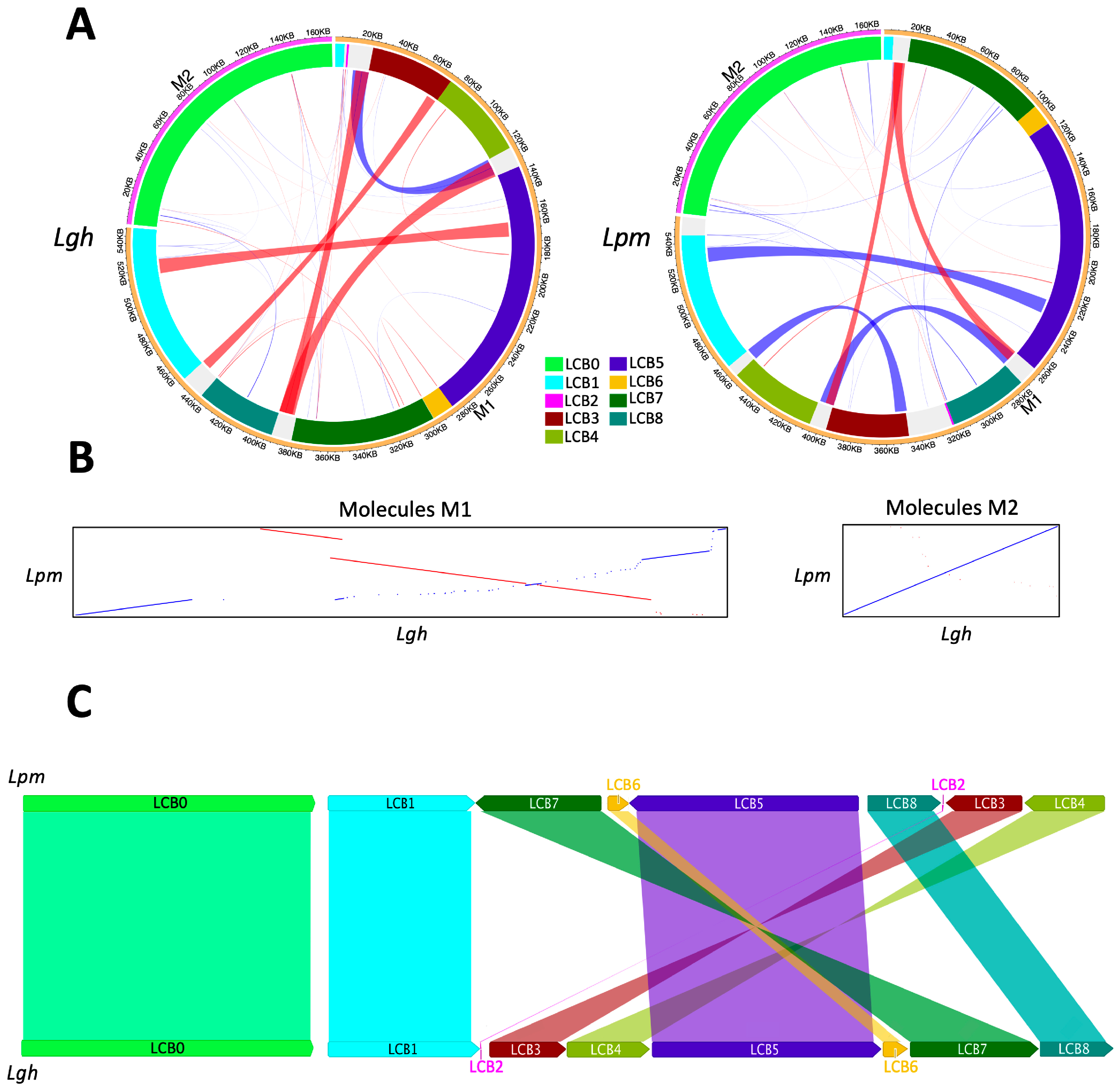
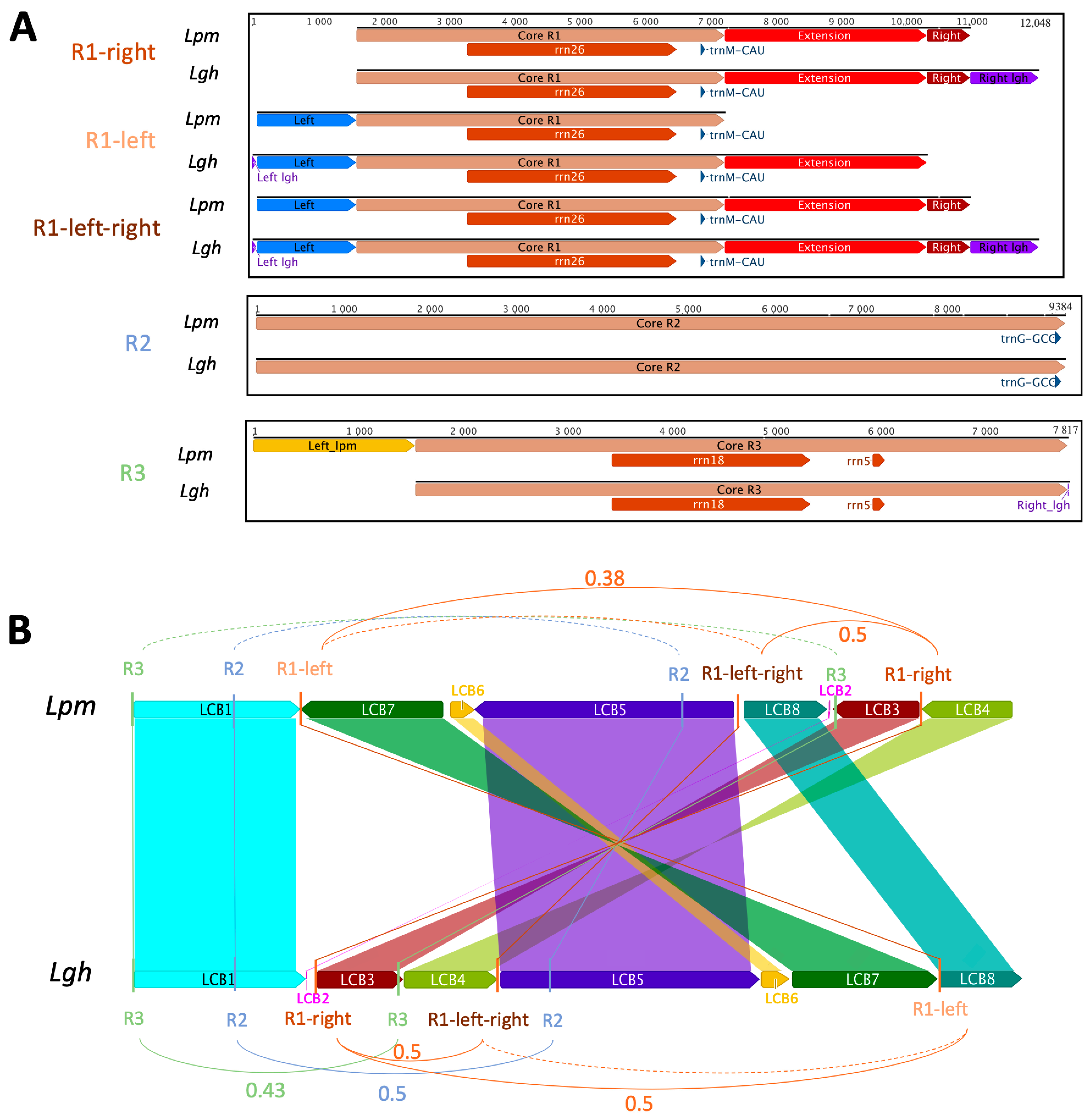
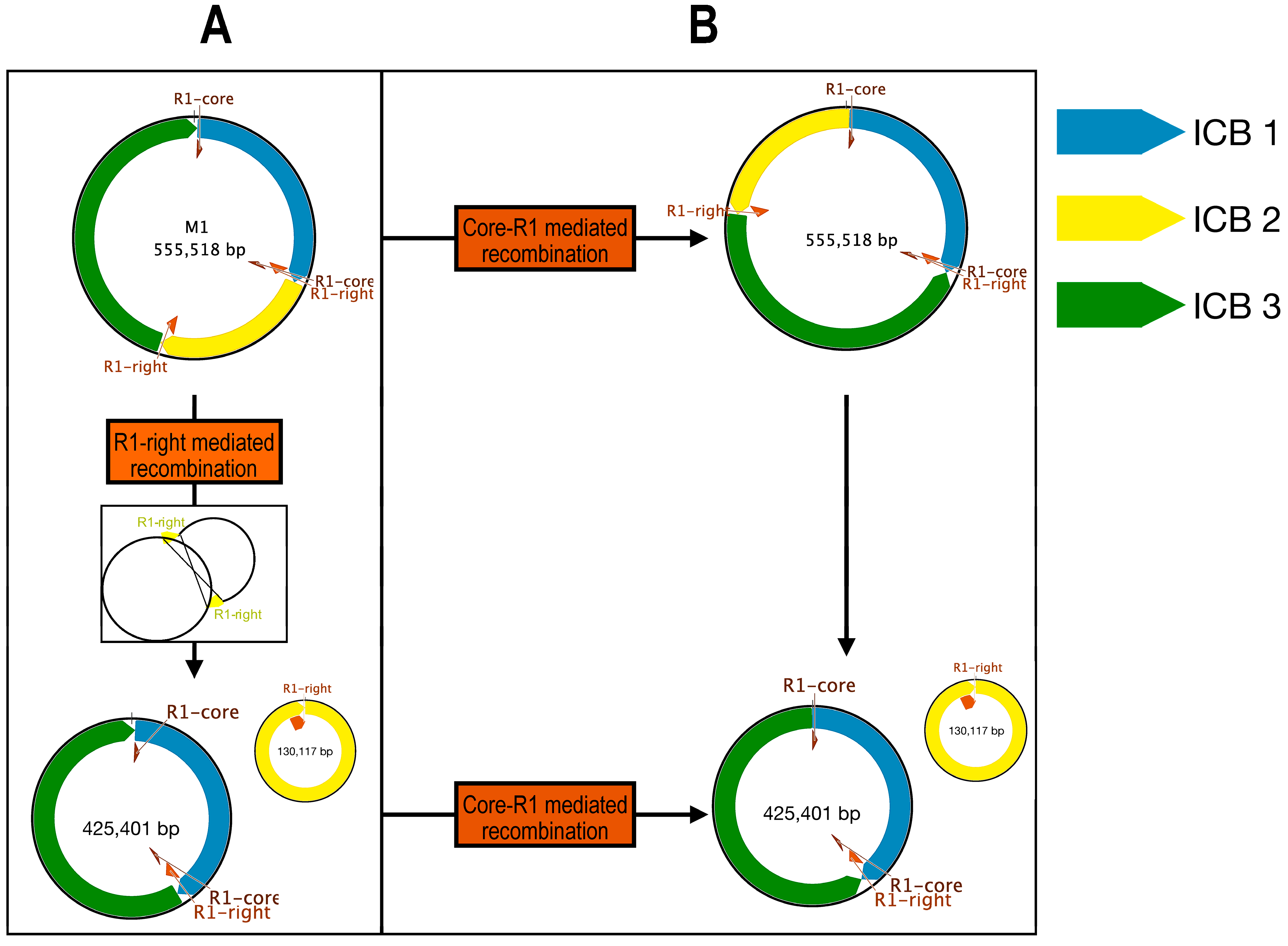
2.2.7. Repeated Sequences and Recombination
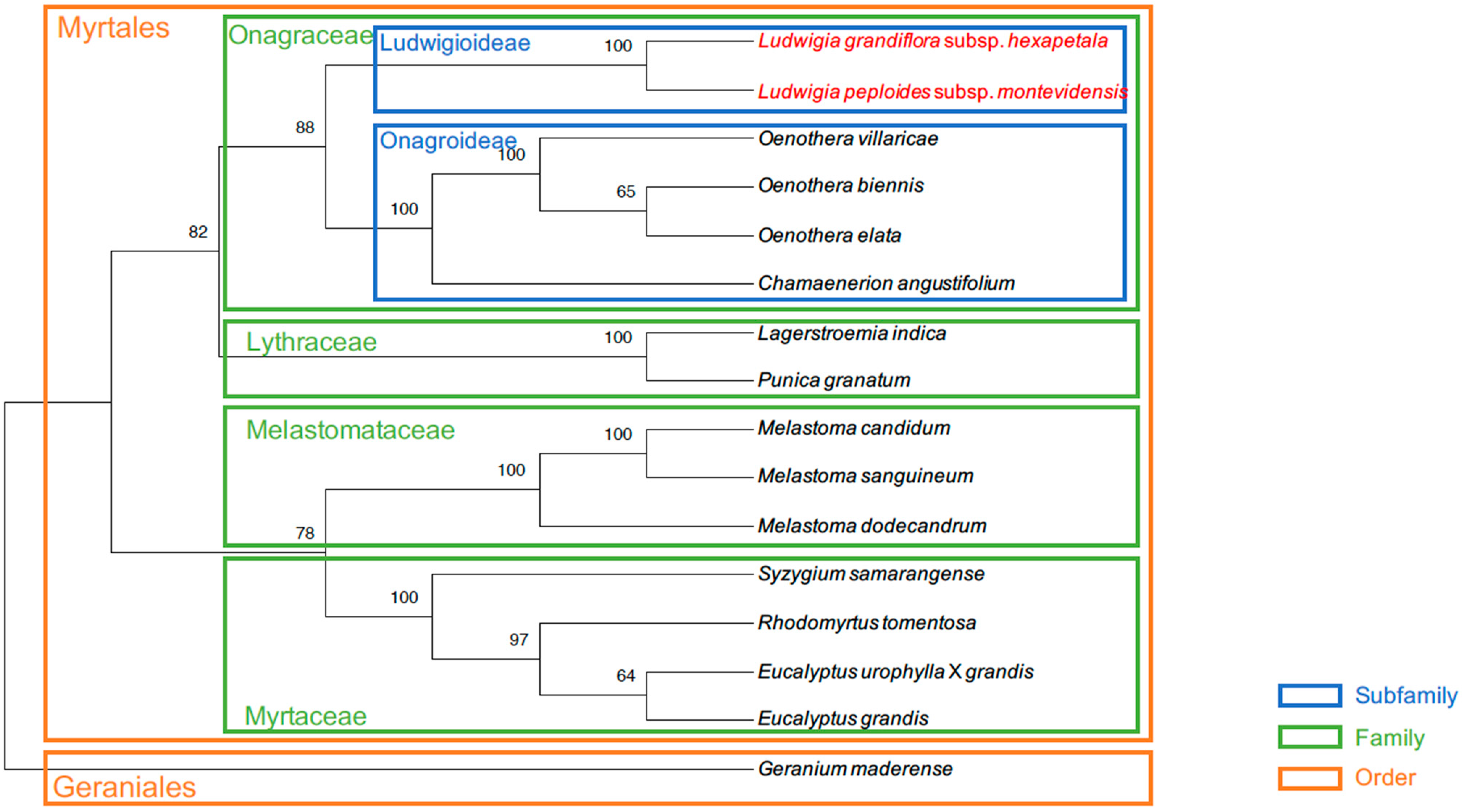
2.3. Gene Comparisons and Phylogeny
3. Discussion
3.1. Comparison of Plant Mitogenomes in Myrtales Order
3.2. Existence of Multi-Chromosomal Mitogenomes
3.3. Plastid DNA Transfer in Lpm and Lgh Mitogenomes
4. Materials and Methods
4.1. Plant Material and Sequencing Data
4.2. Assembly Strategies and Annotations
4.3. Sequence Analysis
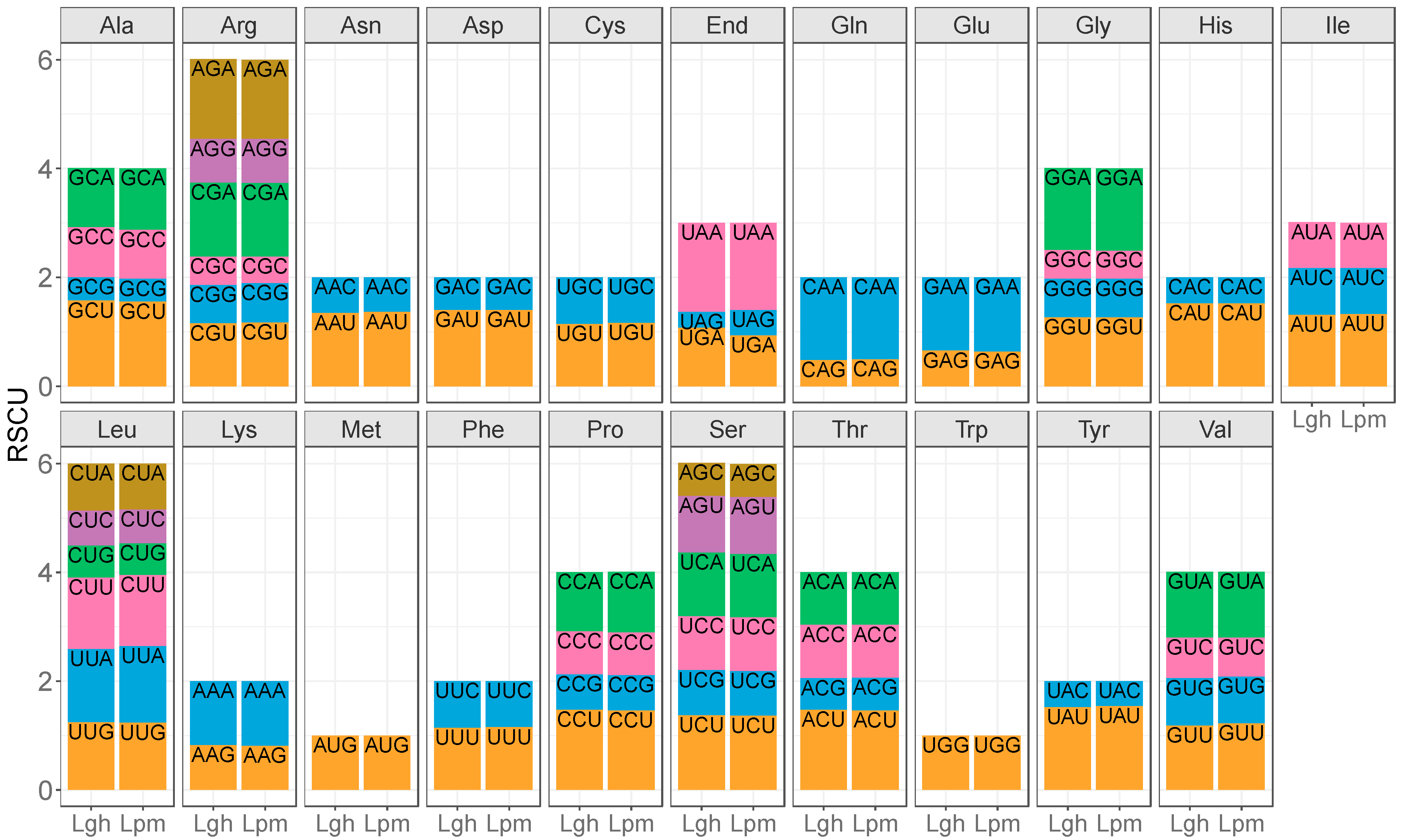
4.4. Codon Usage Bias and Ka/Ks Calculation
4.5. Molecular Mass, Isoelectric Point and Subcellular Localization Predictions
4.6. Phylogenetic and Comparative Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rai, P.K. Environmental Degradation by Invasive Alien Plants in the Anthropocene: Challenges and Prospects for Sustainable Restoration. Anthr. Sci. 2022, 1, 5–28. [Google Scholar] [CrossRef]
- Pyšek, P.; Hulme, P.E.; Simberloff, D.; Bacher, S.; Blackburn, T.M.; Carlton, J.T.; Dawson, W.; Essl, F.; Foxcroft, L.C.; Genovesi, P.; et al. Scientists’ Warning on Invasive Alien Species. Biol. Rev. 2020, 95, 1511–1534. [Google Scholar] [CrossRef]
- Gioria, M.; Hulme, P.E.; Richardson, D.M.; Pyšek, P. Why Are Invasive Plants Successful? Annu. Rev. Plant Biol. 2023, 74, 635–670. [Google Scholar] [CrossRef] [PubMed]
- Mikulyuk, A. Ludwigia grandiflora (Water Primrose). CABI Compend. 2009, 109148. [Google Scholar] [CrossRef]
- Grewell, B.; Netherland, M.; Skaer Thomason, M. Establishing Research and Management Priorities for Invasive Water Primroses (Ludwigia spp.). 2016. Available online: https://www.researchgate.net/publication/295856718_Establishing_Research_and_Management_Priorities_for_Invasive_Water_Primroses_Ludwigia_spp (accessed on 28 June 2024).
- Okada, M.; Grewell, B.J.; Jasieniuk, M. Clonal Spread of Invasive Ludwigia hexapetala and L. Grandiflora in Freshwater Wetlands of California. Aquat. Bot. 2009, 91, 123–129. [Google Scholar] [CrossRef]
- Thouvenot, L.; Haury, J.; Thiebaut, G. A Success Story: Water Primroses, Aquatic Plant Pests. Aquat. Conserv. Mar. Freshw. Ecosyst. 2013, 23, 790–803. [Google Scholar] [CrossRef]
- Dandelot, S.; Matheron, R.; Le Petit, J.; Verlaque, R.; Cazaubon, A. Variations Temporelles des Paramètres Physicochimiques et Microbiologiques de Trois Écosystèmes Aquatiques (Sud-est de la France) Envahis par des Ludwigia. Comptes Rendus Biol. 2005, 328, 991–999. [Google Scholar] [CrossRef]
- Portillo Lemus, L.O.; Bozec, M.; Harang, M.; Coudreuse, J.; Haury, J.; Stoeckel, S.; Barloy, D. Self-Incompatibility Limits Sexual Reproduction Rather than Environmental Conditions in an Invasive Water Primrose. Plant-Environ. Interact. 2021, 2, 74–86. [Google Scholar] [CrossRef]
- Thiébaut, G.; Dutartre, A. Management of invasive aquatic plants in France. In Aquatic Ecosystem Research Trends; Nova Science Pub Inc.: New York, NY, USA, 2009. [Google Scholar]
- Nehring, S.; Kolthoff, D. The Invasive Water Primrose Ludwigia grandiflora (Michaux) Greuter & Burdet (Spermatophyta: Onagraceae) in Germany: First Record and Ecological Risk Assessment. Aquat. Invasions 2011, 6, 83–89. [Google Scholar] [CrossRef]
- Celesti-Grapow, L.; Alessandrini, A.; Arrigoni, P.V.; Banfi, E.; Bernardo, L.; Bovio, M.; Brundu, G.; Cagiotti, M.R.; Camarda, I.; Carli, E.; et al. Inventory of the Non-native Flora of Italy. Plant Biosyst.-Int. J. Deal. All Asp. Plant Biol. 2009, 143, 386–430. [Google Scholar] [CrossRef]
- Bou Manobens, J.; Portillo, L.; Curcó i Massip, A. New Contributions to Allochthonous Ludwigia Species (Onagraceae) on Catalonia. Butlletí Inst. Catalana D’història Nat. 2019, 45–48. [Google Scholar] [CrossRef]
- Lindemann-Matthies, P. Beasts or Beauties? Laypersons’ Perception of Invasive Alien Plant Species in Switzerland and Attitudes towards Their Management. NeoBiota 2016, 29, 15–33. [Google Scholar] [CrossRef]
- Gallardo, B.; Aldridge, D.C. The ‘Dirty Dozen’: Socio-Economic Factors Amplify the Invasion Potential of 12 High-Risk Aquatic Invasive Species in Great Britain and Ireland. J. Appl. Ecol. 2013, 50, 757–766. [Google Scholar] [CrossRef]
- Arslan, Z.F.; Uludag, A.; Uremis, I. Status of Invasive Alien Plants Included in EPPO Lists in Turkey. EPPO Bull. 2015, 45, 66–72. [Google Scholar] [CrossRef]
- Hieda, S.; Kaneko, Y.; Nakagawa, M.; Noma, N. Ludwigia grandiflora (Michx.) Greuter & Burdet subsp. Hexapetala (Hook. & Arn.) GL Nesom & Kartesz, an Invasive Aquatic Plant in Lake Biwa, the Largest Lake in Japan 2020. Acta Phytotaxon. Geobot. 2020, 71, 65–71. [Google Scholar]
- EPPO. Ludwigia grandiflora and L. Peploides onagraceae—Water Primroses. EPPO Bull. 2011, 41, 414–418. [Google Scholar] [CrossRef]
- Sarika, M. Ludwigia peploides subsp. Montevidensis, a New Allien Taxon for the Flora of Greece and the Balkans. J. Biol. Res. 2006, 5, 71–78. [Google Scholar]
- Dandelot, S. Les Ludwigia spp. invasives du Sud de la France: Historique, Biosystématique, Biologie et Ecologie. Ph.D. Thesis, Université Paul Cézanne: Aix-Marseille III, Marseille, France, 2004. [Google Scholar]
- Muller, S. Plantes invasives en France; Muséum National d’Histoire Naturelle: Paris, France, 2004. [Google Scholar]
- Rolon, A.S.; Lacerda, T.; Maltchik, L.; Guadagnin, D.L. Influence of Area, Habitat and Water Chemistry on Richness and Composition of Macrophyte Assemblages in Southern Brazilian Wetlands. J. Veg. Sci. 2008, 19, 221–228. [Google Scholar] [CrossRef]
- Hernández-R, J.; Rangel-Ch, J.O. Vegetation of the Wetland Jaboque (Bogotá, DC). Caldasia 2009, 31, 355–379. [Google Scholar]
- Haury, J.; Druel, A.; Cabral, T.; Paulet, Y.; Bozec, M.; Coudreuse, J. Which Adaptations of Some Invasive Ludwigia Spp. (Rosidae, Onagraceae) Populations Occur in Contrasting Hydrological Conditions in Western France? Hydrobiologia 2014, 737, 45–56. [Google Scholar] [CrossRef]
- Billet, K.; Genitoni, J.; Bozec, M.; Renault, D.; Barloy, D. Aquatic and Terrestrial Morphotypes of the Aquatic Invasive Plant, Ludwigia grandiflora, Show Distinct Morphological and Metabolomic Responses. Ecol. Evol. 2018, 8, 2568–2579. [Google Scholar] [CrossRef] [PubMed]
- Moravcová, L.; Pyšek, P.; Jarošík, V.; Pergl, J. Getting the Right Traits: Reproductive and Dispersal Characteristics Predict the Invasiveness of Herbaceous Plant Species. PLoS ONE 2015, 10, e0123634. [Google Scholar] [CrossRef] [PubMed]
- Mounger, J.; Ainouche, M.L.; Bossdorf, O.; Cavé-Radet, A.; Li, B.; Parepa, M.; Salmon, A.; Yang, J.; Richards, C.L. Epigenetics and the Success of Invasive Plants. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2021, 376, 20200117. [Google Scholar] [CrossRef] [PubMed]
- Hodkinson, T.R.; Perdereau, A.; Klaas, M.; Cormican, P.; Barth, S. Genotyping by Sequencing and Plastome Analysis Finds High Genetic Variability and Geographical Structure in Dactylis glomerata L. in Northwest Europe Despite Lack of Ploidy Variation. Agronomy 2019, 9, 342. [Google Scholar] [CrossRef]
- Chown, S.L.; Hodgins, K.A.; Griffin, P.C.; Oakeshott, J.G.; Byrne, M.; Hoffmann, A.A. Biological Invasions, Climate Change and Genomics. Evol. Appl. 2015, 8, 23–46. [Google Scholar] [CrossRef]
- Hamelin, R.; Roe, A. Genomic Biosurveillance of Forest Invasive Alien Enemies: A Story Written in Code. Evol. Appl. 2019, 13, 12853. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-H.; Edwards, C.; Hoch, P.C.; Raven, P.H.; Barber, J.C. Complete Plastome Sequence of Ludwigia octovalvis (Onagraceae), a Globally Distributed Wetland Plant. Genome Announc. 2016, 4, e01274-16. [Google Scholar] [CrossRef]
- Barloy-Hubler, F.; Gac, A.-L.L.; Boury, C.; Guichoux, E.; Barloy, D. Sequencing, de Novo Assembly of Ludwigia Plastomes, and Comparative Analysis within the Onagraceae Family. bioRxiv 2023. [Google Scholar] [CrossRef]
- Barloy, D.; Portillo-Lemus, L.; Krueger-Hadfield, S.; Huteau, V.; Coriton, O. Genomic relationships among diploid and polyploid species of the genus Ludwigia L. section Jussiaea using a combination of molecular cytogenetic, morphological, and crossing investigations. Peer Community J. 2024, 4, e8. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, J.; Jiang, M.; Lei, W.; Zhang, X.; Tang, H. Sequencing and Assembly of Polyploid Genomes. Methods Mol. Biol. 2023, 2545, 429–458. [Google Scholar] [CrossRef]
- Jacoby, R.P.; Li, L.; Huang, S.; Pong Lee, C.; Millar, A.H.; Taylor, N.L. Mitochondrial Composition, Function and Stress Response in PlantsF. J. Integr. Plant Biol. 2012, 54, 887–906. [Google Scholar] [CrossRef]
- Mackenzie, S.; McIntosh, L. Higher Plant Mitochondria. Plant Cell 1999, 11, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Dotzek, J.; Walther, D.; Greiner, S. Graph-Based Models of the Oenothera Mitochondrial Genome Capture the Enormous Complexity of Higher Plant Mitochondrial DNA Organization. NAR Genom. Bioinform. 2022, 4, lqac027. [Google Scholar] [CrossRef]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Schranz, M.E.; Michelmore, R.W.; Christensen, A.C. The Alternative Reality of Plant Mitochondrial DNA: One Ring Does Not Rule Them All. PLoS Genet. 2019, 15, e1008373. [Google Scholar] [CrossRef]
- Liu, D.; Zeng, M.; Wu, Y.; Du, Y.; Liu, J.; Luo, S.; Zeng, Y. Comparative Transcriptomic Analysis Provides Insights into the Molecular Basis Underlying Pre-Harvest Sprouting in Rice. BMC Genom. 2022, 23, 771. [Google Scholar] [CrossRef] [PubMed]
- Varré, J.-S.; D’Agostino, N.; Touzet, P.; Gallina, S.; Tamburino, R.; Cantarella, C.; Ubrig, E.; Cardi, T.; Drouard, L.; Gualberto, J.M.; et al. Complete Sequence, Multichromosomal Architecture and Transcriptome Analysis of the Solanum Tuberosum Mitochondrial Genome. Int. J. Mol. Sci. 2019, 20, 4788. [Google Scholar] [CrossRef]
- Li, J.; Xu, Y.; Shan, Y.; Pei, X.; Yong, S.; Liu, C.; Yu, J. Assembly of the Complete Mitochondrial Genome of an Endemic Plant, Scutellaria Tsinyunensis, Revealed the Existence of Two Conformations Generated by a Repeat-Mediated Recombination. Planta 2021, 254, 36. [Google Scholar] [CrossRef] [PubMed]
- Tsujimura, M.; Kaneko, T.; Sakamoto, T.; Kimura, S.; Shigyo, M.; Yamagishi, H.; Terachi, T. Multichromosomal Structure of the Onion Mitochondrial Genome and a Transcript Analysis. Mitochondrion 2019, 46, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Lloyd Evans, D.; Hlongwane, T.T.; Joshi, S.V.; Riaño Pachón, D.M. The Sugarcane Mitochondrial Genome: Assembly, Phylogenetics and Transcriptomics. PeerJ 2019, 7, e7558. [Google Scholar] [CrossRef]
- Kim, C.-K.; Kim, Y.-K. The Multipartite Mitochondrial Genome of Fallopia multiflora (Caryophyllales: Polygonaceae). Mitochondrial DNA B Resour. 2018, 3, 155–156. [Google Scholar] [CrossRef]
- Yang, H.; Chen, H.; Ni, Y.; Li, J.; Cai, Y.; Wang, J.; Liu, C. Mitochondrial Genome Sequence of Salvia officinalis (Lamiales: Lamiaceae) Suggests Diverse Genome Structures in Cogeneric Species and Finds the Stop Gain of Genes through RNA Editing Events. Int. J. Mol. Sci. 2023, 24, 5372. [Google Scholar] [CrossRef]
- Guo, Y.; Li, Z.; Jin, S.; Chen, S.; Li, F.; Wu, H. Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Two Species of Calla Lilies (Zantedeschia, Araceae). Int. J. Mol. Sci. 2023, 24, 9566. [Google Scholar] [CrossRef]
- Kang, S.-H.; Oh, J.-H.; Kim, H.-J.; Kim, C.-K. The Multipartite Mitochondrial Genome of Cynanchum wilfordii (Gentianales: Apocynaceae). Mitochondrial DNA B Resour. 2017, 2, 720–721. [Google Scholar] [CrossRef]
- Chang, S.; Yang, T.; Du, T.; Huang, Y.; Chen, J.; Yan, J.; He, J.; Guan, R. Mitochondrial Genome Sequencing Helps Show the Evolutionary Mechanism of Mitochondrial Genome Formation in Brassica. BMC Genom. 2011, 12, 497. [Google Scholar] [CrossRef]
- Alverson, A.J.; Rice, D.W.; Dickinson, S.; Barry, K.; Palmer, J.D. Origins and Recombination of the Bacterial-Sized Multichromosomal Mitochondrial Genome of Cucumber. Plant Cell 2011, 23, 2499–2513. [Google Scholar] [CrossRef]
- Rice, D.W.; Alverson, A.J.; Richardson, A.O.; Young, G.J.; Sanchez-Puerta, M.V.; Munzinger, J.; Barry, K.; Boore, J.L.; Zhang, Y.; dePamphilis, C.W.; et al. Horizontal Transfer of Entire Genomes via Mitochondrial Fusion in the Angiosperm Amborella. Science 2013, 342, 1468–1473. [Google Scholar] [CrossRef]
- Yang, J.-X.; Dierckxsens, N.; Bai, M.-Z.; Guo, Y.-Y. Multichromosomal Mitochondrial Genome of Paphiopedilum Micranthum: Compact and Fragmented Genome, and Rampant Intracellular Gene Transfer. Int. J. Mol. Sci. 2023, 24, 3976. [Google Scholar] [CrossRef]
- Sanchez-Puerta, M.V.; García, L.E.; Wohlfeiler, J.; Ceriotti, L.F. Unparalleled Replacement of Native Mitochondrial Genes by Foreign Homologs in a Holoparasitic Plant. New Phytol. 2017, 214, 376–387. [Google Scholar] [CrossRef]
- Wu, Z.; Cuthbert, J.M.; Taylor, D.R.; Sloan, D.B. The Massive Mitochondrial Genome of the Angiosperm Silene Noctiflora Is Evolving by Gain or Loss of Entire Chromosomes. Proc. Natl. Acad. Sci. USA 2015, 112, 10185–10191. [Google Scholar] [CrossRef]
- Sloan, D.B.; Alverson, A.J.; Wu, M.; Palmer, J.D.; Taylor, D.R. Recent Acceleration of Plastid Sequence and Structural Evolution Coincides with Extreme Mitochondrial Divergence in the Angiosperm Genus Silene. Genome Biol. Evol. 2012, 4, 294–306. [Google Scholar] [CrossRef]
- Skippington, E.; Barkman, T.J.; Rice, D.W.; Palmer, J.D. Miniaturized Mitogenome of the Parasitic Plant Viscum Scurruloideum Is Extremely Divergent and Dynamic and Has Lost All Nad Genes. Proc. Natl. Acad. Sci. USA 2015, 112, E3515–E3524. [Google Scholar] [CrossRef] [PubMed]
- Putintseva, Y.A.; Bondar, E.I.; Simonov, E.P.; Sharov, V.V.; Oreshkova, N.V.; Kuzmin, D.A.; Konstantinov, Y.M.; Shmakov, V.N.; Belkov, V.I.; Sadovsky, M.G.; et al. Siberian larch (Larix sibirica Ledeb.) mitochondrial genome assembled using both short and long nucleotide sequence reads is currently the largest known mitogenome. BMC Genom. 2020, 21, 654. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P.; Sloan, D.B.; Alverson, A.J. Plant Mitochondrial Genome Diversity: The Genomics Revolution. In Plant Genome; Wendel, J.F., Greilhuber, J., Dolezel, J., Leitch, I.J., Eds.; Springer: Vienna, Austria, 2012; Volume 1, pp. 123–144. ISBN 978-3-7091-1129-1. [Google Scholar]
- Wynn, E.L.; Christensen, A.C. Repeats of Unusual Size in Plant Mitochondrial Genomes: Identification, Incidence and Evolution. G3 Genes Genomes Genet. 2019, 9, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Bruenn, J.A.; Warner, B.E.; Yerramsetty, P. Widespread Mitovirus Sequences in Plant Genomes. PeerJ 2015, 3, e876. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.-S.; Wojciechowski, M.F.; Ruhlman, T.A.; Jansen, R.K. In and out: Evolution of Viral Sequences in the Mitochondrial Genomes of Legumes (Fabaceae). Mol. Phylogenet. Evol. 2021, 163, 107236. [Google Scholar] [CrossRef]
- Knoop, V.; Unseld, M.; Marienfeld, J.; Brandt, P.; Sünkel, S.; Ullrich, H.; Brennicke, A. Copia-, Gypsy- and LINE-like Retrotransposon Fragments in the Mitochondrial Genome of Arabidopsis Thaliana. Genetics 1996, 142, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Jiang, W.; Tan, W.; Zhang, X.; Tian, X. Deciphering the Organelle Genomes and Transcriptomes of a Common Ornamental Plant Ligustrum Quihoui Reveals Multiple Fragments of Transposable Elements in the Mitogenome. Int. J. Biol. Macromol. 2020, 165, 1988–1999. [Google Scholar] [CrossRef]
- de Abreu, V.A.C.; Moysés Alves, R.; Silva, S.R.; Ferro, J.A.; Domingues, D.S.; Miranda, V.F.O.; Varani, A.M. Comparative Analyses of Theobroma Cacao and T. Grandiflorum Mitogenomes Reveal Conserved Gene Content Embedded within Complex and Plastic Structures. Gene 2023, 849, 146904. [Google Scholar] [CrossRef]
- Sloan, D.B. One Ring to Rule Them All? Genome Sequencing Provides New Insights into the ‘Master Circle’ Model of Plant Mitochondrial DNA Structure. New Phytol. 2013, 200, 978–985. [Google Scholar] [CrossRef]
- Arrieta-Montiel, M.P.; Mackenzie, S.A. Plant Mitochondrial Genomes and Recombination. In Plant Mitochondria; Kempken, F., Ed.; Springer: New York, NY, USA, 2011; pp. 65–82. ISBN 978-0-387-89781-3. [Google Scholar]
- Zhang, X.; Shan, Y.; Li, J.; Qin, Q.; Yu, J.; Deng, H. Assembly of the Complete Mitochondrial Genome of Pereskia Aculeata Revealed That Two Pairs of Repetitive Elements Mediated the Recombination of the Genome. Int. J. Mol. Sci. 2023, 24, 8366. [Google Scholar] [CrossRef]
- Yang, Z.; Ni, Y.; Lin, Z.; Yang, L.; Chen, G.; Nijiati, N.; Hu, Y.; Chen, X. De Novo Assembly of the Complete Mitochondrial Genome of Sweet Potato (Ipomoea batatas [L.] Lam) Revealed the Existence of Homologous Conformations Generated by the Repeat-Mediated Recombination. BMC Plant Biol. 2022, 22, 285. [Google Scholar] [CrossRef]
- Sullivan, A.R.; Eldfjell, Y.; Schiffthaler, B.; Delhomme, N.; Asp, T.; Hebelstrup, K.H.; Keech, O.; Öberg, L.; Møller, I.M.; Arvestad, L.; et al. The Mitogenome of Norway Spruce and a Reappraisal of Mitochondrial Recombination in Plants. Genome Biol. Evol. 2020, 12, 3586–3598. [Google Scholar] [CrossRef]
- Lonsdale, D.M.; Brears, T.; Hodge, T.P.; Melville, S.E.; Rottmann, W.H.; Leaver, C.J.; Lonsdale, D.M. The Plant Mitochondrial Genome: Homologous Recombination as a Mechanism for Generating Heterogeneity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1997, 319, 149–163. [Google Scholar] [CrossRef]
- Gualberto, J.M.; Newton, K.J. Plant Mitochondrial Genomes: Dynamics and Mechanisms of Mutation. Annu. Rev. Plant Biol. 2017, 68, 225–252. [Google Scholar] [CrossRef]
- Kubo, T.; Newton, K.J. Angiosperm Mitochondrial Genomes and Mutations. Mitochondrion 2008, 8, 5–14. [Google Scholar] [CrossRef]
- Marienfeld, J.; Unseld, M.; Brennicke, A. The Mitochondrial Genome of Arabidopsis Is Composed of Both Native and Immigrant Information. Trends Plant Sci. 1999, 4, 495–502. [Google Scholar] [CrossRef]
- Hao, W.; Palmer, J.D. Fine-Scale Mergers of Chloroplast and Mitochondrial Genes Create Functional, Transcompartmentally Chimeric Mitochondrial Genes. Proc. Natl. Acad. Sci. USA 2009, 106, 16728–16733. [Google Scholar] [CrossRef]
- Marechal-Drouard, L.; Weil, J.H.; Dietrich, A. Transfer RNAs and Transfer RNA Genes in Plants. Annu. Rev. Plant Biol. 1993, 44, 13–32. [Google Scholar] [CrossRef]
- Nakazono, M.; Nishiwaki, S.; Tsutsumi, N.; Hirai, A. A Chloroplast-Derived Sequence Is Utilized as a Source of Promoter Sequences for the Gene for Subunit 9 of NADH Dehydrogenase (Nad9) in Rice Mitochondria. Mol. Gen. Genet. 1996, 252, 371–378. [Google Scholar] [CrossRef]
- Wang, D.; Rousseau-Gueutin, M.; Timmis, J.N. Plastid Sequences Contribute to Some Plant Mitochondrial Genes. Mol. Biol. Evol. 2012, 29, 1707–1711. [Google Scholar] [CrossRef] [PubMed]
- Fox, T.D.; Leaver, C.J. The Zea Mays Mitochondrial Gene Coding Cytochrome Oxidase Subunit II Has an Intervening Sequence and Does Not Contain TGA Codons. Cell 1981, 26, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Chao, S.; Sederoff, R.R.; Levings, C.S. Partial Sequence Analysis of the 5S to 18S rRNA Gene Region of the Maize Mitochondrial Genome. Plant Physiol. 1983, 71, 190–193. [Google Scholar] [CrossRef]
- Sloan, D.B.; Alverson, A.J.; Chuckalovcak, J.P.; Wu, M.; McCauley, D.E.; Palmer, J.D.; Taylor, D.R. Rapid Evolution of Enormous, Multichromosomal Genomes in Flowering Plant Mitochondria with Exceptionally High Mutation Rates. PLoS Biol. 2012, 10, e1001241. [Google Scholar] [CrossRef]
- Knoop, V. The Mitochondrial DNA of Land Plants: Peculiarities in Phylogenetic Perspective. Curr. Genet. 2004, 46, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Unseld, M.; Marienfeld, J.R.; Brandt, P.; Brennicke, A. The Mitochondrial Genome of Arabidopsis Thaliana Contains 57 Genes in 366,924 Nucleotides. Nat. Genet. 1997, 15, 57–61. [Google Scholar] [CrossRef]
- Adams, K.L.; Palmer, J.D. Evolution of Mitochondrial Gene Content: Gene Loss and Transfer to the Nucleus. Mol. Phylogenet. Evol. 2003, 29, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, M.; Sugita, M. RNA Editing and Its Molecular Mechanism in Plant Organelles. Genes 2016, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Small, I.D.; Schallenberg-Rüdinger, M.; Takenaka, M.; Mireau, H.; Ostersetzer-Biran, O. Plant Organellar RNA Editing: What 30 Years of Research Has Revealed. Plant J. 2020, 101, 1040–1056. [Google Scholar] [CrossRef]
- Gualberto, J.M.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotfi, F.; Dietrich, A. The Plant Mitochondrial Genome: Dynamics and Maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef]
- Wang, S.; Song, Q.; Li, S.; Hu, Z.; Dong, G.; Song, C.; Huang, H.; Liu, Y. Assembly of a Complete Mitogenome of Chrysanthemum Nankingense Using Oxford Nanopore Long Reads and the Diversity and Evolution of Asteraceae mitogenomes. Genes 2018, 9, 547. [Google Scholar] [CrossRef]
- Kovar, L.; Nageswara-Rao, M.; Ortega-Rodriguez, S.; Dugas, D.V.; Straub, S.; Cronn, R.; Strickler, S.R.; Hughes, C.E.; Hanley, K.A.; Rodriguez, D.N.; et al. PacBio-Based Mitochondrial Genome Assembly of Leucaena trichandra (Leguminosae) and an Intrageneric Assessment of Mitochondrial RNA Editing. Genome Biol. Evol. 2018, 10, 2501–2517. [Google Scholar] [CrossRef] [PubMed]
- The Angiosperm Phylogeny Group; Chase, M.W.; Christenhusz, M.J.M.; Fay, M.F.; Byng, J.W.; Judd, W.S.; Soltis, D.E.; Mabberley, D.J.; Sennikov, A.N.; Soltis, P.S.; et al. An Update of the Angiosperm Phylogeny Group Classification for the Orders and Families of Flowering Plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 12385. [Google Scholar] [CrossRef]
- Lu, G.; Zhang, K.; Que, Y.; Li, Y. Assembly and Analysis of the First Complete Mitochondrial Genome of Punica Granatum and the Gene Transfer from Chloroplast Genome. Front. Plant Sci. 2023, 14, 1132551. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Ma, Z.; Liu, S.; Duan, Z. Analysis of the Rhodomyrtus tomentosa Mitochondrial Genome: Insights into Repeat-Mediated Recombination and Intra-Cellular DNA Transfer. Gene 2024, 909, 148288. [Google Scholar] [CrossRef] [PubMed]
- Pinard, D.; Myburg, A.A.; Mizrachi, E. The Plastid and Mitochondrial Genomes of Eucalyptus Grandis. BMC Genom. 2019, 20, 132. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, Y.; Driguez, P.; Bougouffa, S.; Carty, K.; Putra, A.; Cheung, M.-S.; Ermini, L. Plasticity of Repetitive Sequences Demonstrated by the Complete Mitochondrial Genome of Eucalyptus Camaldulensis. Front. Plant Sci. 2024, 15, 1339594. [Google Scholar] [CrossRef]
- Wu, H.-H.; Zhao, X.-H.; Zong, X.-Y.; Ding, R.; Chen, X.-H. Complete Mitochondrial Genome of Medinilla magnifica (Myrtales, Melastomataceae). Mitochondrial DNA Part B 2020, 5, 1716–1717. [Google Scholar] [CrossRef]
- Zhou, Y.; Zheng, R.; Peng, Y.; Chen, J.; Zhu, X.; Xie, K.; Ahmad, S.; Chen, J.; Wang, F.; Shen, M.; et al. The First Mitochondrial Genome of Melastoma dodecandrum Resolved Structure Evolution in Melastomataceae and Micro Inversions from Inner Horizontal Gene Transfer. Ind. Crops Prod. 2023, 205, 117390. [Google Scholar] [CrossRef]
- Wu, Z.-Q.; Liao, X.-Z.; Zhang, X.-N.; Tembrock, L.R.; Broz, A. Genomic Architectural Variation of Plant Mitochondria—A Review of Multichromosomal Structuring. J. Syst. Evol. 2020, 60, 160–168. [Google Scholar] [CrossRef]
- Cole, L.W.; Guo, W.; Mower, J.P.; Palmer, J.D. High and Variable Rates of Repeat-Mediated Mitochondrial Genome Rearrangement in a Genus of Plants. Mol. Biol. Evol. 2018, 35, 2773–2785. [Google Scholar] [CrossRef]
- Bi, C.; Qu, Y.; Hou, J.; Wu, K.; Ye, N.; Yin, T. Deciphering the Multi-Chromosomal Mitochondrial Genome of Populus simonii. Front. Plant Sci. 2022, 13, 914635. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chen, H.; Ni, Y.; Li, J.; Cai, Y.; Ma, B.; Yu, J.; Wang, J.; Liu, C. De Novo Hybrid Assembly of the Salvia miltiorrhiza Mitochondrial Genome Provides the First Evidence of the Multi-Chromosomal Mitochondrial DNA Structure of Salvia Species. Int. J. Mol. Sci. 2022, 23, 14267. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Alverson, A.J.; Štorchová, H.; Palmer, J.D.; Taylor, D.R. Extensive Loss of Translational Genes in the Structurally Dynamic Mitochondrial Genome of the Angiosperm Silene Latifolia. BMC Evol. Biol. 2010, 10, 274. [Google Scholar] [CrossRef] [PubMed]
- You, C.; Cui, T.; Zhang, C.; Zang, S.; Su, Y.; Que, Y. Assembly of the Complete Mitochondrial Genome of Gelsemium Elegans Revealed the Existence of Homologous Conformations Generated by a Repeat Mediated Recombination. Int. J. Mol. Sci. 2023, 24, 527. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-C.; Chen, H.; Yang, D.; Liu, C. Diversity of Mitochondrial Plastid DNAs (MTPTs) in Seed Plants. Mitochondrial DNA Part A 2018, 29, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Luo, Z.; Zhang, J.; Chen, L.; Li, L. Multi-Chromosomal Mitochondrial Genome of Medicinal Plant Acorus Tatarinowii (Acoraceae): Firstly Reported from Acorales Order. Gene 2024, 892, 147847. [Google Scholar] [CrossRef]
- Yang, L.; Liu, J.; Guo, W.; Zheng, Z.; Xu, Y.; Xia, H.; Xiao, T. Insights into the Multi-Chromosomal Mitochondrial Genome Structure of the Xero-Halophytic Plant Haloxylon ammodendron (C.A.Mey.) Bunge Ex Fenzl. BMC Genom. 2024, 25, 123. [Google Scholar] [CrossRef]
- Hao, W.; Liu, G.; Wang, W.; Shen, W.; Zhao, Y.; Sun, J.; Yang, Q.; Zhang, Y.; Fan, W.; Pei, S.; et al. RNA Editing and Its Roles in Plant Organelles. Front. Genet. 2021, 12, 757109. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, Q.; Yin, P. RNA Editing Machinery in Plant Organelles. Sci. China Life Sci. 2018, 61, 162–169. [Google Scholar] [CrossRef]
- Gommans, W.M.; Mullen, S.P.; Maas, S. RNA Editing: A Driving Force for Adaptive Evolution? BioEssays 2009, 31, 1137–1145. [Google Scholar] [CrossRef]
- Lu, G.; Li, Q. Complete Mitochondrial Genome of Syzygium Samarangense Reveals Genomic Recombination, Gene Transfer, and RNA Editing Events. Front. Plant Sci. 2024, 14, 1301164. [Google Scholar] [CrossRef]
- Xiong, J.; Tao, T.; Luo, Z.; Yan, S.; Liu, Y.; Yu, X.; Liu, G.; Xia, H.; Luo, L. RNA Editing Responses to Oxidative Stress between a Wild Abortive Type Male-Sterile Line and Its Maintainer Line. Front. Plant Sci. 2017, 8, 2023. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate Paired Shotgun Read Merging via Overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef]
- Holley, G.; Beyter, D.; Ingimundardottir, H.; Møller, P.L.; Kristmundsdottir, S.; Eggertsson, H.P.; Halldorsson, B.V. Ratatosk: Hybrid Error Correction of Long Reads Enables Accurate Variant Calling and Assembly. Genome Biol. 2021, 22, 28. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of Long, Error-Prone Reads Using Repeat Graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and Accurate Annotation of Organelle Genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) Version 1.3.1: Expanded Toolkit for the Graphical Visualization of Organellar Genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [PubMed]
- Edera, A.A.; Small, I.; Milone, D.H.; Sanchez-Puerta, M.V. Deepred-Mt: Deep Representation Learning for Predicting C-to-U RNA Editing in Plant Mitochondria. Comput. Biol. Med. 2021, 136, 104682. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Neumann, P.; Pech, J.; Steinhaisl, J.; Macas, J. RepeatExplorer: A Galaxy-Based Web Server for Genome-Wide Characterization of Eukaryotic Repetitive Elements from next-Generation Sequence Reads. Bioinformatics 2013, 29, 792–793. [Google Scholar] [CrossRef]
- Kurtz, S.; Schleiermacher, C. REPuter: Fast Computation of Maximal Repeats in Complete Genomes. Bioinformatics 1999, 15, 426–427. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-Web: A Web Server for Microsatellite Prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Benson, G. Tandem Repeats Finder: A Program to Analyze DNA Sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef]
- Yu, Y.; Ouyang, Y.; Yao, W. shinyCircos: An R/Shiny Application for Interactive Creation of Circos Plot. Bioinformatics 2018, 34, 1229–1231. [Google Scholar] [CrossRef]
- Harris, R.S. Improved Pairwise Alignment of Genomic DNA; The Pennsylvania State University: University Park, PA, USA, 2007. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Stothard, P. The Sequence Manipulation Suite: JavaScript Programs for Analyzing and Formatting Protein and DNA Sequences. BioTechniques 2000, 28, 1102–1104. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.-C.; Shen, H.-B. Plant-mPLoc: A Top-Down Strategy to Augment the Power for Predicting Plant Protein Subcellular Localization. PLoS ONE 2010, 5, e11335. [Google Scholar] [CrossRef]
- Kriebel, R.; Khabbazian, M.; Sytsma, K.J. A Continuous Morphological Approach to Study the Evolution of Pollen in a Phylogenetic Context: An Example with the Order Myrtales. PLoS ONE 2017, 12, e0187228. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | LCB | Molecule | Size (in bp) | Molecular Weight (in kDa) | Protein Isoelectric Point | Hypothetical Localization |
|---|---|---|---|---|---|---|
| Complex I (NADH dehydrogenase) | ||||||
| nad1 (5*) | LCB0 | M2 | 1008 | 36.81 | 10.10 | Mitochondrion |
| LCB1 | M1 | |||||
| LCB8 | M1 | |||||
| nad2 (5*) | LCB0 | M2 | 1518 | 55.34 | 9.28 | Mitochondrion |
| nad3 | LCB3 | M1 | 357 | 13.39 | 4.33 | Mitochondrion |
| nad4 (4) | LCB5 | M1 | 1473 | 54.27 | 8.97 | Mitochondrion |
| nad4L | LCB5 | M1 | 303 | 10.94 | 6.70 | Mitochondrion |
| nad5 (5*) | LCB1 | M1 | 1794 | 74.55 | 7.90 | Mitochondrion |
| LCB5 | M1 | |||||
| nad6 | LCB1 | M1 | 618 | 23.45 | 10.53 | Mitochondrion |
| nad7 (5) | LCB8 | M1 | 1182 | 44.32 | 7.87 | Mitochondrion |
| nad9 | LCB5 | M1 | 573 | 22.63 | 8.20 | Mitochondrion |
| Complex II (succinate dehydrogenase) | ||||||
| sdh4 | LCB7 | M1 | 393 | 15.25 | 10.76 | Mitochondrion |
| Complex III (cytochrome c reductase) | ||||||
| cob | LCB5 | M1 | 1182 | 44.04 | 7.52 | Cell membrane |
| Complex IV (cytochrome c oxidase) | ||||||
| cox1 | LCB0 | M2 | 1623 | 58.75 | 9.25 | Mitochondrion |
| cox2 (2) | LCB7 | M1 | 780 | 29.09 | 5.53 | Mitochondrion |
| cox3 | LCB7 | M1 | 798 | 29.54 | 7.53 | Mitochondrion |
| Complex V (ATP synthase) | ||||||
| atp1 | LCB3 | M1 | 1533 | 55.51 | 6.21 | Mitochondrion |
| atp4 | LCB0 | M2 | 573 | 21.25 | 10.26 | Mitochondrion |
| atp6 | LCB8 | M1 | 1044 | 38.30 | 4.80 | Mitochondrion |
| atp8 | LCB7 | M1 | 480 | 18.19 | 9.50 | Mitochondrion |
| atp9 | LCB0 | M2 | 330 | 11.61 | 9.09 | Mitochondrion |
| Cytochrome c biogenesis | ||||||
| ccmB | LCB4 | M1 | 621 | 23.07 | 8.62 | Mitochondrion |
| ccmC | LCB6 | M1 | 723 | 26.92 | 10.98 | Mitochondrion |
| ccmFc (2) | LCB4 | M1 | 1500 | 56.40 | 10.74 | Mitochondrion |
| ccmFn | LCB7 | M1 | 1983 | 74.21 | 10.45 | Mitochondrion |
| Maturase | ||||||
| matR | LCB8 | M1 | 1983 | 73.76 | 10.71 | Chloroplast |
| Protein translocase | ||||||
| mttb | LCB1 | M1 | 768 | 29.07 | 7.75 | Mitochondrion |
| Ribosomal large subunit | ||||||
| rpl2 (3) | LCB0 | M2 | 1389 | 50.69 | 11.28 | Mitochondrion |
| rpl5 | LCB0 | M2 | 564 | 21.55 | 5.89 | Mitochondrion |
| rpl10 | LCB4 | M1 | 486 | 18.53 | 7.45 | Cell membrane, chloroplast, and nucleus |
| rpl16 | LCB0 | M2 | 435 | 15.98 | 11.81 | Mitochondrion |
| Ribosomal small subunit | ||||||
| rps1 | LCB0 | M2 | 693 | 25.87 | 11.89 | Mitochondrion |
| rps3 (2) | LCB0 | M2 | 1659 | 63.57 | 10.96 | Mitochondrion |
| rps4 | LCB0 | M2 | 1047 | 41.18 | 11.63 | Mitochondrion |
| rps7 | LCB5 | M1 | 447 | 17.04 | 11.21 | Chloroplast and mitochondrion |
| rps10 (2) | LCB7 | M1 | 408 | 16.40 | 11.25 | Mitochondrion |
| rps12 | LCB3 | M1 | 378 | 14.30 | 11.80 | Chloroplast and mitochondrion |
| rps13 | LCB0 | M2 | 351 | 13.27 | 11.21 | Mitochondrion |
| rps14 | LCB0 | M2 | 303 | 11.96 | 12.04 | Mitochondrion |
| rps19 | LCB0 | M2 | 285 | 11.20 | 12.32 | Chloroplast |
| LCB7 | M1 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doré, G.; Barloy, D.; Barloy-Hubler, F. De Novo Hybrid Assembly Unveils Multi-Chromosomal Mitochondrial Genomes in Ludwigia Species, Highlighting Genomic Recombination, Gene Transfer, and RNA Editing Events. Int. J. Mol. Sci. 2024, 25, 7283. https://doi.org/10.3390/ijms25137283
Doré G, Barloy D, Barloy-Hubler F. De Novo Hybrid Assembly Unveils Multi-Chromosomal Mitochondrial Genomes in Ludwigia Species, Highlighting Genomic Recombination, Gene Transfer, and RNA Editing Events. International Journal of Molecular Sciences. 2024; 25(13):7283. https://doi.org/10.3390/ijms25137283
Chicago/Turabian StyleDoré, Guillaume, Dominique Barloy, and Frédérique Barloy-Hubler. 2024. "De Novo Hybrid Assembly Unveils Multi-Chromosomal Mitochondrial Genomes in Ludwigia Species, Highlighting Genomic Recombination, Gene Transfer, and RNA Editing Events" International Journal of Molecular Sciences 25, no. 13: 7283. https://doi.org/10.3390/ijms25137283