
Hypomethylation of the RUNX2 Gene Is a New Potential Biomarker of Primary Osteoporosis in Men and Women


Abstract
:1. Introduction
2. Results
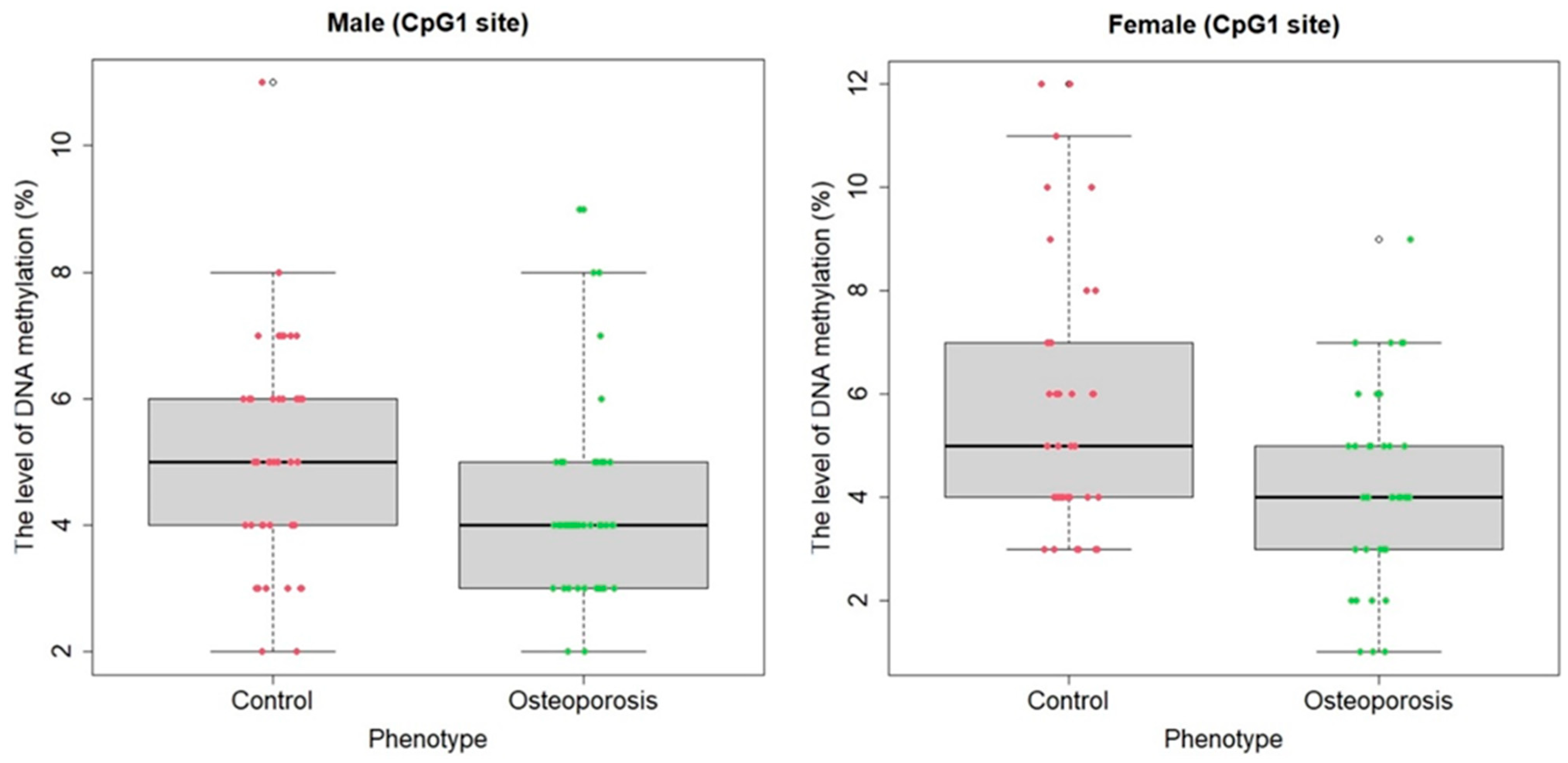
2.1. Associative Analysis
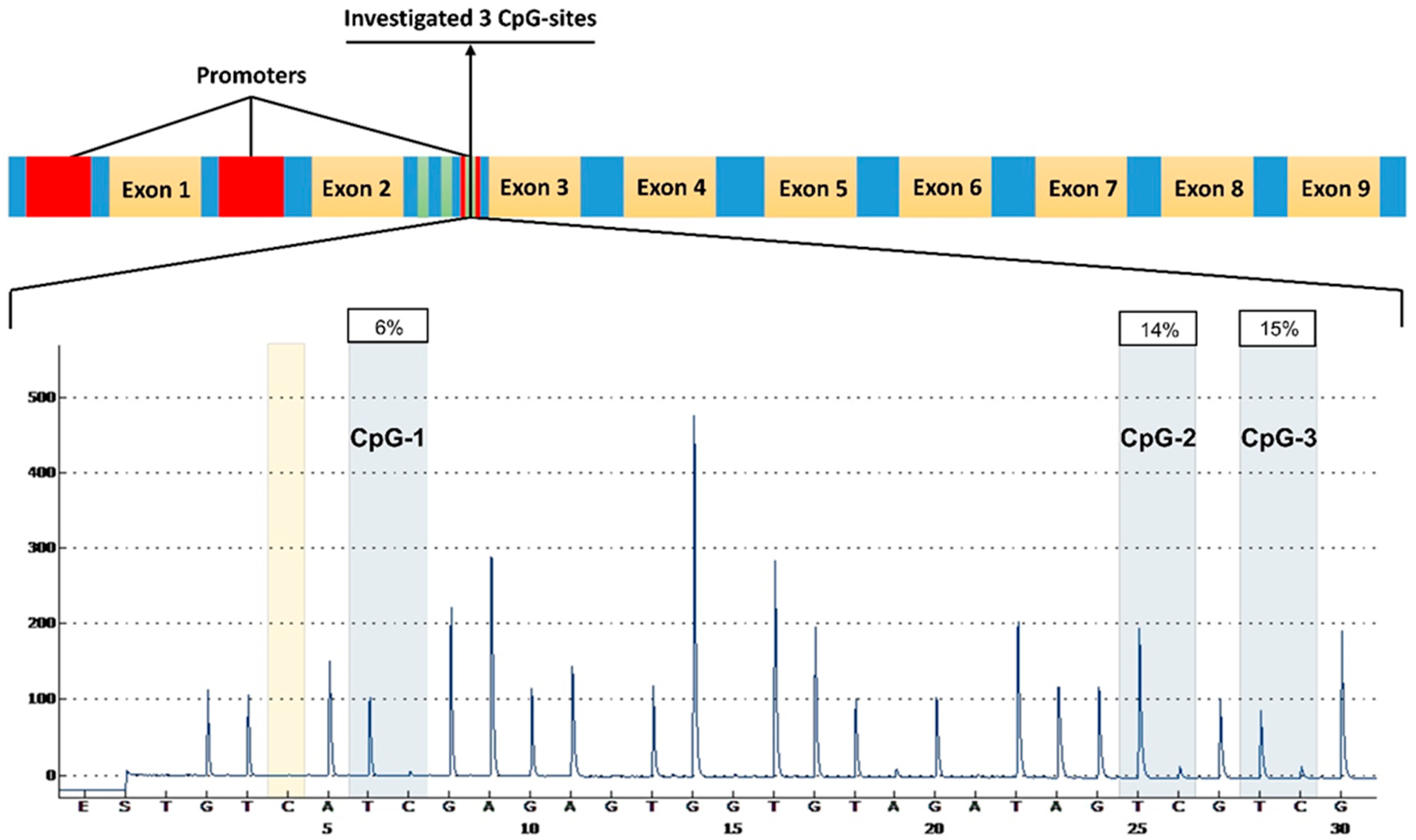
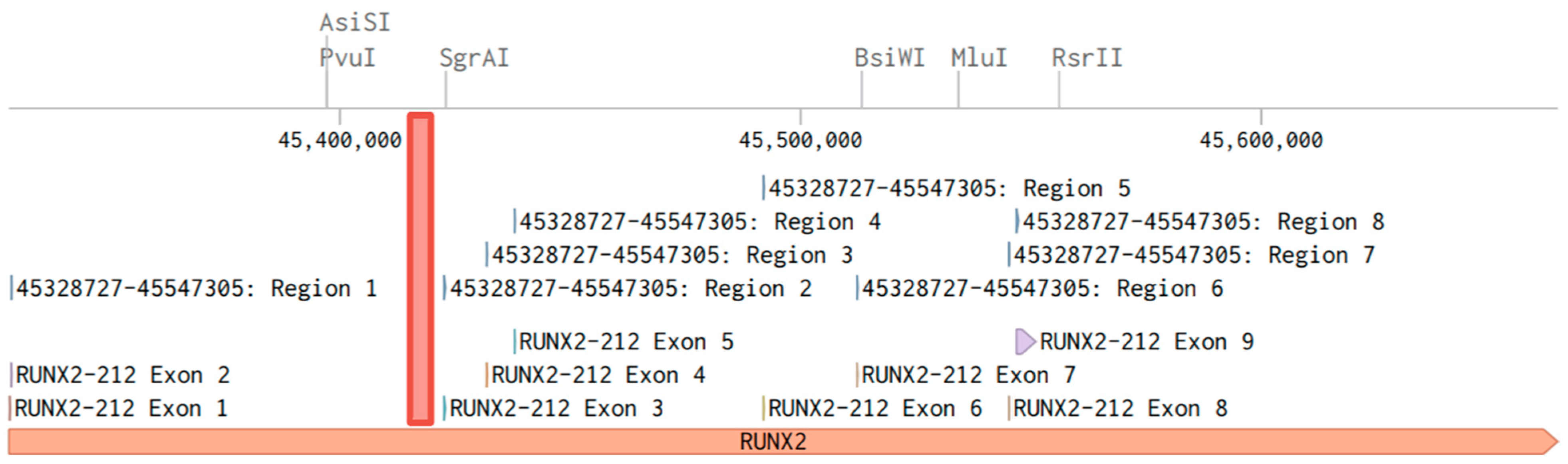
2.2. Functional Significance of Methylation in the Studied CpG Sites of RUNX2
3. Discussion
4. Materials and Methods
4.1. Study Sample
4.2. DNA Methylation Analysis
- The bisulfite conversion of the DNA samples was performed using the EpiTect Fast DNA Bisulfite Kit (QIAGEN®, Germany).
- The purification of the bisulfite-converted DNA was carried out on MinElute spin columns using the EpiTect Fast DNA Bisulfite Kit (QIAGEN®, Germany).
- The polymerase chain reaction (PCR) of the converted DNA was performed using 2 primers, one of which was biotinylated (PyroMark PCR Kit (QIAGEN®, Germany)).
- Pyrosequencing with the sequencing primer was performed using the PyroMark Gold Q24 Reagents Kit (QIAGEN®, Germany).
4.3. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Limitations of the Study
Conflicts of Interest
List of Abbreviations
| OP | osteoporosis |
| RUNX2 | a transcription factor 2 associated with RUNT |
| UCSC | an online genome browser from the University of California, Santa Cruz |
| FRAX | fracture risk assessment tool |
| METTL3 | methyltransferase-like protein 3 |
| DEXA | dual-energy X-ray absorptiometry |
References
- Chin, K.Y.; Ng, B.N.; Rostam, M.K.I.; Muhammad Fadzil, N.F.D.; Raman, V.; Mohamed Yunus, F.; Syed Hashim, S.A.; Ekeuku, S.O. A Mini Review on Osteoporosis: From Biology to Pharmacological Management of Bone Loss. J. Clin. Med. 2022, 11, 6434. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sun, Y.; Xue, X.; Ma, H. Comprehensive Analysis of Epigenetics Mechanisms in Osteoporosis. Front. Genet. 2023, 14, 1153585. [Google Scholar] [CrossRef] [PubMed]
- Salari, N.; Ghasemi, H.; Mohammadi, L.; Behzadi, M.h.; Rabieenia, E.; Shohaimi, S.; Mohammadi, M. The Global Prevalence of Osteoporosis in the World: A Comprehensive Systematic Review and Meta-Analysis. J. Orthop. Surg. Res. 2021, 16, 609. [Google Scholar] [CrossRef] [PubMed]
- Zaigrova, N.K.; Uryasev, O.M.; Shakhanov, A.V.; Tverdova, L.V. Possibility of Frax Tool in the Diagnostics of Osteoporosis. I. P. Pavlov. Russ. Med. Biol. Her. 2017, 25, 62–68. [Google Scholar] [CrossRef]
- Shen, Y.; Huang, X.; Wu, J.; Lin, X.; Zhou, X.; Zhu, Z.; Pan, X.; Xu, J.; Qiao, J.; Zhang, T.; et al. The Global Burden of Osteoporosis, Low Bone Mass, and Its Related Fracture in 204 Countries and Territories, 1990-2019. Front. Endocrinol. 2022, 13, 882241. [Google Scholar] [CrossRef] [PubMed]
- Aibar-Almazán, A.; Voltes-Martínez, A.; Castellote-Caballero, Y.; Afanador-Restrepo, D.F.; Carcelén-Fraile, M.d.C.; López-Ruiz, E. Current Status of the Diagnosis and Management of Osteoporosis. Int. J. Mol. Sci. 2022, 23, 9465. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, C.; Bortoluzzi, S.; Airoldi, C.; Leigheb, F.; Nicolini, D.; Russotto, S.; Vanhaecht, K.; Panella, M. The Early Detection of Osteoporosis in a Cohort of Healthcare Workers: Is There Room for a Screening Program? Int. J. Environ. Res. Public. Health 2021, 18, 1368. [Google Scholar] [CrossRef] [PubMed]
- Yalaev, B.; Tyurin, A.; Prokopenko, I.; Karunas, A.; Khusnutdinova, E.; Khusainova, R. Using a Polygenic Score to Predict the Risk of Developing Primary Osteoporosis. Int. J. Mol. Sci. 2022, 23, 10021. [Google Scholar] [CrossRef]
- Akhiiarova, K.; Khusainova, R.; Minniakhmetov, I.; Mokrysheva, N.; Tyurin, A. Peak Bone Mass Formation: Modern View of the Problem. Biomedicines 2023, 11, 2982. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.C. RUNX2 and Cancer. Int. J. Mol. Sci. 2023, 24, 7001. [Google Scholar] [CrossRef] [PubMed]
- Yalaev, B.I.; Khusainova, R.I. Epigenetic Regulation of Bone Remodeling and Its Role in the Pathogenesis of Primary Osteoporosis. Vavilovskii Zhurnal Genet. I Sel. 2023, 27, 401–410. [Google Scholar] [CrossRef] [PubMed]
- San Martin, I.A.; Varela, N.; Gaete, M.; Villegas, K.; Osorio, M.; Tapia, J.C.; Antonelli, M.; Mancilla, E.E.; Pereira, B.P.; Nathan, S.S.; et al. Impaired Cell Cycle Regulation of the Osteoblast-Related Heterodimeric Transcription Factor RUNX2-Cbfβ in Osteosarcoma Cells. J. Cell. Physiol. 2009, 221, 560–571. [Google Scholar] [CrossRef] [PubMed]
- Jeon, M.J.; Kim, J.A.; Kwon, S.H.; Kim, S.W.; Park, K.S.; Park, S.W.; Kim, S.Y.; Shin, C.S. Activation of Peroxisome Proliferator-Activated Receptor-γ Inhibits the RUNX2-Mediated Transcription of Osteocalcin in Osteoblasts. J. Biol. Chem. 2003, 278, 23270–23277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Li, X.; Qian, S.W.; Guo, L.; Huang, H.Y.; He, Q.; Liu, Y.; Ma, C.G.; Tang, Q.Q. Down-Regulation of Type I RUNX2 Mediated by Dexamethasone Is Required for 3T3-L1 Adipogenesis. Mol. Endocrinol. 2012, 26, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Gersbach, C.A.; Byers, B.A.; Pavlath, G.K.; García, A.J. RUNX2/Cbfa1 Stimulates Transdifferentiation of Primary Skeletal Myoblasts into a Mineralizing Osteoblastic Phenotype. Exp. Cell Res. 2004, 300, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Signaling Networks in RUNX2-Dependent Bone Development. J. Cell. Biochem. 2011, 112, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, T.M.; Jensen, E.D.; Westendorf, J.J. RUNX2: A Master Organizer of Gene Transcription in Developing and Maturing Osteoblasts. Birth Defects Res. Part C—Embryo Today Rev. 2005, 75, 213–225. [Google Scholar] [CrossRef]
- Bradley, E.W.; McGee-Lawrence, M.E.; Westendorf, J.J. Hdac-Mediated Control of Endochondral and Intramembranous Ossification. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, N.; Champagne, N.; Stifani, S.; Yang, X.J. MOZ and MORF Histone Acetyltransferases Interact with the Runt-Domain Transcription Factor RUNX2. Oncogene 2002, 21, 2729–2740. [Google Scholar] [CrossRef]
- Villagra, A.; Cruzat, F.; Carvallo, L.; Paredes, R.; Olate, J.; Van Wijnen, A.J.; Stein, G.S.; Lian, J.B.; Stein, J.L.; Imbalzano, A.N.; et al. Chromatin Remodeling and Transcriptional Activity of the Bone-Specific Osteocalcin Gene Require CCAAT/Enhancer-Binding Protein β-Dependent Recruitment of SWI/SNF Activity. J. Biol. Chem. 2006, 281, 22695–22706. [Google Scholar] [CrossRef]
- Valenti, M.T.; Serafini, P.; Innamorati, G.; Gili, A.; Cheri, S.; Bassi, C.; Carbonare, L.D. RUNX2 Expression: A Mesenchymal Stem Marker for Cancer. Oncol. Lett. 2016, 12, 4167. [Google Scholar] [CrossRef] [PubMed]
- Gomathi, K.; Akshaya, N.; Srinaath, N.; Moorthi, A.; Selvamurugan, N. Regulation of RUNX2 by Post-Translational Modifications in Osteoblast Differentiation. Life Sci. 2020, 245, 117389. [Google Scholar] [CrossRef]
- Chen, J.; Qiu, M.; Dou, C.; Cao, Z.; Dong, S. MicroRNAs in Bone Balance and Osteoporosis. Drug Dev. Res. 2015, 76, 235–245. [Google Scholar] [CrossRef]
- Zhang, W.; Wu, Y.; Shiozaki, Y.; Sugimoto, Y.; Takigawa, T.; Tanaka, M.; Matsukawa, A.; Ozaki, T. MiRNA-133a-5p Inhibits the Expression of Osteoblast Differentiation-Associated Markers by Targeting the 3′ UTR of RUNX2. DNA Cell Biol. 2018, 37, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Xiao, G.; Jiang, D.; Yang, Q.; Hatch, N.E.; Roca, H.; Franceschi, R.T. Identification and Functional Characterization of ERK/MAPK Phosphorylation Sites in the RUNX2 Transcription Factor. J. Biol. Chem. 2009, 284, 32533–32543. [Google Scholar] [CrossRef] [PubMed]
- Afzal, F.; Pratap, J.; Ito, K.; Ito, Y.; Stein, J.L.; Van Wijnen, A.J.; Stein, G.S.; Lian, J.B.; Javed, A. Smad Function and Intranuclear Targeting Share a RUNX2 Motif Required for Osteogenic Lineage Induction and BMP2 Responsive Transcription. J. Cell. Physiol. 2005, 204, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Li, W.; Yang, X.; Na, L.; Chen, L.; Liu, G. The Roles of Epigenetics Regulation in Bone Metabolism and Osteoporosis. Front. Cell Dev. Biol. 2021, 8, 619301. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.A.; Tsai, P.C.; Joehanes, R.; Zheng, J.; Trajanoska, K.; Soerensen, M.; Forgetta, V.; Castillo-Fernandez, J.E.; Frost, M.; Spector, T.D.; et al. Epigenome-wide Association of DNA Methylation in Whole Blood with Bone Mineral Density. J. Bone Miner. Res. 2017, 32, 1644. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Chen, Y.; Kong, J.; Xu, S.; Xu, S.; Shuai, Z.; Cai, G.; Pan, F. Differences of RUNX2 Gene Promoter Methylation and Transcription Level in Ankylosing Spondylitis. Int. J. Rheum. Dis. 2023, 26, 2526–2533. [Google Scholar] [CrossRef] [PubMed]
- Rice, S.J.; Aubourg, G.; Sorial, A.K.; Almarza, D.; Tselepi, M.; Deehan, D.J.; Reynard, L.N.; Loughlin, J. Identification of a Novel, Methylation-Dependent, RUNX2 Regulatory Region Associated with Osteoarthritis Risk. Hum. Mol. Genet. 2018, 27, 3464–3474. [Google Scholar] [CrossRef]
- Hagh, M.F.; Noruzinia, M.; Mortazavi, Y.; Soleimani, M.; Kaviani, S.; Abroun, S.; Fard, A.D.; Maymand, M.M. Different Methylation Patterns of RUNX2, OSX, DLX5 and BSP in Osteoblastic Differentiation of Mesenchymal Stem Cells. Cell J. 2015, 17, 71. [Google Scholar] [CrossRef]
- Alajem, A.; Roth, H.; Ratgauzer, S.; Bavli, D.; Motzik, A.; Lahav, S.; Peled, I.; Ram, O. DNA Methylation Patterns Expose Variations in Enhancer-Chromatin Modifications during Embryonic Stem Cell Differentiation. PLoS Genet. 2021, 17, e1009498. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Wang, L.; Li, G.; Shen, G.; Zhang, P.; Xu, Y. Methylation of Bone SOST Impairs SP7, RUNX2, and ERα Transactivation in Patients with Postmenopausal Osteoporosis. Biochem. Cell Biol. 2019, 97, 369–374. [Google Scholar] [CrossRef]
- Marini, F.; Cianferotti, L.; Brandi, M.L. Epigenetic Mechanisms in Bone Biology and Osteoporosis: Can They Drive Therapeutic Choices? Int. J. Mol. Sci. 2016, 17, 1329. [Google Scholar] [CrossRef] [PubMed]
- Krstic, N.; Bishop, N.; Curtis, B.; Cooper, C.; Harvey, N.; Lilycrop, K.; Murray, R.; Owen, R.; Reilly, G.; Skerry, T.; et al. Early Life Vitamin D Depletion and Mechanical Loading Determine Methylation Changes in the RUNX2, RXRA, and Osterix Promoters in Mice. Genes Nutr. 2022, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J. Epigenetics of Osteoporosis: Critical Analysis of Epigenetic Epidemiology Studies. Curr. Genom. 2015, 16, 405–410. [Google Scholar] [CrossRef] [PubMed]
- de Nigris, F.; Ruosi, C.; Colella, G.; Napoli, C. Epigenetic Therapies of Osteoporosis. Bone 2021, 142, 115680. [Google Scholar] [CrossRef]
- Fontalis, A.; Kenanidis, E.; Kotronias, R.A.; Papachristou, A.; Anagnostis, P.; Potoupnis, M.; Tsiridis, E. Current and Emerging Osteoporosis Pharmacotherapy for Women: State of the Art Therapies for Preventing Bone Loss. Expert. Opin. Pharmacother. 2019, 20, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.C.; Chen, Y.J.; Lian, Y.C.; Chang, B.E.; Huang, C.C.; Huang, W.L.; Pan, Y.H.; Jeng, J.H. Butyrate Stimulates Histone H3 Acetylation, 8-Isoprostane Production, RANKL Expression, and Regulated Osteoprotegerin Expression/Secretion in MG-63 Osteoblastic Cells. Int. J. Mol. Sci. 2018, 19, 4071. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.J.; Ohta, S.; Sheu, K.M.; Spreafico, R.; Adelaja, A.; Taylor, B.; Hoffmann, A. NF-ΚB Dynamics Determine the Stimulus Specificity of Epigenomic Reprogramming in Macrophages. Science 2021, 372, 1349–1353. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Yuan, Y.; He, M.; Gong, R.; Lei, H.; Zhou, H.; Wang, W.; Du, W.; Ma, T.; Liu, S.; et al. M6A Methylation of Precursor-MiR-320/RUNX2 Controls Osteogenic Potential of Bone Marrow-Derived Mesenchymal Stem Cells. Mol. Ther. Nucleic Acids 2020, 19, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Hawkins-Hooker, A.; Visonà, G.; Narendra, T.; Rojas-Carulla, M.; Schölkopf, B.; Schweikert, G. Getting Personal with Epigenetics: Towards Individual-Specific Epigenomic Imputation with Machine Learning. Nat. Commun. 2023, 14, 4750. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, P.; Luthman, H.; McGuigan, F.E.; Akesson, K.E. Epigenome-Wide Cross-Tissue Correlation of Human Bone and Blood DNA Methylation–Can Blood Be Used as a Surrogate for Bone? Epigenetics 2020, 16, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Mathew, C.G.P. The Isolation of High Molecular Weight Eukaryotic DNA. In Nucleic Acids; Methods in Molecular Biology; Humana Press: New York, NY, USA, 1984; pp. 31–34. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Females | |||||||
| Median (case) (%) | Median (control) (%) | ||||||
| CpG1 | CpG2 | CpG3 | Mean | CpG1 | CpG2 | CpG3 | Mean |
| 4 | 8 | 9 | 7 | 5 | 8 | 9 | 7.67 |
| Mean (case) (%) | Mean (control) (%) | ||||||
| CpG1 | CpG2 | CpG3 | Mean | CpG1 | CpG2 | CpG3 | Mean |
| 4.21 | 9.51 | 10.08 | 7.93 | 5.7 | 9.35 | 9.02 | 8.03 |
| Males | |||||||
| Median (case) (%) | Median (control) (%) | ||||||
| CpG1 | CpG2 | CpG3 | Mean | CpG1 | CpG2 | CpG3 | Mean |
| 4 | 8 | 9 | 7 | 5 | 10 | 10 | 8.33 |
| Mean (case) (%) | Mean (control) (%) | ||||||
| CpG1 | CpG2 | CpG3 | Mean | CpG1 | CpG2 | CpG3 | Mean |
| 4.34 | 8.48 | 9.18 | 7.35 | 5.20 | 9.82 | 10.51 | 8.51 |
| General sample | |||||||
| Median (case) (%) | Median (control) (%) | ||||||
| CpG1 | CpG2 | CpG3 | Mean | CpG1 | CpG2 | CpG3 | Mean |
| 4 | 8 | 9 | 7.33 | 5 | 9 | 9 | 7.67 |
| Mean (case) (%) | Mean (control) (%) | ||||||
| CpG1 | CpG2 | CpG3 | Mean | CpG1 | CpG2 | CpG3 | Mean |
| 4.59 | 9.23 | 9.89 | 7.90 | 5.05 | 9.57 | 9.90 | 8.17 |
| Parameters | Estimate | Std. Error | z-Value | p-Value |
|---|---|---|---|---|
| Males | ||||
| Sample | 1.632 | 0.749 | 2.178 | 0.029 |
| CpG1 | −0.325 | 0.152 | −2.130 | 0.033 |
| CpG2 | −0.174 | 0.089 | −1.941 | 0.042 |
| CpG3 | −0.204 | 0.093 | −2.188 | 0.028 |
| Mean | −0.234 | 0.110 | −2.127 | 0.033 |
| Females | ||||
| Sample | 1.653 | 0.714 | 2.315 | 0.020 |
| CpG1 | −0.337 | 0.136 | −2.466 | 0.013 |
| CpG2 | −0.156 | 0.132 | −1.179 | 0.238 |
| CpG3 | 0.300 | 0.166 | 1.807 | 0.070 |
| Mean | −9.017 | 2.135 | −1.990 | 0.067 |
| General sample | ||||
| Sample | 1.798 | 0.535 | 3.356 | 0.0008 |
| CpG1 | −0.370 | 0.108 | −3.421 | 0.0006 |
| CpG2 | 1.741 | 3.879 | 0.910 | 0.363 |
| CpG3 | 1.859 | 3.865 | 0.916 | 0.360 |
| Mean | −0.079 | 0.067 | −1.184 | 0.236 |
| Gene | Genomic Coordinates | Analyzed DNA Sequence | Analyzed DNA Sequence Sequence after Bisulfite Conversion | CpG- Sites |
|---|---|---|---|---|
| RUNX2 | 45420262-45420295 | GCACGGAAGATGGGGGCCTGGTGCCAGTCGCGGA | GTAUGGAAGATGGGGGTTTGGTGTTAGTUGUGGA | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yalaev, B.; Tyurin, A.; Akhiiarova, K.; Khusainova, R. Hypomethylation of the RUNX2 Gene Is a New Potential Biomarker of Primary Osteoporosis in Men and Women. Int. J. Mol. Sci. 2024, 25, 7312. https://doi.org/10.3390/ijms25137312
Yalaev B, Tyurin A, Akhiiarova K, Khusainova R. Hypomethylation of the RUNX2 Gene Is a New Potential Biomarker of Primary Osteoporosis in Men and Women. International Journal of Molecular Sciences. 2024; 25(13):7312. https://doi.org/10.3390/ijms25137312
Chicago/Turabian StyleYalaev, Bulat, Anton Tyurin, Karina Akhiiarova, and Rita Khusainova. 2024. "Hypomethylation of the RUNX2 Gene Is a New Potential Biomarker of Primary Osteoporosis in Men and Women" International Journal of Molecular Sciences 25, no. 13: 7312. https://doi.org/10.3390/ijms25137312