
Anticancer Activity of Metallodrugs and Metallizing Host Defense Peptides—Current Developments in Structure-Activity Relationship

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
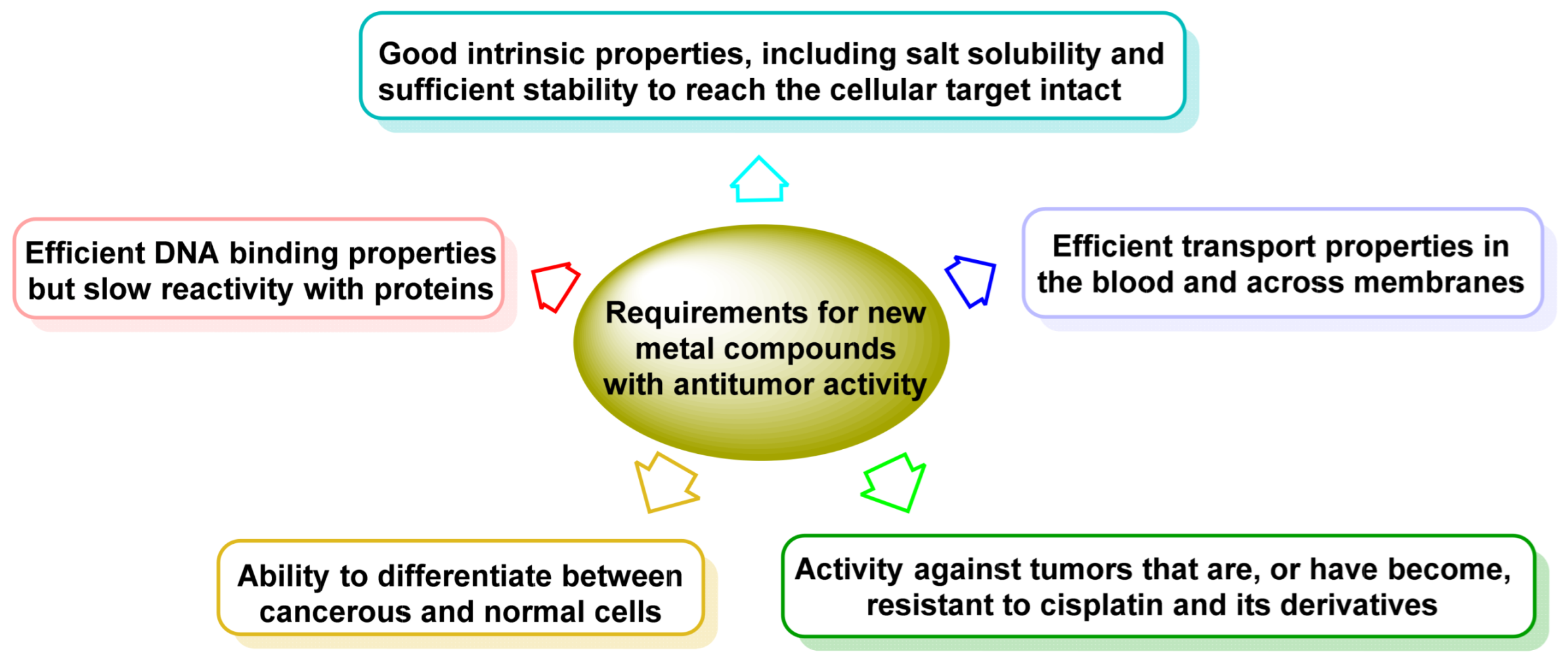
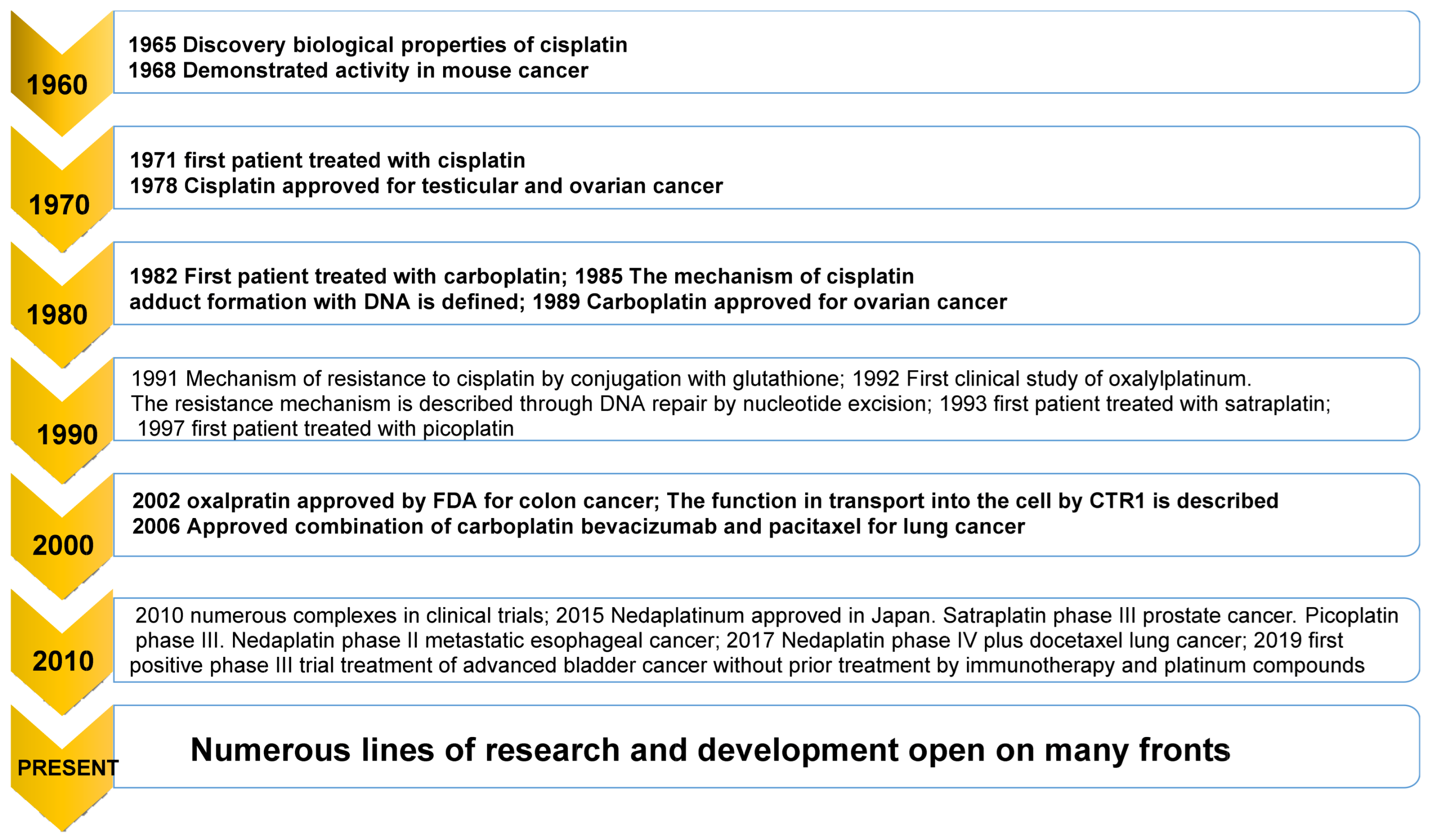
1. Introduction
2. Platinum (II) Complexes
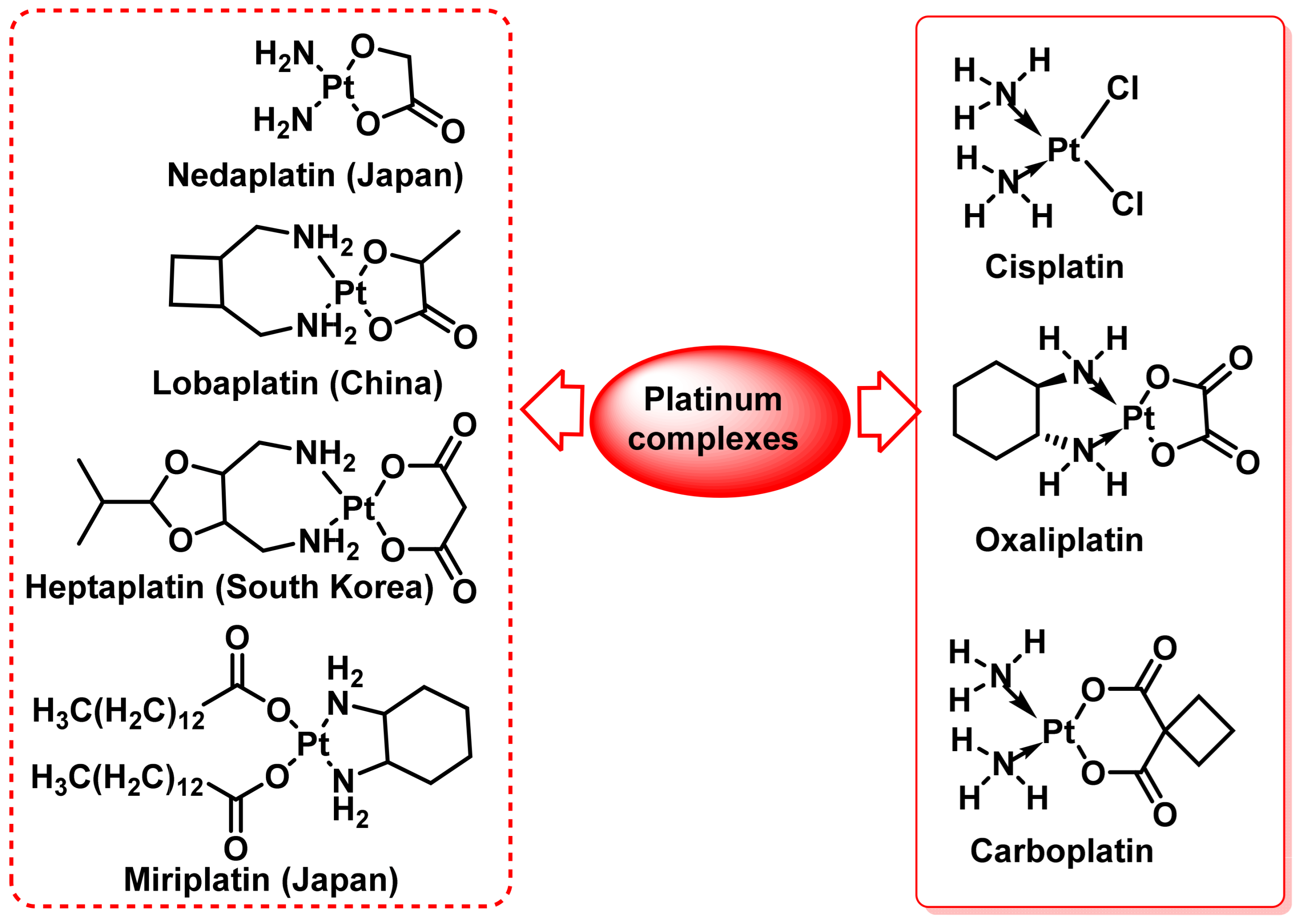
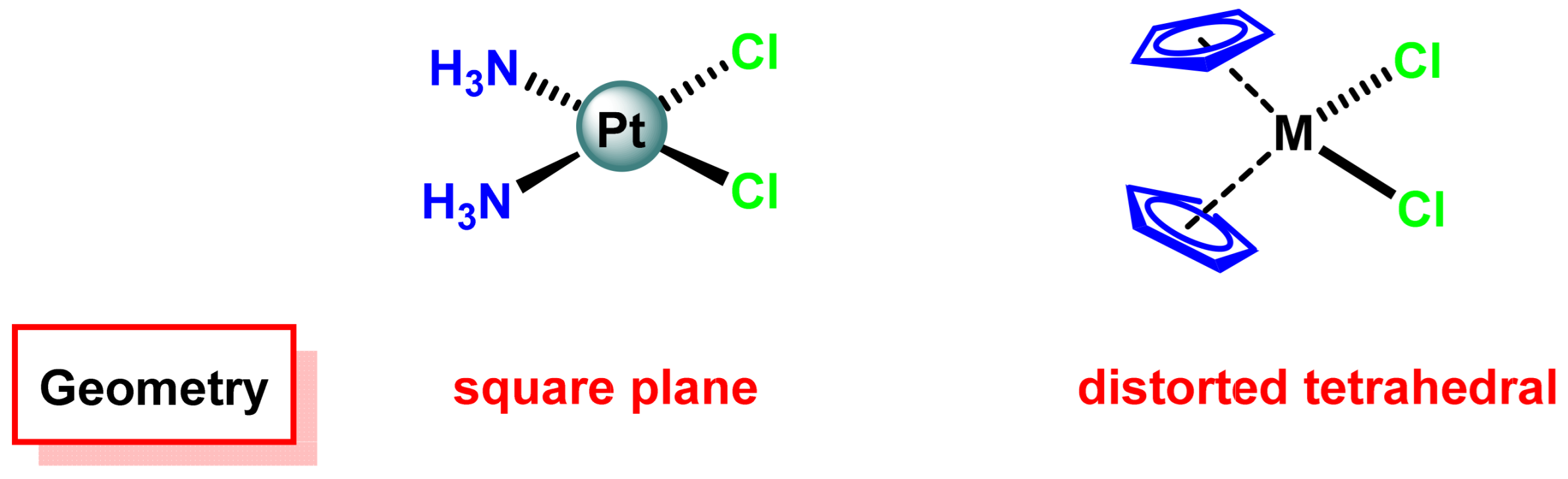
2.1. Cis-Platinum(II) Complexes
2.1.1. Carboplatin
2.1.2. Oxaliplatin
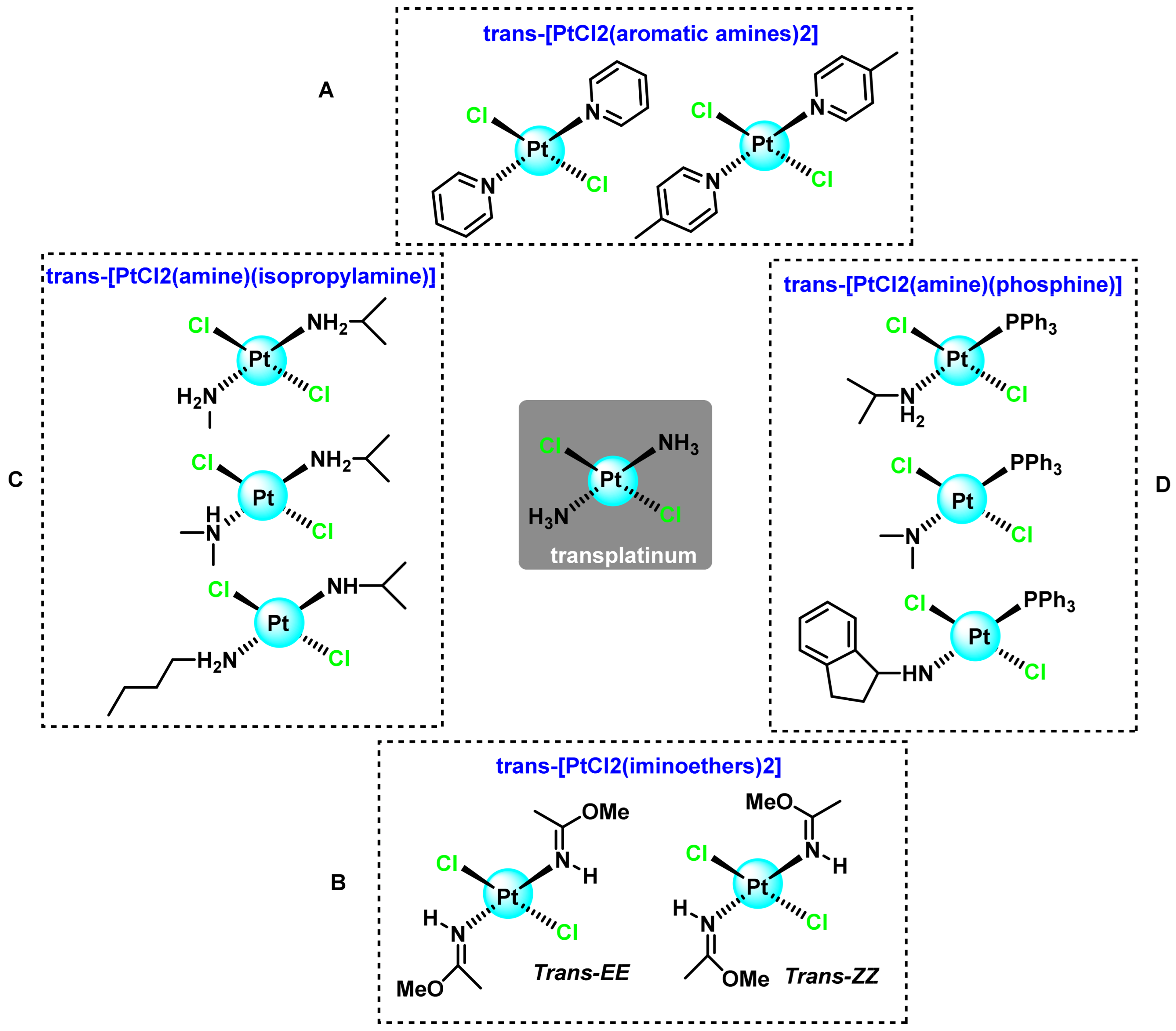
2.2. Trans-Platinum(II) Complexes
2.2.1. Trans-[PtCl2L2] Complexes where L Are Aromatic Amines
2.2.2. Trans-[PtCl2L2] Complexes where L Are Iminoethers
2.2.3. Trans-[PtCl2L2] Complexes where L Are Aliphatic Amines
2.2.4. Trans-[PtCl2(Triphenylphosphine Aliphatic Amine)] Complexes
2.3. Polynuclear Platinum(II) Compounds
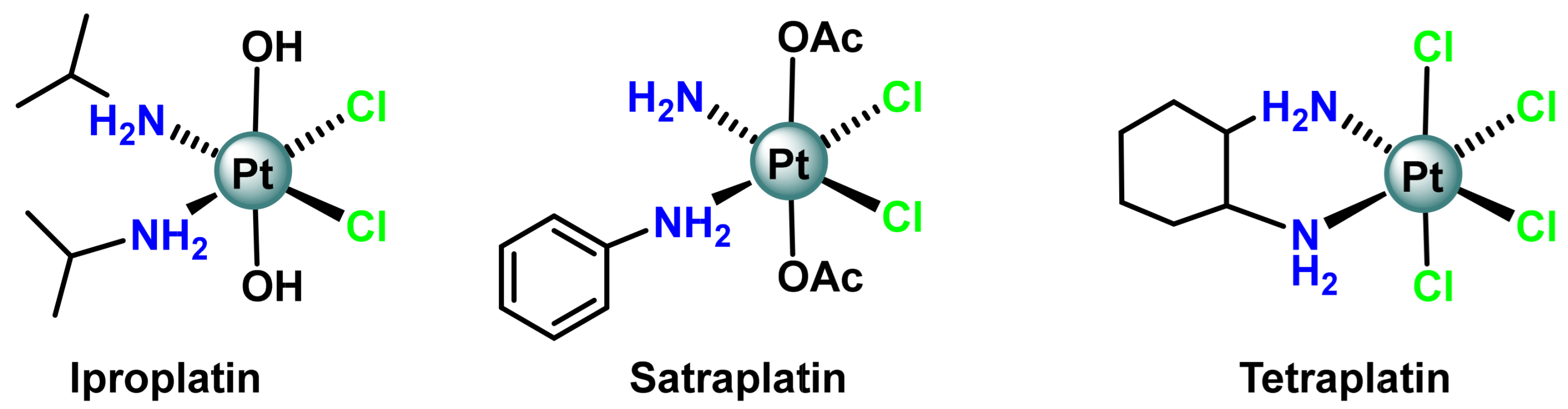
3. Platinum (IV) Prodrugs
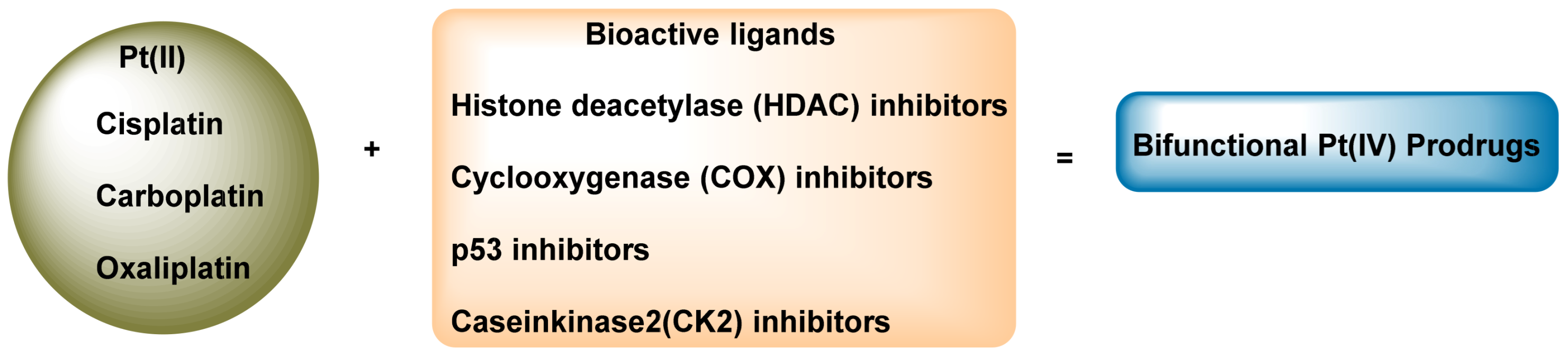
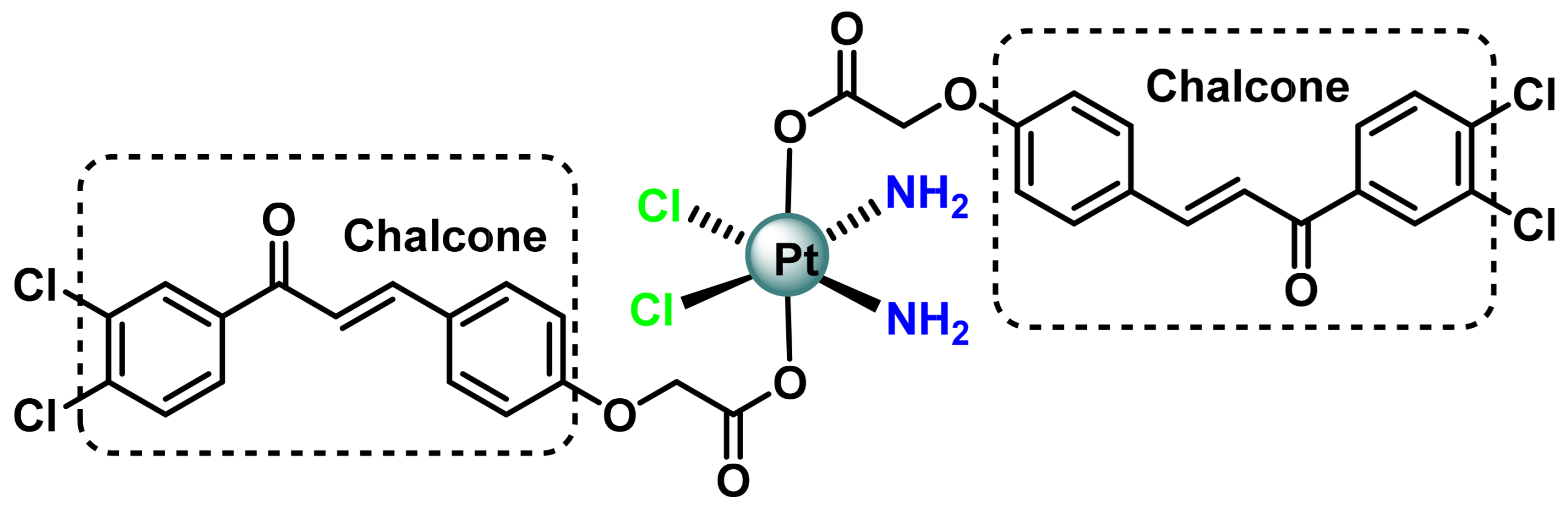
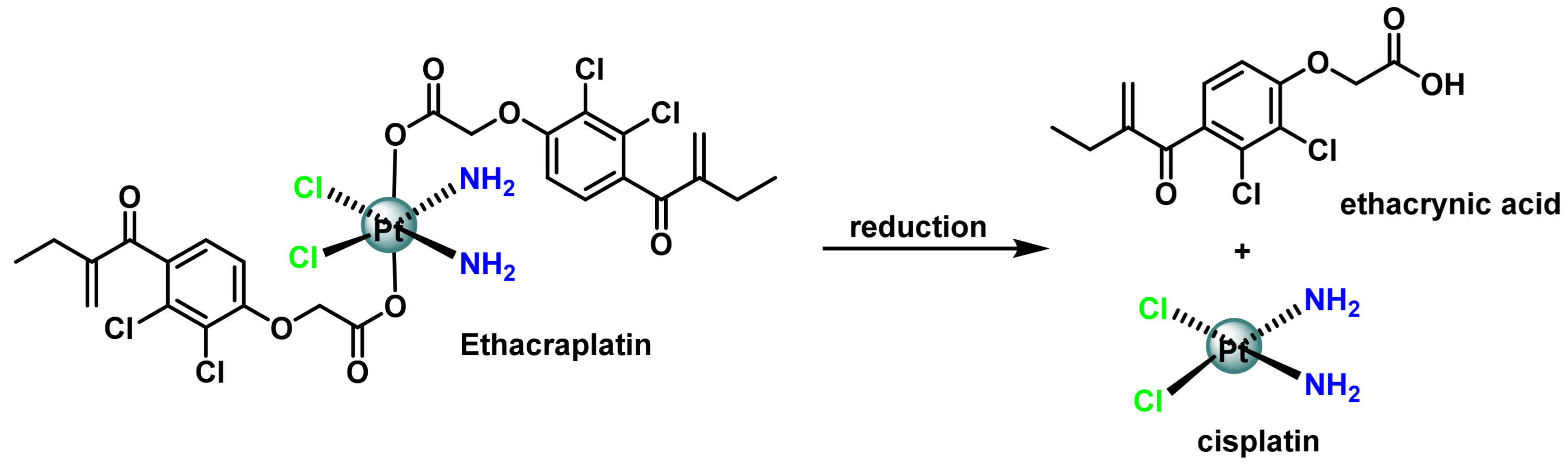
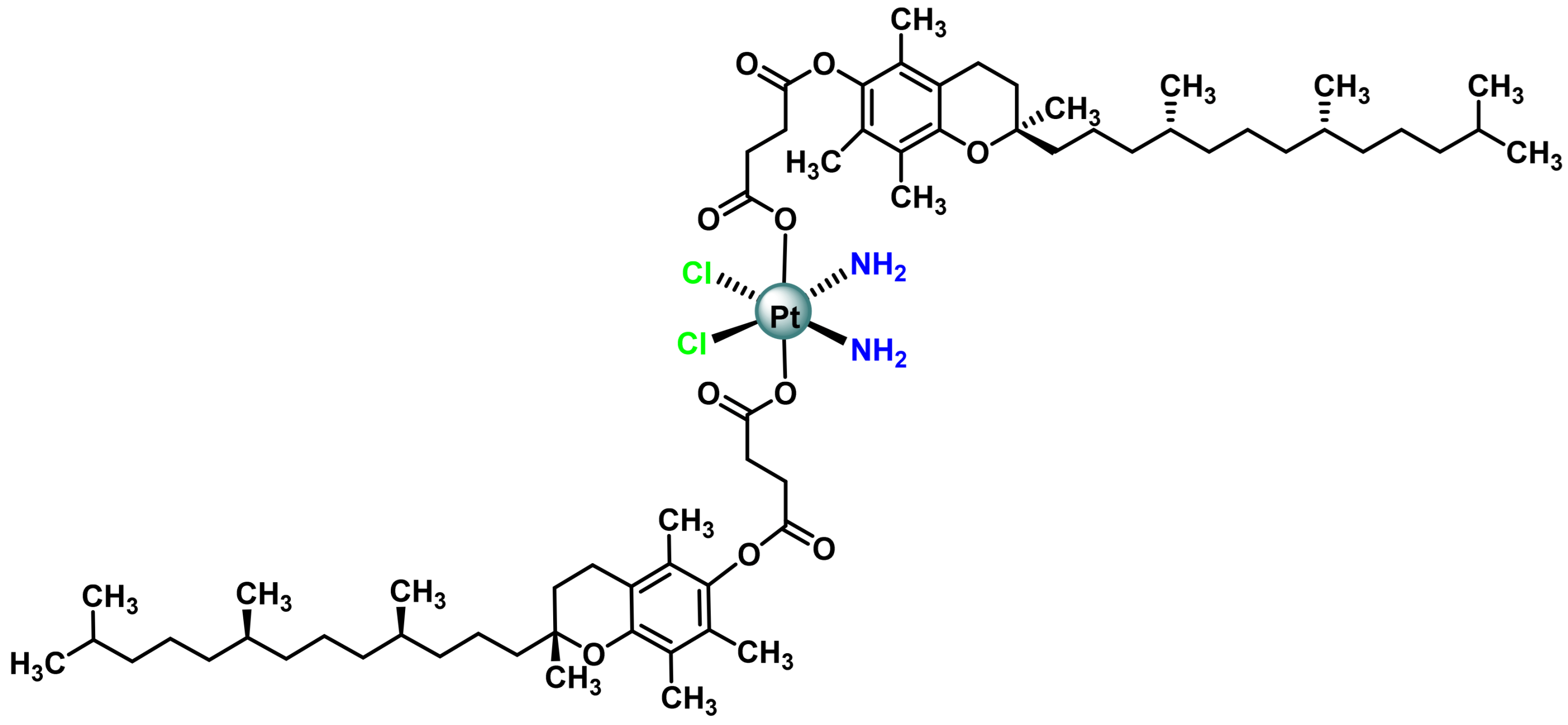
3.1. Platinum (IV) Complexes with Bioactive Ligands
3.2. Pt(IV) Prodrugs with Histone Deacetylase (HDAC) Inhibitory Ligands
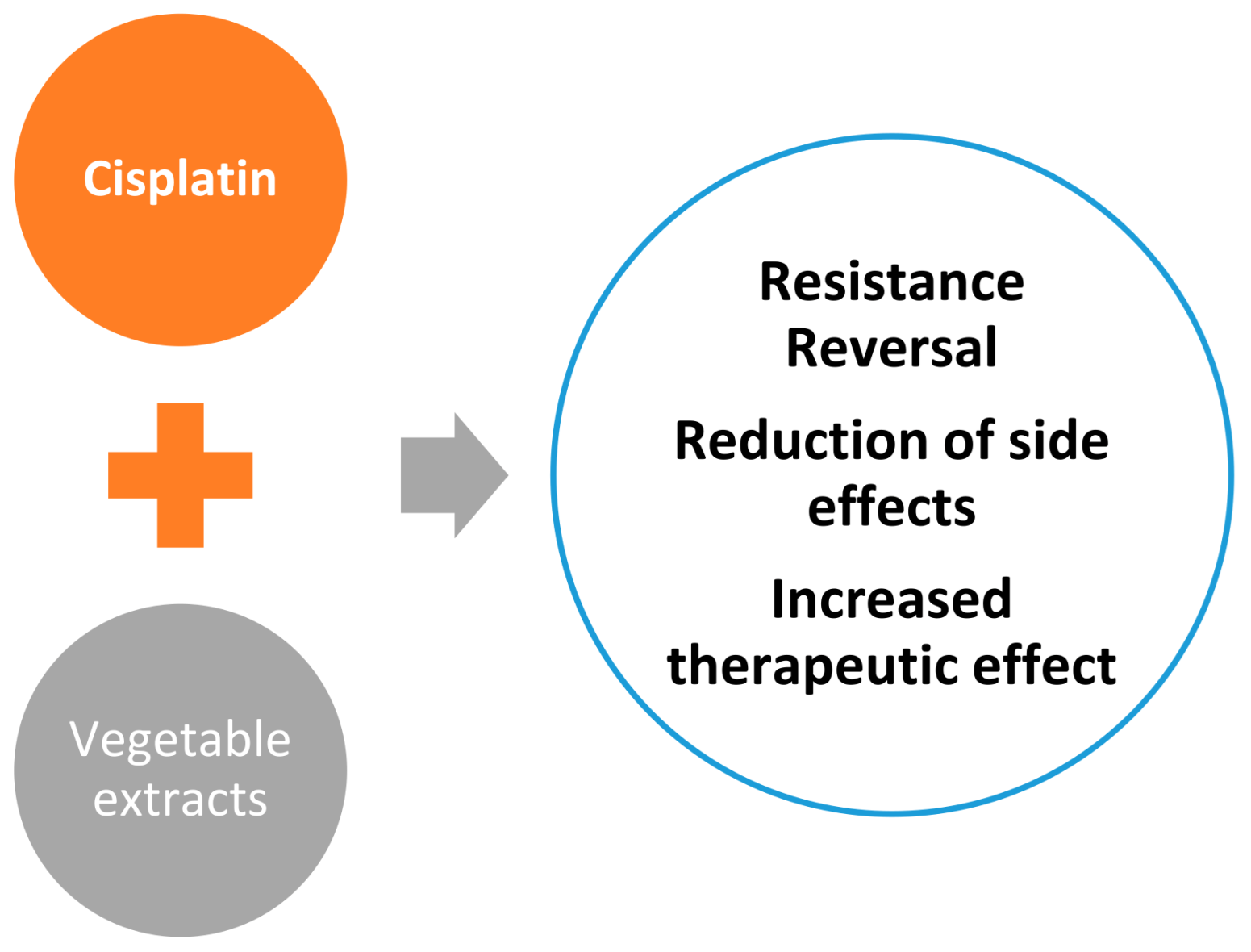
4. Plant Extract-Cisplatin Combination
5. Combining Metallodrugs with Host Defense Peptides for Enhanced Anticancer Therapy
5.1. Combining Metallodrugs with HDPs
5.2. Piscidins with Cu2⁺
5.3. Cisplatin and Tachyplesin I
5.4. Combined Cytotoxic Effects
6. Metallocene Complexes in Cancer Therapies
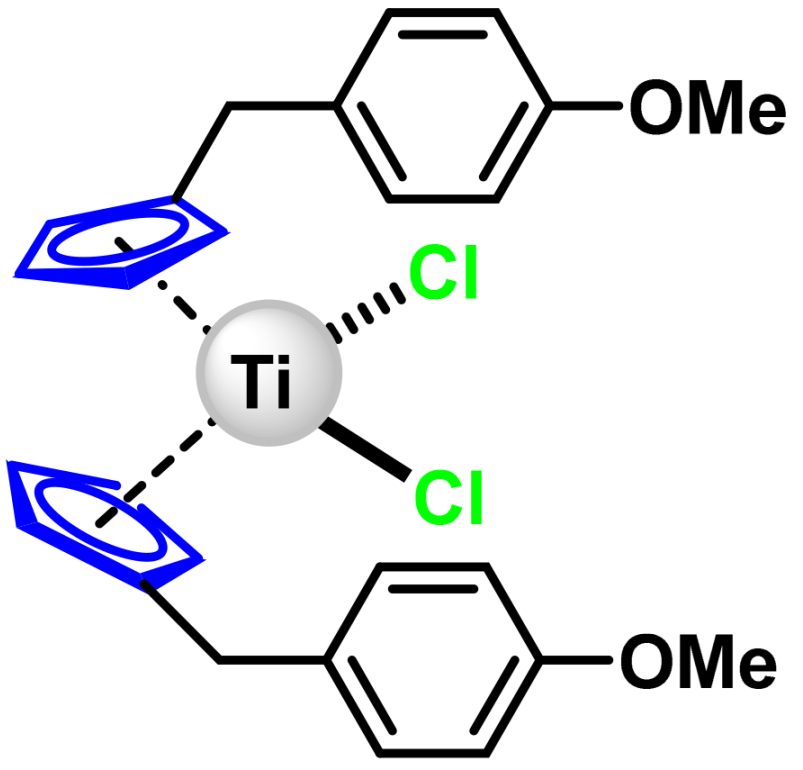
6.1. Titanocene Dichloride
6.2. Titanocene Y
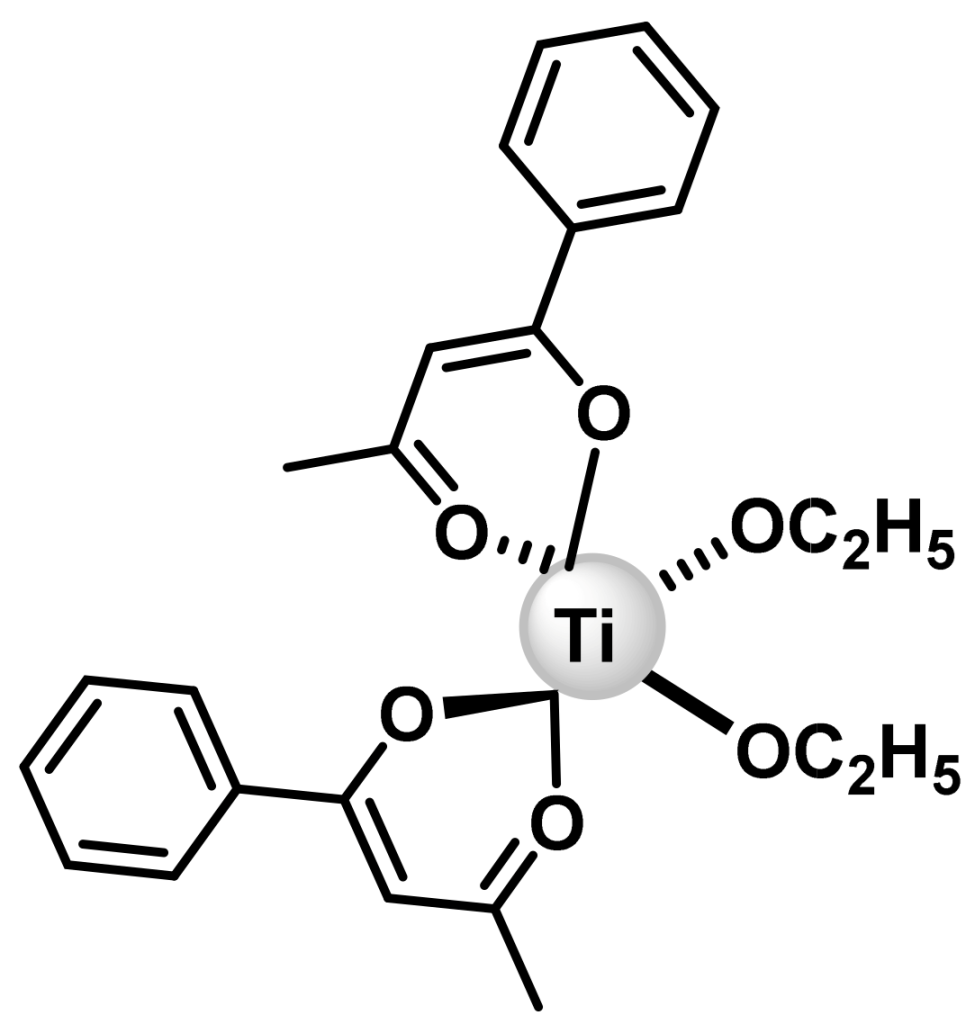
6.3. Budotitane
7. Ruthenium Complexes
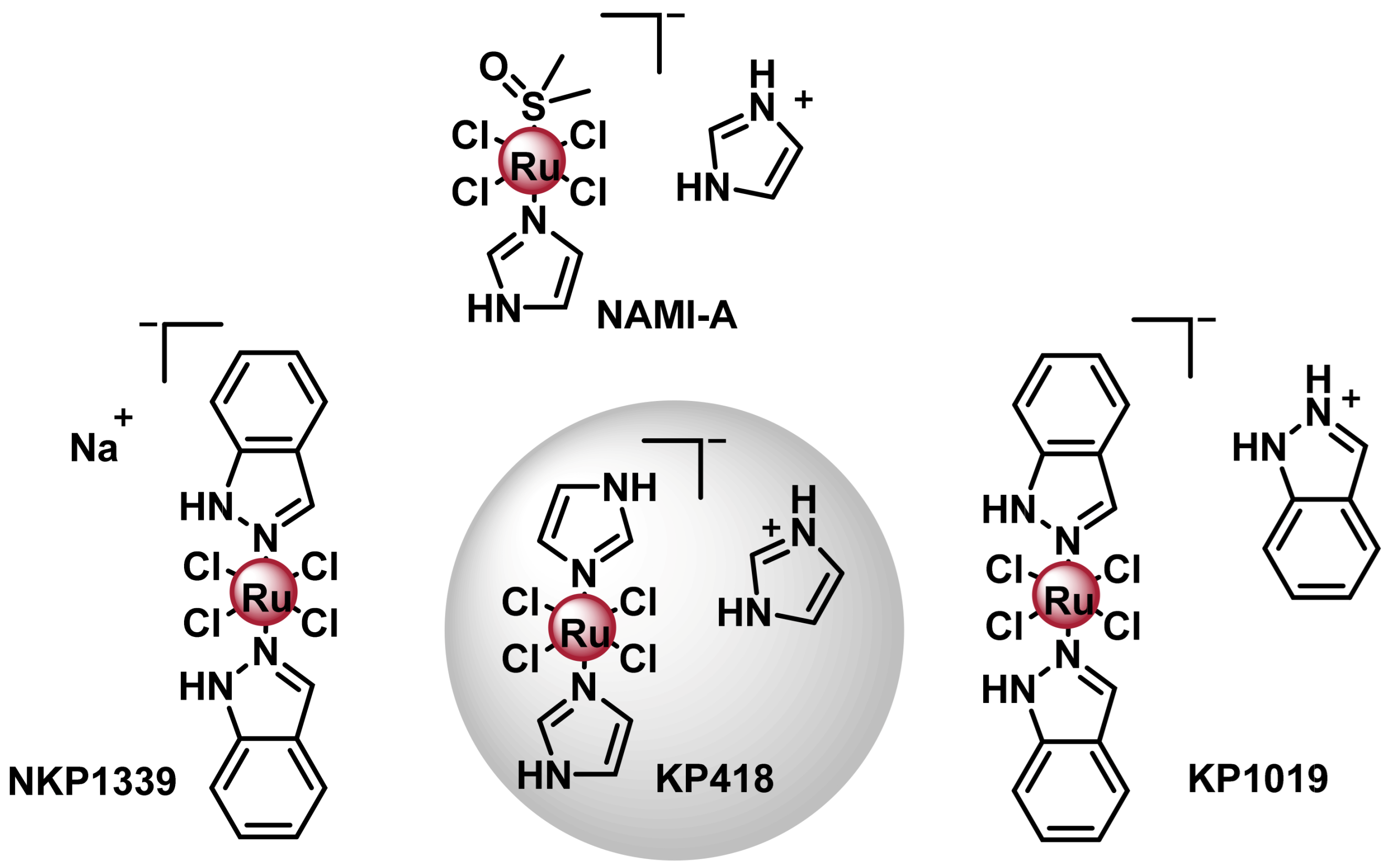
7.1. Ru(III) Complexes
7.1.1. NAMI- A
7.1.2. KP-1019/NKP1339
7.2. Ruthenium(II) Complexes
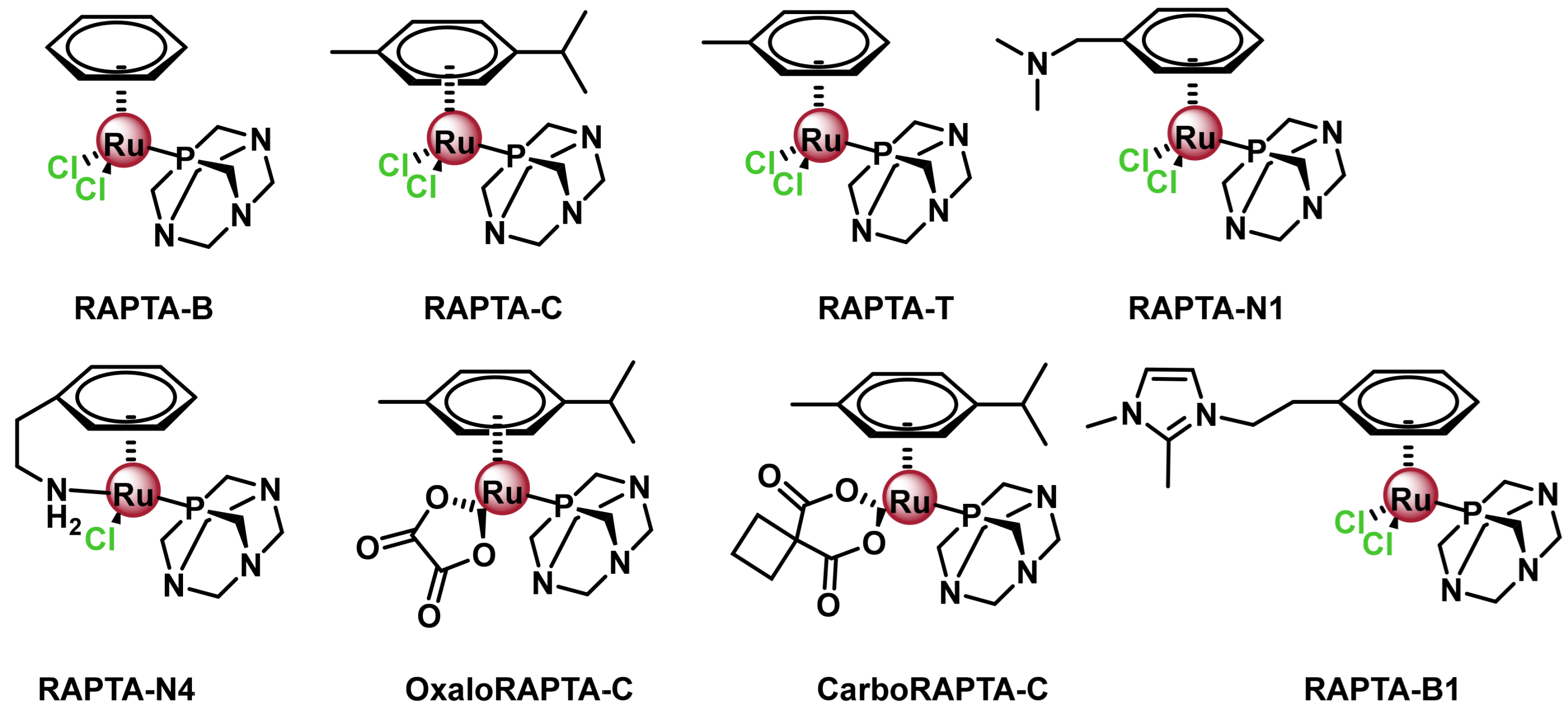
7.2.1. Ruthenium(II) Complexes with Arene Ligands
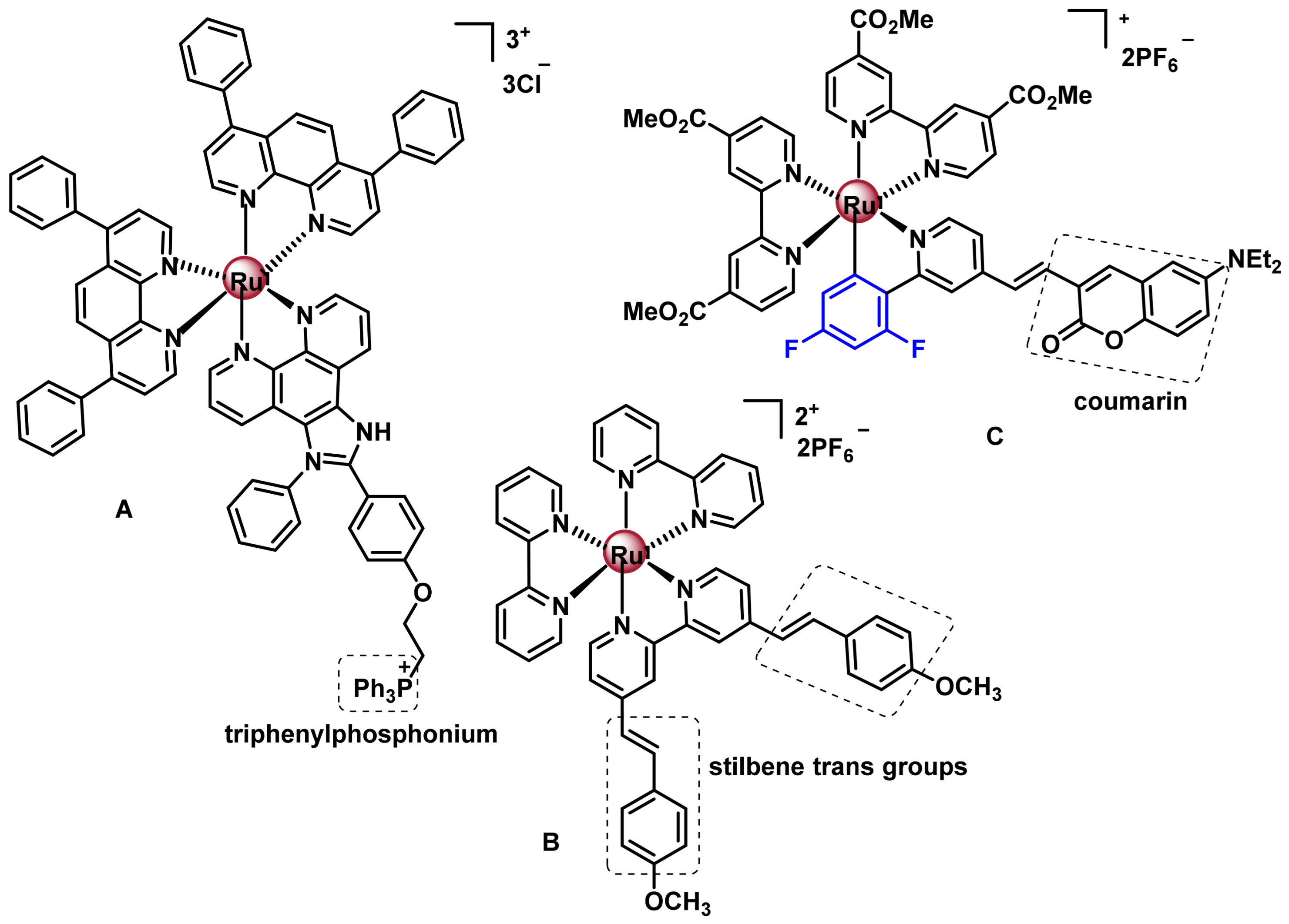
7.2.2. Octahedral Ruthenium(II) Complexes with Polypyridine Ligands
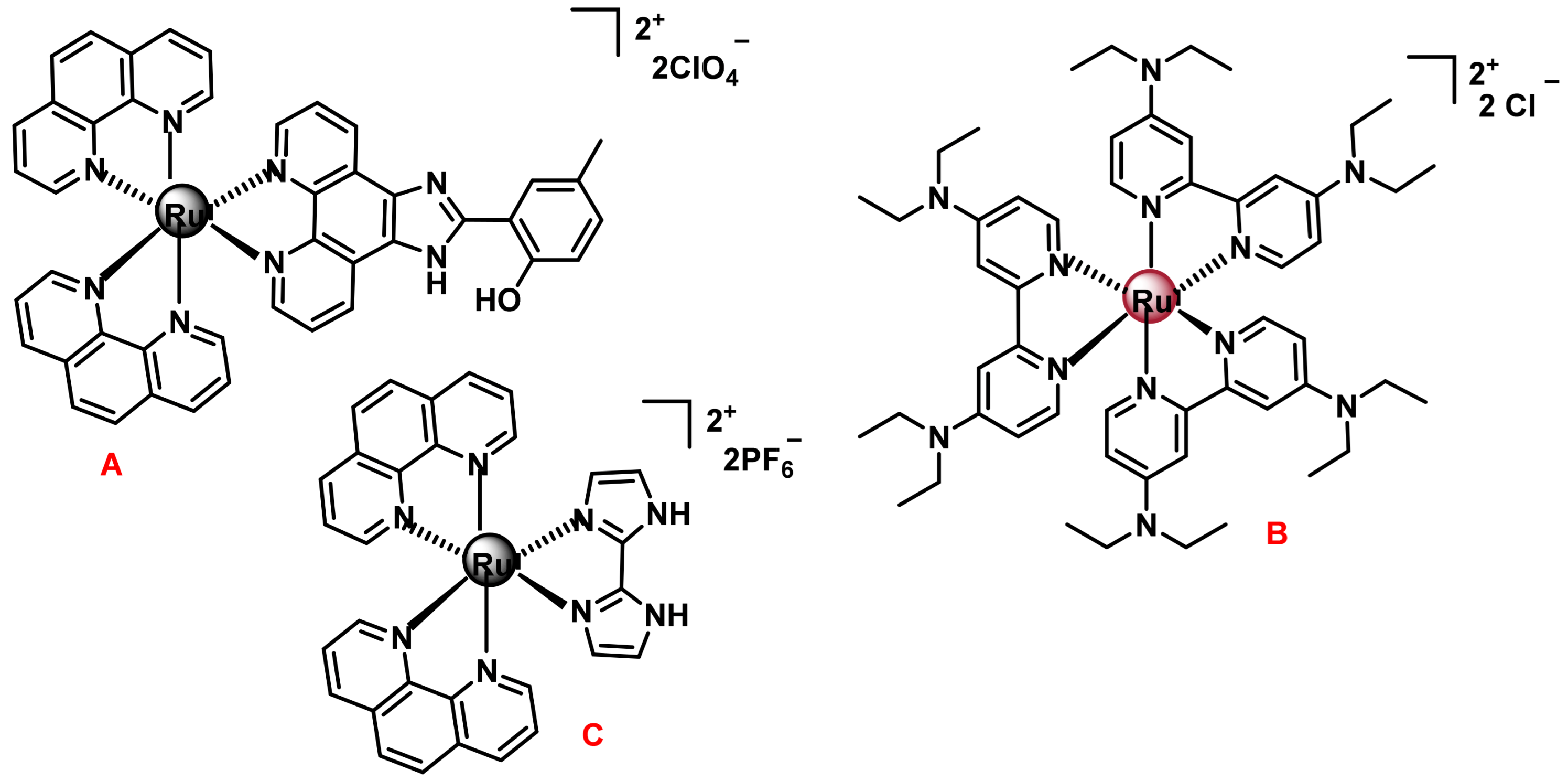
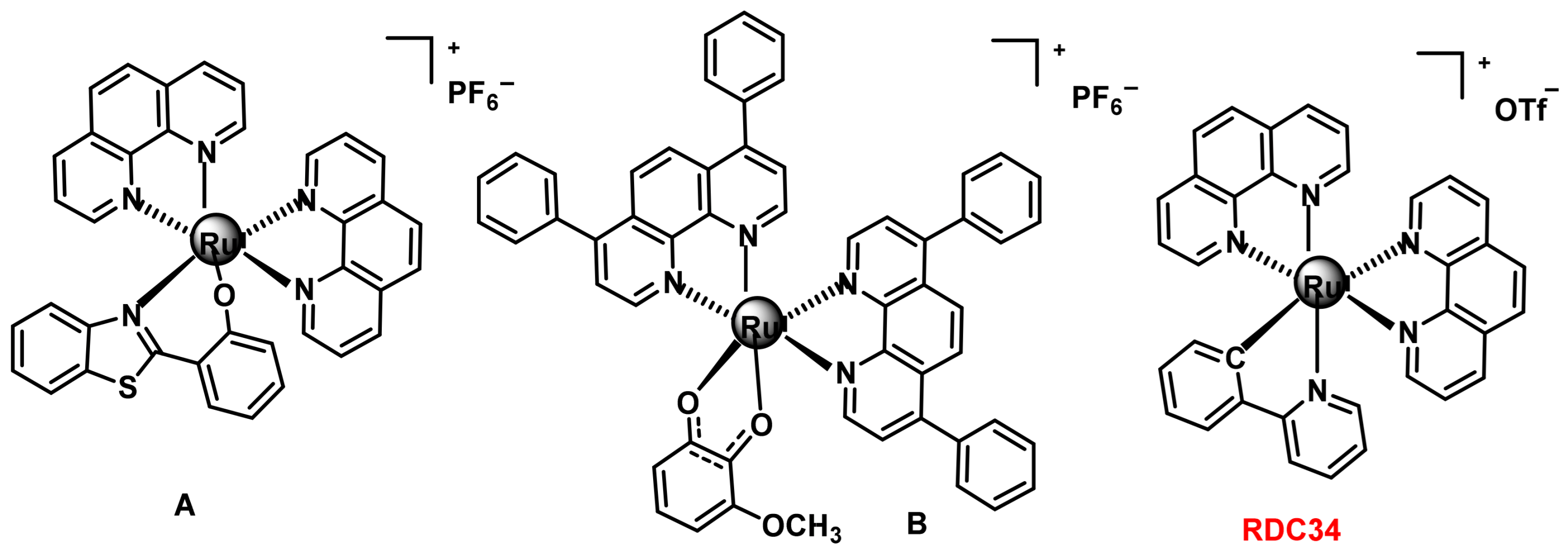
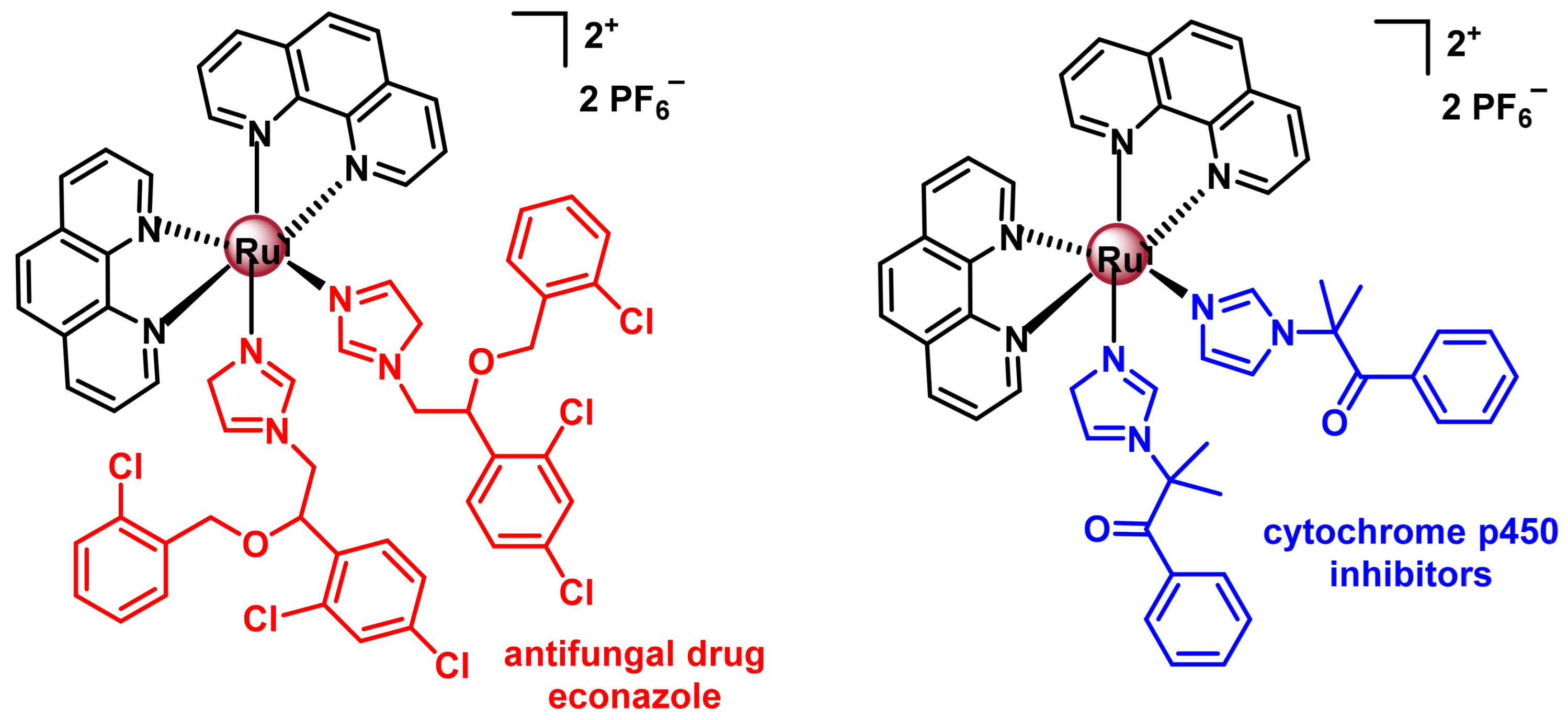
7.2.3. Polypyridyl Ru(II) Complexes as Photosensitisers (PS) in Photodynamic Therapy (PDT)
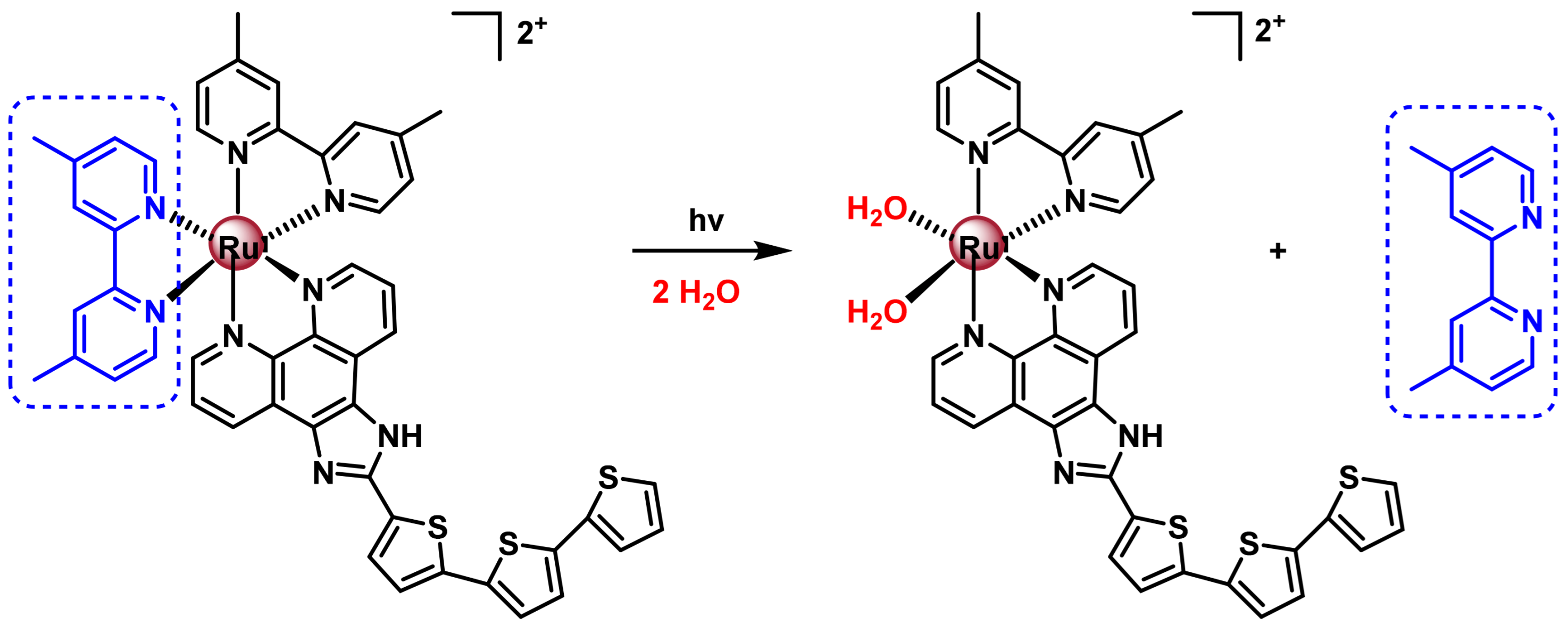
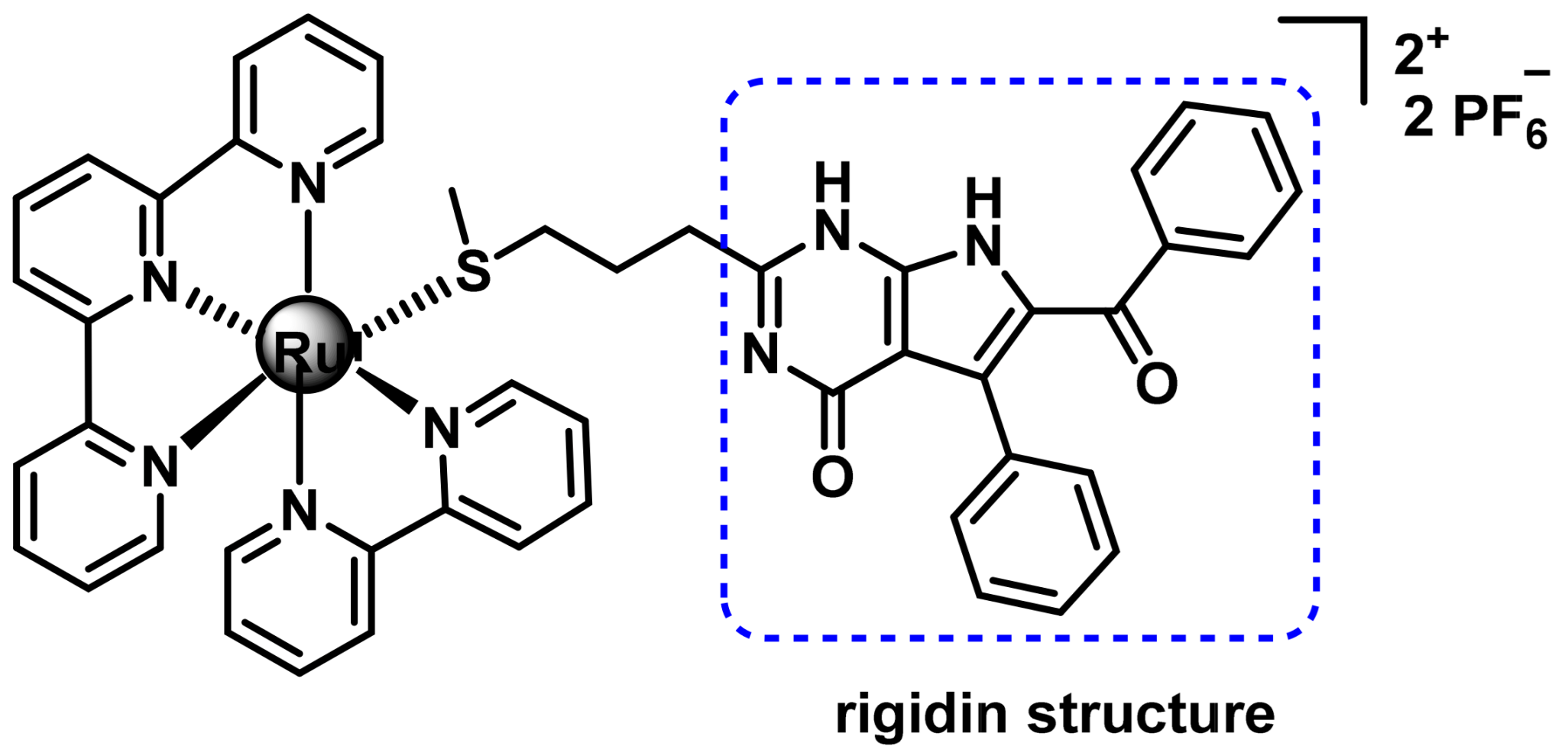
7.2.4. Ru(II) Complexes for Photoactivated Chemotherapy (PACT)
8. Gold Complexes
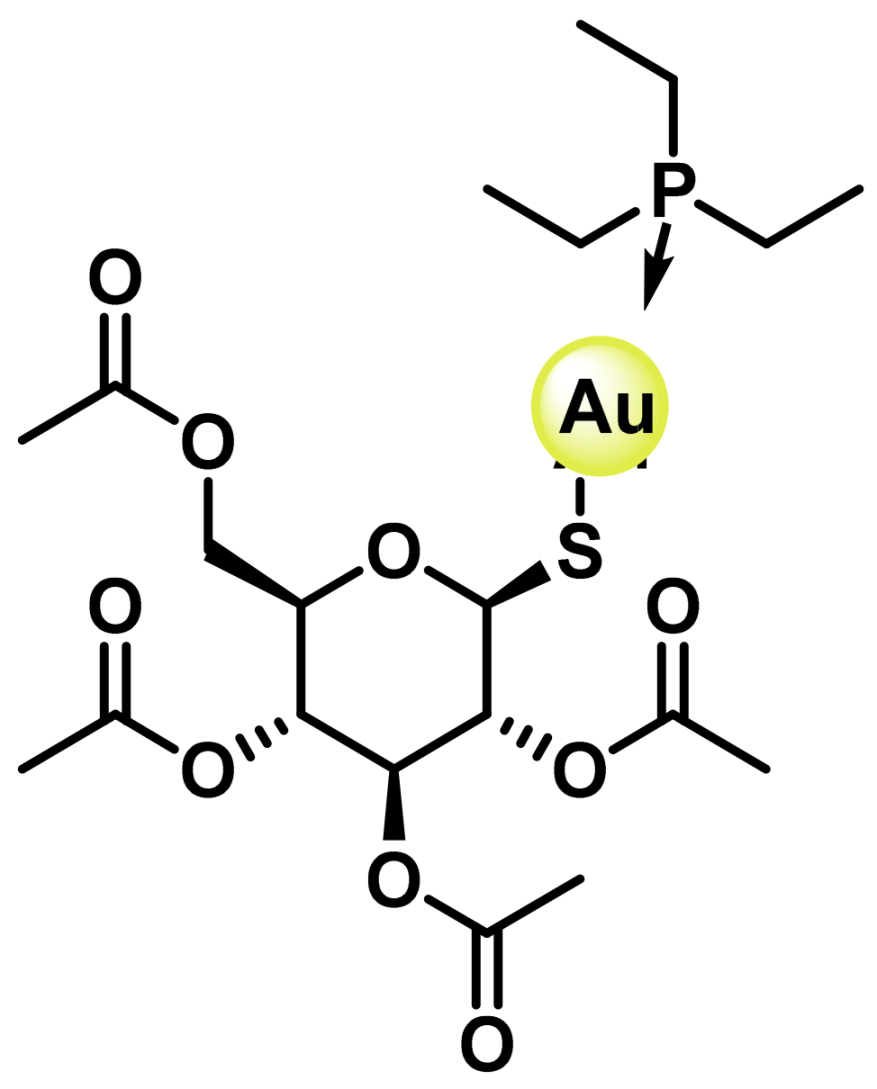
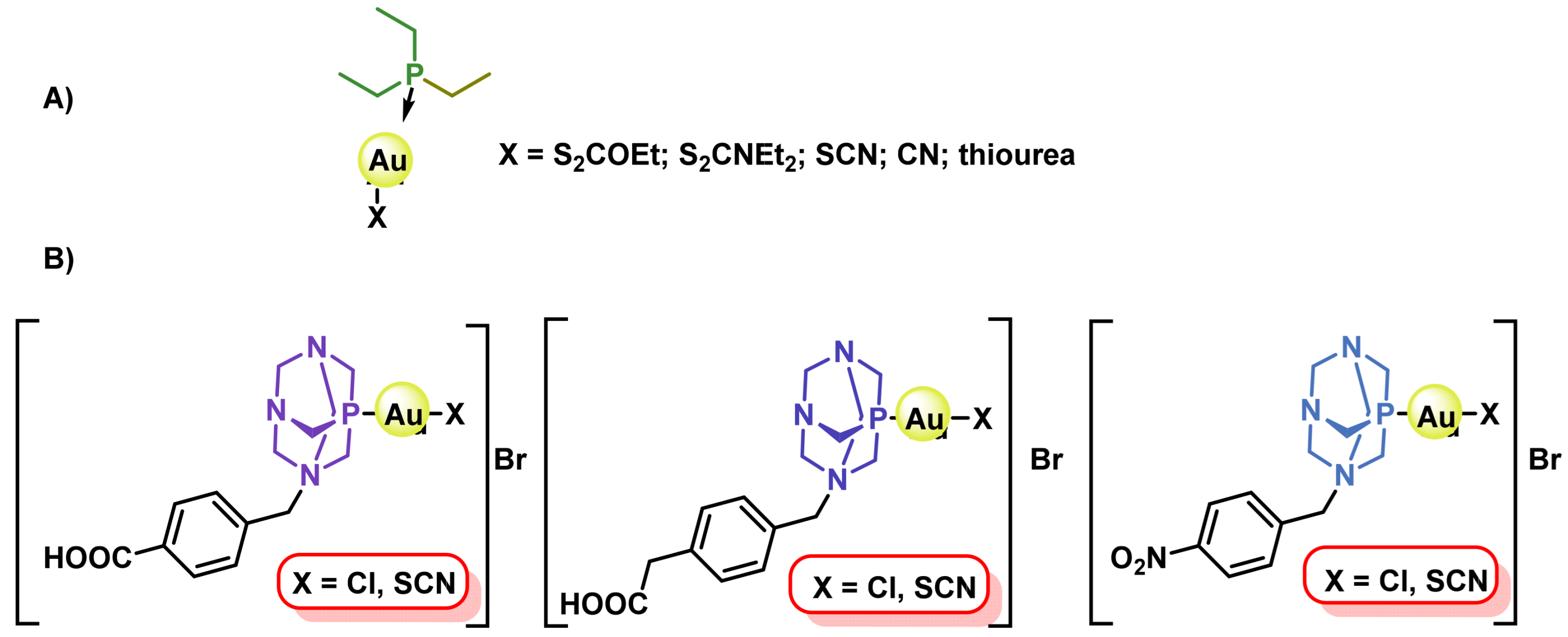
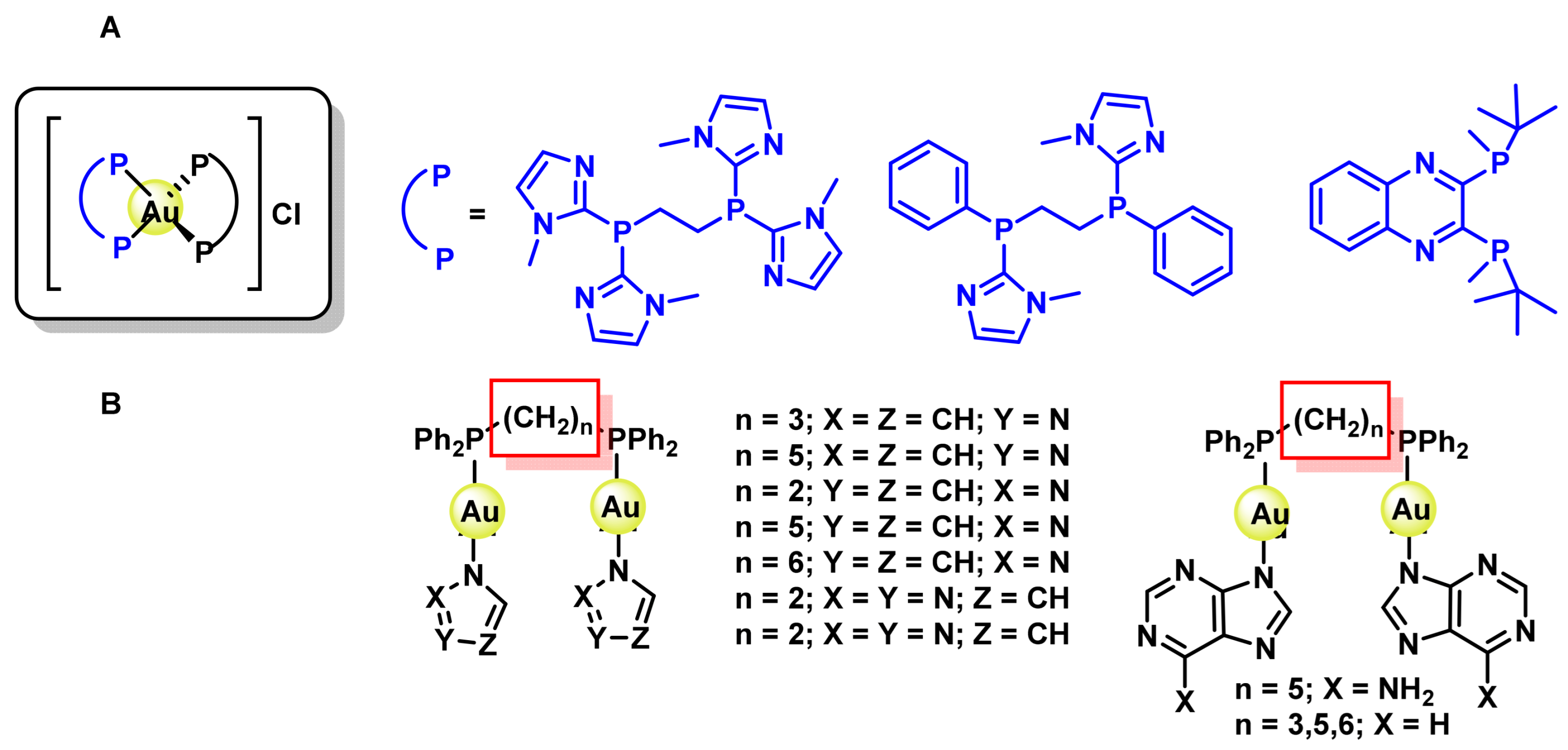
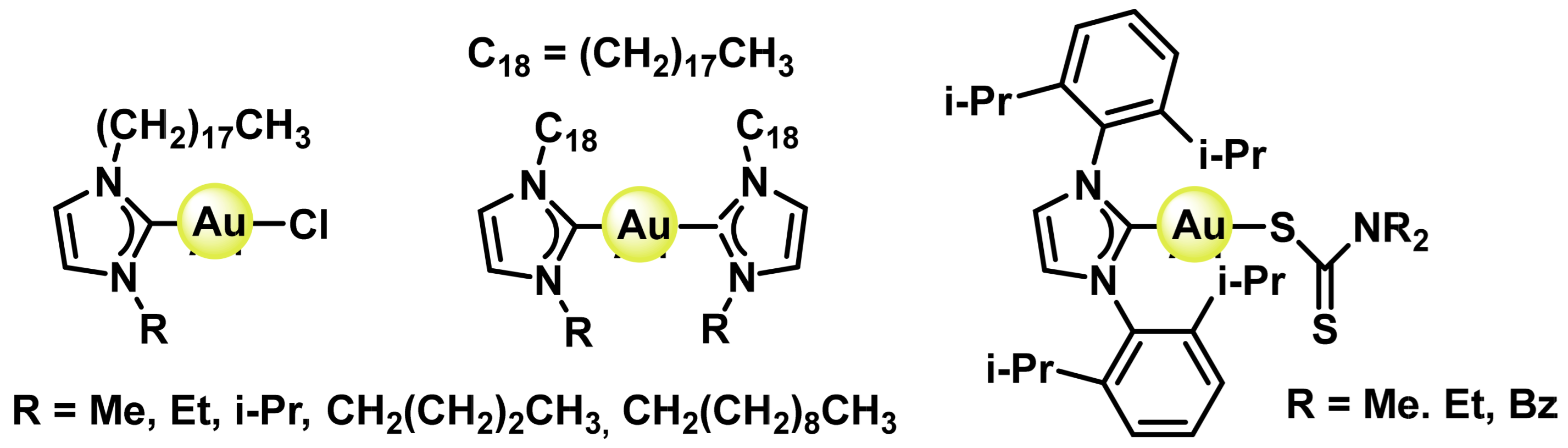
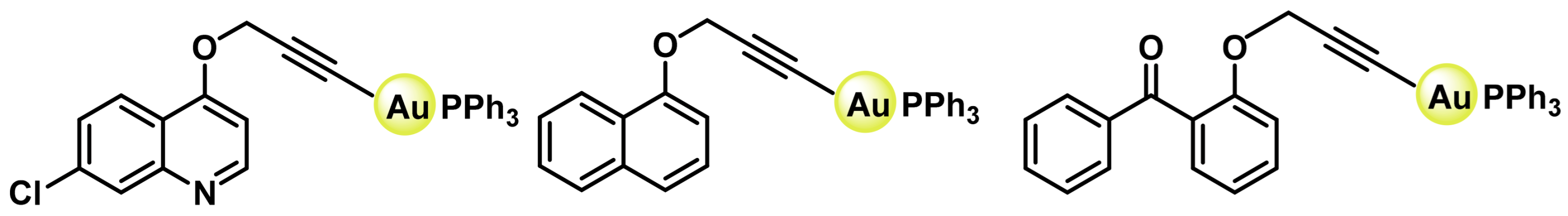
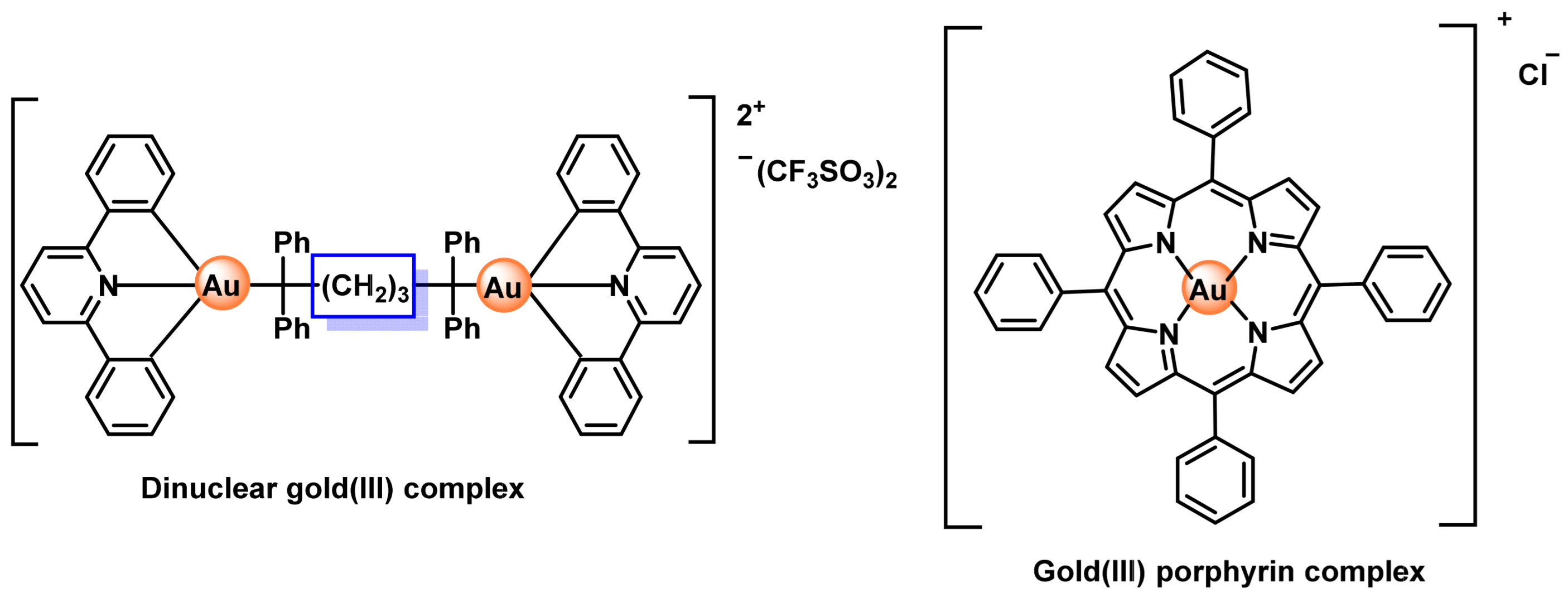
8.1. Gold(I) Complexes
8.2. Gold(III) Complexes
9. Drug Delivery Systems and Nanotechnology
- (i).
- improved biodistribution,
- (ii).
- improved bioavailability and solubility of drugs,
- (iii).
- decreased toxicity,
- (iv).
- prevention of multi-drug resistance,
- (v).
- increased drug stability, prevent degradation of the encapsulated cargo,
- (vi).
- allow encapsulation of a larger amount of drugs, [246]
- (vii).
- cross barriers (such as the blood-brain barrier) due to their small size, and
- (viii).
- to carry out therapies directed at specific targets, which translates into the use of lower doses of drugs and therefore lower toxicity by facilitating their accumulation in tumours through the effect of improved permeability and retention [247].
10. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Debela, D.T.; Muzazu, S.G.; Heraro, K.D.; Ndalama, M.T.; Mesele, B.W.; Haile, D.C.; Kitui, S.K.; Manyazewal, T. New approaches and procedures for cancer treatment: Current perspectives. SAGE Open Med. 2021, 9, 20503121211034366. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B. Some biological effects of platinum compounds. Platin. Met. Rev. 1971, 15, 42–51. [Google Scholar] [CrossRef]
- Bratsos, I.; Jedner, S.; Gianferrara, T.; Alessio, E. Ruthenium anticancer compounds: Challenges and expectations. Chimia 2007, 61, 692–697. [Google Scholar] [CrossRef]
- Abu-Surrah, A.S.; Kettunen, M. Platinum group antitumor chemistry: Design and development of new anticancer drugs complementary to cisplatin. Curr. Med. Chem. 2006, 13, 1337–1357. [Google Scholar] [CrossRef] [PubMed]
- Farrell, N. Metal Complexes as Drugs and Chemotherapeutic Agents; Elsevier: Boston, MA, USA, 2003. [Google Scholar]
- Bruijnincx, P.C.; Sadler, P.J. New trends for metal complexes with anticancer activity. Curr. Opin. Chem. Biol. 2008, 12, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Levina, A.; Mitra, A.; Lay, P.A. Recent developments in ruthenium anticancer drugs. Metallomics 2009, 1, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Meléndez, E. Titanium complexes in cancer treatment. Crit. Rev. Oncol. Hematol. 2002, 42, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Afzal, M.; Zaki, M.; Ahmad, M.; Tabassum, S.; Bharadwaj, P.K. Synthesis, structure elucidation and DFT studies of a new coumarin-derived Zn (ii) complex: In vitro DNA/HSA binding profile and pBR322 cleavage pathway. RSC Adv. 2014, 4, 43504–43515. [Google Scholar] [CrossRef]
- Tardito, S.; Marchio, L. Copper compounds in anticancer strategies. Curr. Med. Chem. 2009, 16, 1325–1348. [Google Scholar] [CrossRef]
- Tabassum, S.; Zaki, M.; Ahmad, M.; Afzal, M.; Srivastav, S.; Srikrishna, S.; Arjmand, F. Synthesis and crystal structure determination of copper (II)-complex: In vitro DNA and HSA binding, pBR322 plasmid cleavage, cell imaging and cytotoxic studies. Eur. J. Med. Chem. 2014, 83, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, S.; Zaki, M.; Afzal, M.; Arjmand, F. New modulated design and synthesis of quercetin–Cu II/Zn II–Sn 2 IV scaffold as anticancer agents: In vitro DNA binding profile, DNA cleavage pathway and Topo-I activity. Dalton Trans. 2013, 42, 10029–10041. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Guo, Z. Copper in medicine: Homeostasis, chelation therapy and antitumor drug design. Curr. Med. Chem. 2006, 13, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, S.; Zaki, M.; Afzal, M.; Arjmand, F. Synthesis and characterization of Cu (II)-based anticancer chemotherapeutic agent targeting topoisomerase Iα: In vitro DNA binding, pBR322 cleavage, molecular docking studies and cytotoxicity against human cancer cell lines. Eur. J. Med. Chem. 2014, 74, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, S.; Zaki, M.; Arjmand, F.; Ahmad, I. Synthesis of heterobimetallic complexes: In vitro DNA binding, cleavage and antimicrobial studies. J. Photochem. Photobiol. B Biol. 2012, 114, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.; White, A.R. Copper complexes as therapeutic agents. Metallomics 2012, 4, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, S.; Ahmad, M.; Afzal, M.; Zaki, M.; Bharadwaj, P.K. Synthesis and structure elucidation of a copper (II) Schiff-base complex: In vitro DNA binding, pBR322 plasmid cleavage and HSA binding studies. J. Photochem. Photobiol. B Biol. 2014, 140, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, S.; Asim, A.; Arjmand, F.; Afzal, M.; Bagchi, V. Synthesis and characterization of copper (II) and zinc (II)-based potential chemotherapeutic compounds: Their biological evaluation viz. DNA binding profile, cleavage and antimicrobial activity. Eur. J. Med. Chem. 2012, 58, 308–316. [Google Scholar] [CrossRef]
- Comert, F.; Heinrich, F.; Chowdhury, A.; Schoeneck, M.; Darling, C.; Anderson, K.W.; Libardo, M.D.J.; Angeles-Boza, A.M.; Silin, V.; Cotten, M.L.; et al. Copper-binding anticancer peptides from the piscidin family: An expanded mechanism that encompasses physical and chemical bilayer disruption. Sci. Rep. 2021, 11, 12620. [Google Scholar] [CrossRef]
- Ray, B.; Gupta, B.; Mehrotra, R. Binding of platinum derivative, oxaliplatin to deoxyribonucleic acid: Structural insight into antitumor action. J. Biomol. Struct. Dyn. 2019, 37, 3838–3847. [Google Scholar] [CrossRef]
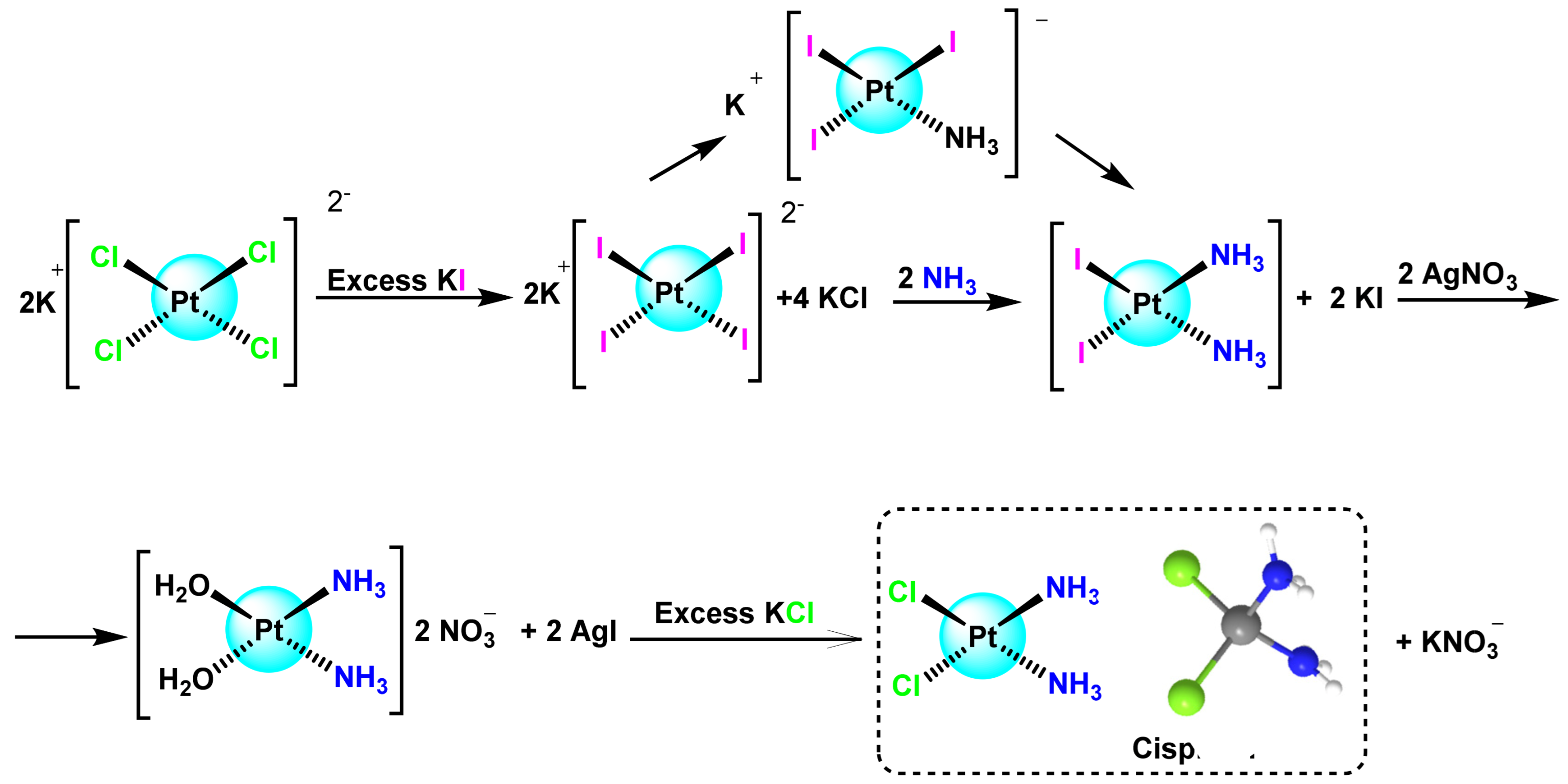
- Dhara, S. A rapid method for the synthesis of cis-[Pt (NH3) 2Cl2]. Indian J. Chem. 1970, 8, 193–194. [Google Scholar]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
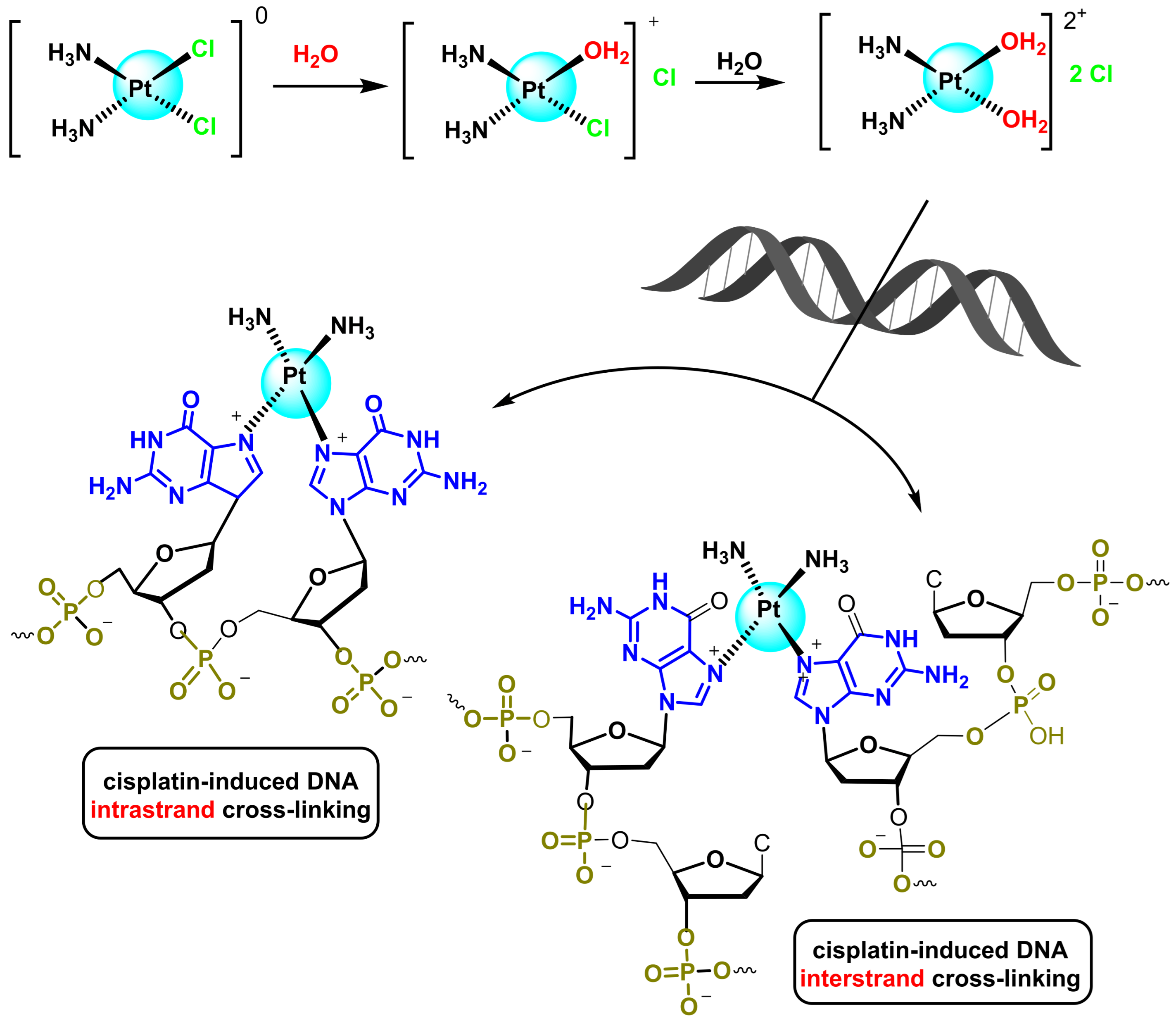
- Chválová, K.; Brabec, V.; Kaspárková, J. Mechanism of the formation of DNA-protein cross-links by antitumor cisplatin. Nucleic Acids Res. 2007, 35, 1812–1821. [Google Scholar] [CrossRef]
- Hambley, T.W. Platinum binding to DNA: Structural controls and consequences. J. Chem. Soc. Dalton Trans. 2001, 24, 2711–2718. [Google Scholar] [CrossRef]
- Hannon, M.J. Metal-based anticancer drugs: From a past anchored in platinum chemistry to a post-genomic future of diverse chemistry and biology. Pure Appl. Chem. 2007, 79, 2243–2261. [Google Scholar] [CrossRef]
- Galanski, M.S.; Jakupec, M.A.; Keppler, B.K. Update of the Preclinical Situation of Anticancer Platinum Complexes: Novel Design Strategies and Innovative Analytical Approaches. Curr. Med. Chem. 2005, 12, 2075–2094. [Google Scholar] [CrossRef]
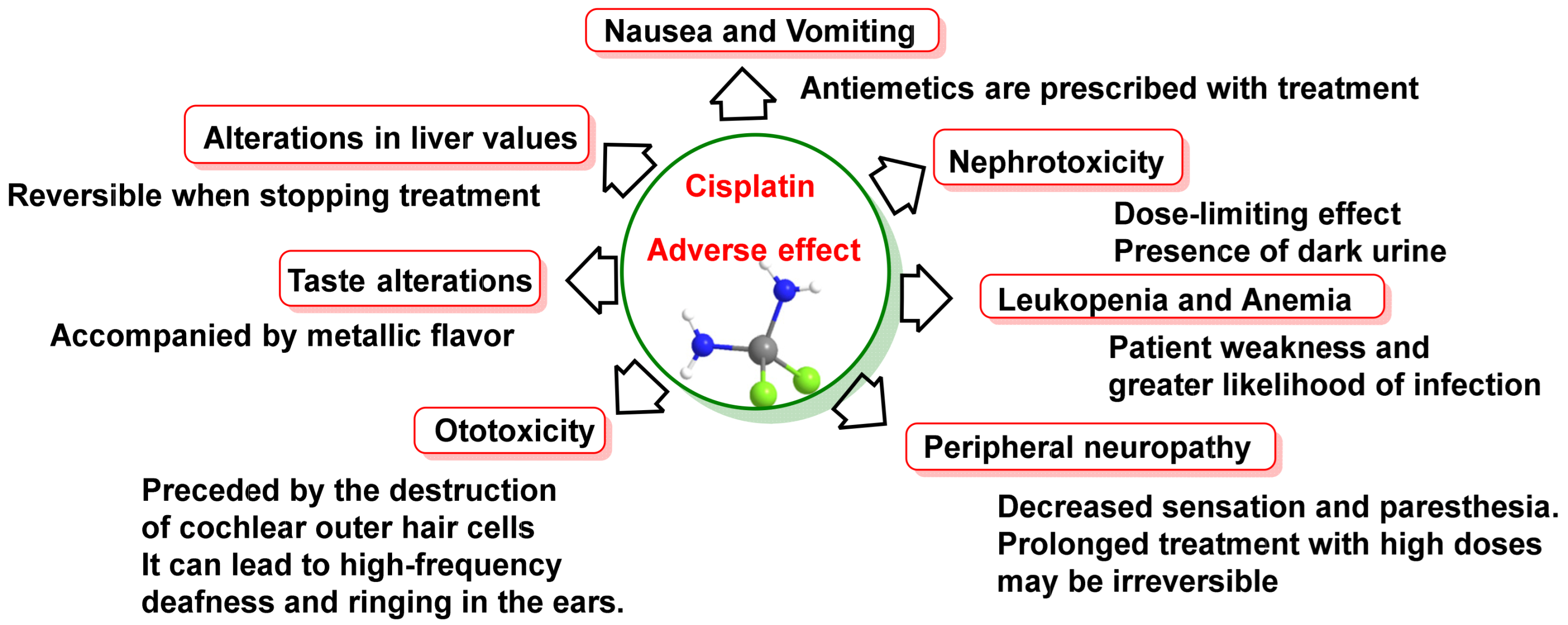
- Dammeyer, P.; Hellberg, V.; Wallin, I.; Laurell, G.; Shoshan, M.; Ehrsson, H.; Arnér, E.S.; Kirkegaard, M. Cisplatin and oxaliplatin are toxic to cochlear outer hair cells and both target thioredoxin reductase in organ of Corti cultures. Acta Otolaryngol. 2014, 134, 448–454. [Google Scholar] [CrossRef]
- Hartmann, J.T.; Lipp, H.P. Toxicity of platinum compounds. Expert Opin. Pharmacother. 2003, 4, 889–901. [Google Scholar] [CrossRef]
- Giaccone, G. Clinical perspectives on platinum resistance. Drugs 2000, 59 (Suppl. S4), 9–17, discussion 37–38. [Google Scholar] [CrossRef]
- Astolfi, L.; Ghiselli, S.; Guaran, V.; Chicca, M.; Simoni, E.; Olivetto, E.; Lelli, G.; Martini, A. Correlation of adverse effects of cisplatin administration in patients affected by solid tumours: A retrospective evaluation. Oncol. Rep. 2013, 29, 1285–1292. [Google Scholar] [CrossRef]
- Gómez-Ruiz, S.; Maksimović-Ivanić, D.; Mijatović, S.; Kaluđerović, G.N. On the discovery, biological effects, and use of Cisplatin and metallocenes in anticancer chemotherapy. Bioinorg. Chem. Appl. 2012, 2012, 140284. [Google Scholar] [CrossRef]
- Kartalou, M.; Essigmann, J.M. Mechanisms of resistance to cisplatin. Mutat. Res. 2001, 478, 23–43. [Google Scholar] [CrossRef]
- Lippard, S.J. Chemistry and molecular biology of platinum anticancer drugs. Pure Appl. Chem. 1987, 59, 731–742. [Google Scholar] [CrossRef]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef]
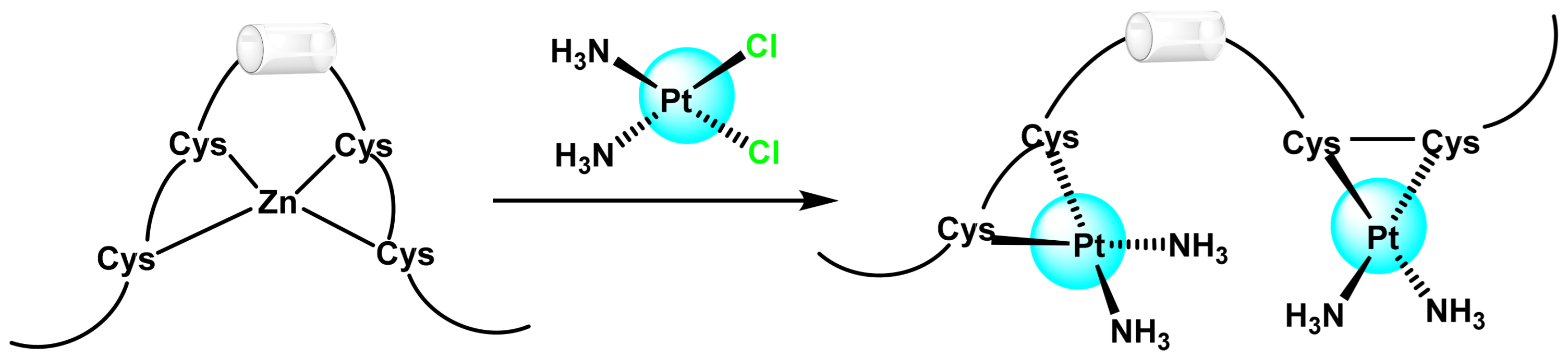
- Ogo, O.A.; Tyson, J.; Cockell, S.J.; Howard, A.; Valentine, R.A.; Ford, D. The zinc finger protein ZNF658 regulates the transcription of genes involved in zinc homeostasis and affects ribosome biogenesis through the zinc transcriptional regulatory element. Mol. Cell Biol. 2015, 35, 977–987. [Google Scholar] [CrossRef]
- Hajdu, B.; Hunyadi-Gulyás, É.; Kato, K.; Kawaguchi, A.; Nagata, K.; Gyurcsik, B. Zinc binding of a Cys2His2-type zinc finger protein is enhanced by the interaction with DNA. J. Biol. Inorg. Chem. 2023, 28, 301–315. [Google Scholar] [CrossRef]
- Chen, B.; Yu, P.; Chan, W.N.; Xie, F.; Zhang, Y.; Liang, L.; Leung, K.T.; Lo, K.W.; Yu, J.; Tse, G.M.K.; et al. Cellular zinc metabolism and zinc signaling: From biological functions to diseases and therapeutic targets. Signal Transduct. Target. Ther. 2024, 9, 6. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef]
- Abuetabh, Y.; Wu, H.H.; Chai, C.; Al Yousef, H.; Persad, S.; Sergi, C.M.; Leng, R. DNA damage response revisited: The p53 family and its regulators provide endless cancer therapy opportunities. Exp. Mol. Med. 2022, 54, 1658–1669. [Google Scholar] [CrossRef]
- Monneret, C. Platinum anticancer drugs. From serendipity to rational design. Ann. Pharm. Fr. 2011, 69, 286–295. [Google Scholar] [CrossRef]
- Avendaño, C.; Menéndez, J.C. Chapter 5—DNA alkylating agents. In Medicinal Chemistry of Anticancer Drugs, 3rd ed.; Avendaño, C., Menéndez, J.C., Eds.; Elsevier: Boston, MA, USA, 2023; pp. 237–290. [Google Scholar]
- Tsvetkova, D.; Ivanova, S. Application of Approved Cisplatin Derivatives in Combination Therapy against Different Cancer Diseases. Molecules 2022, 27, 2466. [Google Scholar] [CrossRef]
- Neidle, S.; Ismail, I.M.; Sadler, P.J. The structure of the antitumor complex cis-(diammino)(1, 1-cyclobutanedicarboxylato)-Pt (II): X ray and nmr studies. J. Inorg. Biochem. 1980, 13, 205–212. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- McWhinney, S.R.; Goldberg, R.M.; McLeod, H.L. Platinum neurotoxicity pharmacogenetics. Mol. Cancer Ther. 2009, 8, 10–16. [Google Scholar] [CrossRef]
- van der Vijgh, W.J. Clinical pharmacokinetics of carboplatin. Clin. Pharmacokinet. 1991, 21, 242–261. [Google Scholar] [CrossRef]
- O’Dowd, P.D.; Sutcliffe, D.F.; Griffith, D.M. Oxaliplatin and its derivatives—An overview. Coord. Chem. Rev. 2023, 497, 215439. [Google Scholar] [CrossRef]
- Misset, J.L.; Bleiberg, H.; Sutherland, W.; Bekradda, M.; Cvitkovic, E. Oxaliplatin clinical activity: A review. Crit. Rev. Oncol. Hematol. 2000, 35, 75–93. [Google Scholar] [CrossRef]
- Martinez-Balibrea, E.; Martínez-Cardús, A.; Ginés, A.; Ruiz de Porras, V.; Moutinho, C.; Layos, L.; Manzano, J.L.; Bugés, C.; Bystrup, S.; Esteller, M.; et al. Tumor-Related Molecular Mechanisms of Oxaliplatin Resistance. Mol. Cancer Ther. 2015, 14, 1767–1776. [Google Scholar] [CrossRef]
- Alcindor, T.; Beauger, N. Oxaliplatin: A review in the era of molecularly targeted therapy. Curr. Oncol. 2011, 18, 18–25. [Google Scholar] [CrossRef]
- Ranasinghe, R.; Mathai, M.L.; Zulli, A. Cisplatin for cancer therapy and overcoming chemoresistance. Heliyon 2022, 8, e10608. [Google Scholar] [CrossRef]
- Natile, G.; Coluccia, M. Current status of trans-platinum compounds in cancer therapy. Coord. Chem. Rev. 2001, 216, 383–410. [Google Scholar] [CrossRef]
- Wilson, J.J.; Lippard, S.J. Synthetic methods for the preparation of platinum anticancer complexes. Chem. Rev. 2014, 114, 4470–4495. [Google Scholar] [CrossRef]
- Dabrowiak, J.C. Metals in Medicine; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Lippert, B. Trans-diammineplatinum (II): What makes it different from cis-DDP? Coordination chemistry of a neglected relative of cisplatin and its interaction with nucleic acids. Met. Ions Biol. Syst. 1996, 33, 105–141. [Google Scholar]
- Farrell, N.; Ha, T.T.; Souchard, J.P.; Wimmer, F.L.; Cros, S.; Johnson, N.P. Cytostatic trans-platinum (II) complexes. J. Med. Chem. 1989, 32, 2240–2241. [Google Scholar] [CrossRef]
- Van Beusichem, M.; Farrell, N. Activation of the trans geometry in platinum antitumor complexes. Synthesis, characterization, and biological activity of complexes with the planar ligands pyridine, N-methylimidazole, thiazole, and quinoline. Crystal and molecular structure of trans-dichlorobis(thiazole)platinum(II). Inorg. Chem. 1992, 31, 634–639. [Google Scholar] [CrossRef]
- Leng, M.; Locker, D.; Giraud-Panis, M.-J.; Schwartz, A.; Intini, F.P.; Natile, G.; Pisano, C.; Boccarelli, A.; Giordano, D.; Coluccia, M. Replacement of an NH3 by an iminoether in transplatin makes an antitumor drug from an inactive compound. Mol. Pharmacol. 2000, 58, 1525–1535. [Google Scholar] [CrossRef]
- Coluccia, M.; Nassi, A.; Loseto, F.; Boccarelli, A.; Mariggio, M.A.; Giordano, D.; Intini, F.P.; Caputo, P.; Natile, G. A trans-platinum complex showing higher antitumor activity than the cis congeners. J. Med. Chem. 1993, 36, 510–512. [Google Scholar] [CrossRef]
- Coluccia, M.; Nassi, A.; Boccarelli, A.; Giordano, D.; Cardellicchio, N.; Locker, D.; Leng, M.; Sivo, M.; Intini, F.P.; Natile, G. In vitro and in vivo antitumour activity and cellular pharmacological properties of new platinum–iminoether complexes with different configuration at the iminoether ligands. J. Inorg. Biochem. 1999, 77, 31–35. [Google Scholar] [CrossRef]
- Novakova, O.; Kasparkova, J.; Malina, J.; Natile, G.; Brabec, V. DNA–protein cross-linking by trans-[PtCl2(E-iminoether)2]. A concept for activation of the trans geometry in platinum antitumor complexes. Nucleic Acids Res. 2003, 31, 6450–6460. [Google Scholar] [CrossRef] [PubMed]
- Pantoja, E.; Álvarez-Valdés, A.; Pérez, J.M.; Navarro-Ranninger, C.; Reedijk, J. Synthesis and characterization of new cis-[PtCl2 (isopropylamine)(amine′)] compounds: Cytotoxic activity and reactions with 5′-GMP compared with their trans-platinum isomers. Inorganica Chim. Acta 2002, 339, 525–531. [Google Scholar] [CrossRef]
- Coluccia, M.; Natile, G. Trans-platinum complexes in cancer therapy. Anticancer Agents Med. Chem. 2007, 7, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Todd, R.C.; Lippard, S.J. Inhibition of transcription by platinum antitumor compounds. Metallomics 2009, 1, 280–291. [Google Scholar] [CrossRef]
- Billecke, C.; Finniss, S.; Tahash, L.; Miller, C.; Mikkelsen, T.; Farrell, N.P.; Bögler, O. Polynuclear platinum anticancer drugs are more potent than cisplatin and induce cell cycle arrest in glioma. Neuro-Oncol. 2006, 8, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Farrell, N.; Qu, Y.; Bierbach, U.; Valsecchi, M.; Mentab, E. Structure-activity relationships within di-and trinuclear platinum phase-I clinical anticancer agents. In Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug; John Wiley & Sons: Hoboken, NJ, USA, 1999; pp. 477–496. [Google Scholar]
- Farrell, N.; Qu, Y.; Hacker, M.P. Cytotoxicity and antitumor activity of bis(platinum) complexes. A novel class of platinum complexes active in cell lines resistant to both cisplatin and 1,2-diaminocyclohexane complexes. J. Med. Chem. 1990, 33, 2179–2184. [Google Scholar] [CrossRef] [PubMed]
- Farrell, N. Nonclassical platinum antitumor agents: Perspectives for design and development of new drugs complementary to cisplatin. Cancer Investig. 1993, 11, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Summa, N.; Maigut, J.; Puchta, R.; van Eldik, R. Possible biotransformation reactions of polynuclear Pt(II) complexes. Inorg. Chem. 2007, 46, 2094–2104. [Google Scholar] [CrossRef]
- Prisecaru, A.; Molphy, Z.; Kipping, R.G.; Peterson, E.J.; Qu, Y.; Kellett, A.; Farrell, N.P. The phosphate clamp: Sequence selective nucleic acid binding profiles and conformational induction of endonuclease inhibition by cationic Triplatin complexes. Nucleic Acids Res. 2014, 42, 13474–13487. [Google Scholar] [CrossRef]
- Zou, Y.; Van Houten, B.; Farrell, N. Sequence specificity of DNA-DNA interstrand cross-link formation by cisplatin and dinuclear platinum complexes. Biochemistry 1994, 33, 5404–5410. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; van Boom, S.S.; Reedijk, J.; van Boom, J.H.; Farrell, N.; Wang, A.H.-J. A novel DNA structure induced by the anticancer bisplatinum compound crosslinked to a GpC site in DNA. Nat. Struct. Biol. 1995, 2, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xu, C.; Gao, X.; Yao, Q. Platinum-based drugs for cancer therapy and anti-tumor strategies. Theranostics 2022, 12, 2115–2132. [Google Scholar] [CrossRef]
- Song, X.Q.; Liu, R.P.; Wang, S.Q.; Li, Z.; Ma, Z.Y.; Zhang, R.; Xie, C.Z.; Qiao, X.; Xu, J.Y. Anticancer Melatplatin Prodrugs: High Effect and Low Toxicity, MT1-ER-Target and Immune Response In vivo. J. Med. Chem. 2020, 63, 6096–6106. [Google Scholar] [CrossRef]
- Wang, Q.; Xiao, M.; Wang, D.; Hou, X.; Gao, J.; Liu, J.; Liu, J. In situ supramolecular self-assembly of Pt (IV) prodrug to conquer cisplatin resistance. Adv. Funct. Mater. 2021, 31, 2101826. [Google Scholar] [CrossRef]
- Huang, J.; Ding, W.; Zhu, X.; Li, B.; Zeng, F.; Wu, K.; Wu, X.; Wang, F. Ligand Evolution in the Photoactivatable Platinum(IV) Anticancer Prodrugs. Front. Chem. 2022, 10, 876410. [Google Scholar] [CrossRef]
- Hall, M.D.; Hambley, T.W. Platinum (IV) antitumour compounds: Their bioinorganic chemistry. Coord. Chem. Rev. 2002, 232, 49–67. [Google Scholar] [CrossRef]
- Deng, Z.; Zhu, G. Beyond mere DNA damage: Recent progress in platinum(IV) anticancer complexes containing multi-functional axial ligands. Curr. Opin. Chem. Biol. 2023, 74, 102303. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, S.A.; Kerwood, D.J.; Goodisman, J.; Dabrowiak, J.C. Pt(IV) complexes as prodrugs for cisplatin. J. Inorg. Biochem. 2012, 107, 6–14. [Google Scholar] [CrossRef]
- Xiao, H.; Yan, L.; Dempsey, E.M.; Song, W.; Qi, R.; Li, W.; Huang, Y.; Jing, X.; Zhou, D.; Ding, J.; et al. Recent progress in polymer-based platinum drug delivery systems. Prog. Polym. Sci. 2018, 87, 70–106. [Google Scholar] [CrossRef]
- Wang, X.; Guo, Z. Targeting and delivery of platinum-based anticancer drugs. Chem. Soc. Rev. 2013, 42, 202–224. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y.; Tian, H. Current Developments in Pt(IV) Prodrugs Conjugated with Bioactive Ligands. Bioinorg. Chem. Appl. 2018, 2018, 8276139. [Google Scholar] [CrossRef] [PubMed]
- Ravera, M.; Gabano, E.; McGlinchey, M.J.; Osella, D. A view on multi-action Pt (IV) antitumor prodrugs. Inorganica Chim. Acta 2019, 492, 32–47. [Google Scholar] [CrossRef]
- Yang, J.; Sun, X.; Mao, W.; Sui, M.; Tang, J.; Shen, Y. Conjugate of Pt(IV)-histone deacetylase inhibitor as a prodrug for cancer chemotherapy. Mol. Pharm. 2012, 9, 2793–2800. [Google Scholar] [CrossRef] [PubMed]
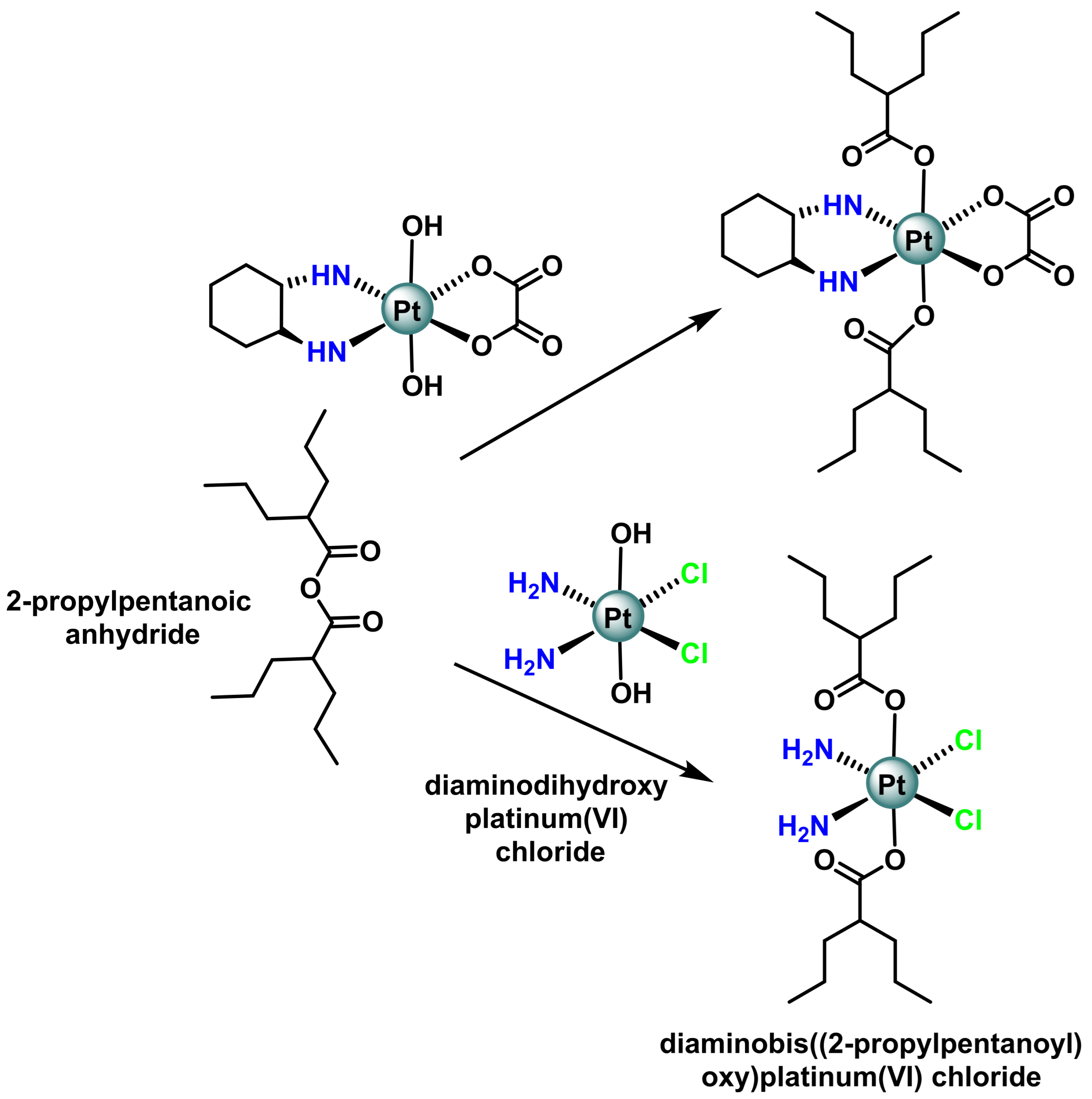
- Novohradsky, V.; Zerzankova, L.; Stepankova, J.; Vrana, O.; Raveendran, R.; Gibson, D.; Kasparkova, J.; Brabec, V. Antitumor platinum(IV) derivatives of oxaliplatin with axial valproato ligands. J. Inorg. Biochem. 2014, 140, 72–79. [Google Scholar] [CrossRef]
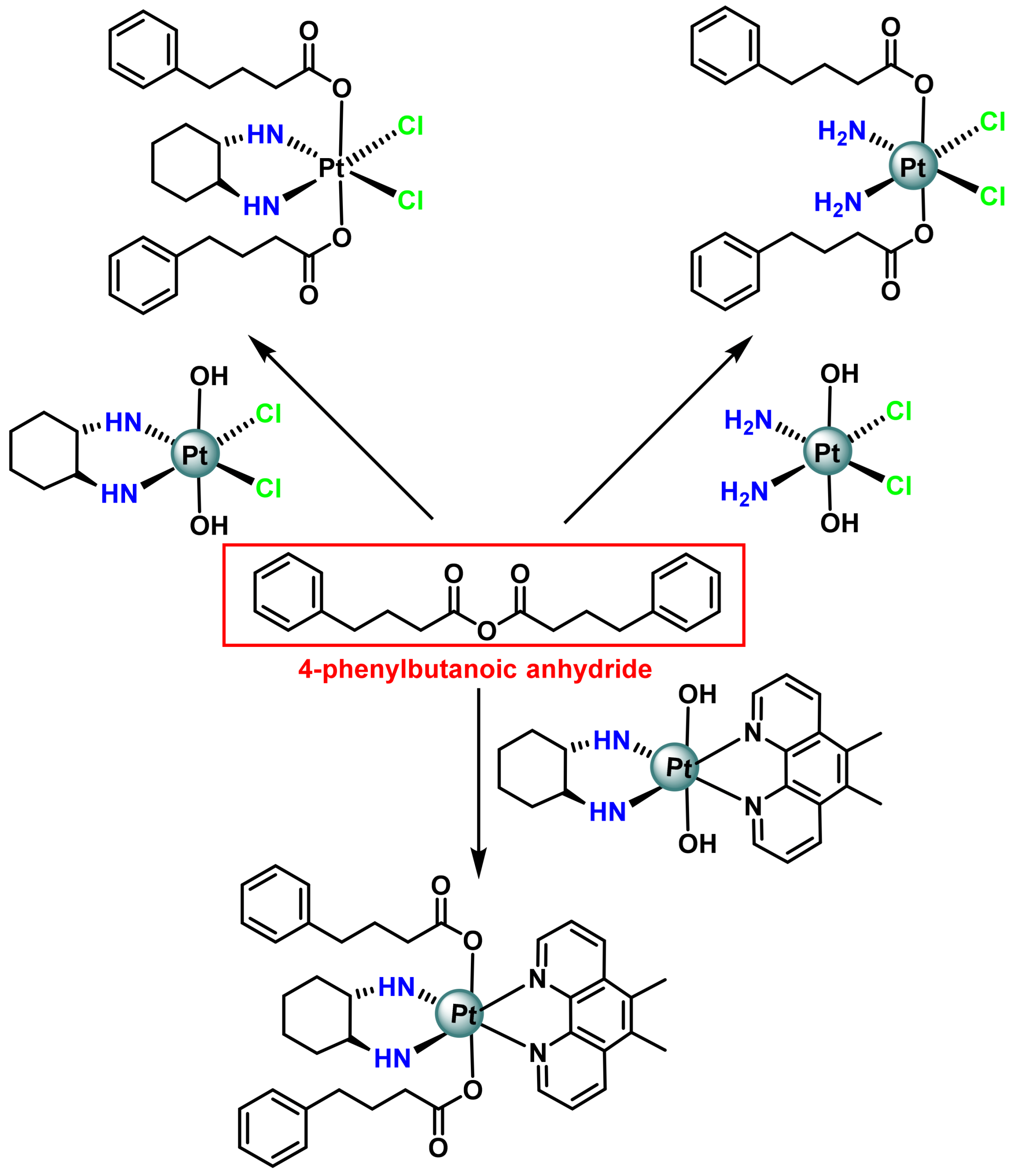
- Raveendran, R.; Braude, J.P.; Wexselblatt, E.; Novohradsky, V.; Stuchlikova, O.; Brabec, V.; Gandin, V.; Gibson, D. Pt(iv) derivatives of cisplatin and oxaliplatin with phenylbutyrate axial ligands are potent cytotoxic agents that act by several mechanisms of action. Chem. Sci. 2016, 7, 2381–2391. [Google Scholar] [CrossRef] [PubMed]
- Kasparkova, J.; Kostrhunova, H.; Novakova, O.; Křikavová, R.; Vančo, J.; Trávníček, Z.; Brabec, V. A Photoactivatable Platinum(IV) Complex Targeting Genomic DNA and Histone Deacetylases. Angew. Chem. Int. Ed. 2015, 54, 14478–14482. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Wang, Z. Photoactivatable Platinum-Based Anticancer Drugs: Mode of Photoactivation and Mechanism of Action. Molecules 2020, 25, 5167. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Ayub, W.; Butler, I.S.; Zia-ur-Rehman. Photoactivated platinum-based anticancer drugs. Coord. Chem. Rev. 2018, 376, 405–429. [Google Scholar] [CrossRef]
- Shi, H.; Imberti, C.; Sadler, P.J. Diazido platinum(iv) complexes for photoactivated anticancer chemotherapy. Inorg. Chem. Front. 2019, 6, 1623–1638. [Google Scholar] [CrossRef]
- Imberti, C.; Zhang, P.; Huang, H.; Sadler, P.J. New Designs for Phototherapeutic Transition Metal Complexes. Angew. Chem. Int. Ed. Engl. 2020, 59, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Ma, R.; Wang, Y.; Zhu, X.; Zhang, J.; Chan, H.C.; Chen, X.; Zhang, W.; Chiu, S.-K.; Zhu, G. Chalcoplatin, a dual-targeting and p53 activator-containing anticancer platinum(iv) prodrug with unique mode of action. Chem. Commun. 2015, 51, 6301–6304. [Google Scholar] [CrossRef] [PubMed]
- Ang, W.H.; Khalaila, I.; Allardyce, C.S.; Juillerat-Jeanneret, L.; Dyson, P.J. Rational Design of Platinum(IV) Compounds to Overcome Glutathione-S-Transferase Mediated Drug Resistance. J. Am. Chem. Soc. 2005, 127, 1382–1383. [Google Scholar] [CrossRef]
- Parker, L.J.; Italiano, L.C.; Morton, C.J.; Hancock, N.C.; Ascher, D.B.; Aitken, J.B.; Harris, H.H.; Campomanes, P.; Rothlisberger, U.; De Luca, A. Studies of glutathione transferase P1-1 bound to a platinum (IV)-based anticancer compound reveal the molecular basis of its activation. Chem. Eur. J. 2011, 17, 7806–7816. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Pan, Q.; Gao, W.; Pu, Y.; He, B. Reversal of cisplatin chemotherapy resistance by glutathione-resistant copper-based nanomedicine via cuproptosis. J. Mater. Chem. B 2022, 10, 6296–6306. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Lippard, S.J. Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate. Proc. Natl. Acad. Sci. USA 2009, 106, 22199–22204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, M.; Zhao, H.; Wang, Z.; Shi, Y.; Dong, J.; Wang, K.; Wang, X.; Li, X.; Qi, H.; et al. Neuroprotective Effects and Therapeutic Potential of Dichloroacetate: Targeting Metabolic Disorders in Nervous System Diseases. Int. J. Nanomed. 2023, 18, 7559–7581. [Google Scholar] [CrossRef] [PubMed]
- Suntharalingam, K.; Song, Y.; Lippard, S.J. Conjugation of vitamin E analog α-TOS to Pt(iv) complexes for dual-targeting anticancer therapy. Chem. Commun. 2014, 50, 2465–2468. [Google Scholar] [CrossRef] [PubMed]
- Barnes, K.R.; Kutikov, A.; Lippard, S.J. Synthesis, characterization, and cytotoxicity of a series of estrogen-tethered platinum(IV) complexes. Chem. Biol. 2004, 11, 557–564. [Google Scholar] [CrossRef]
- Boulikas, T.; Vougiouka, M. Recent clinical trials using cisplatin, carboplatin and their combination chemotherapy drugs. Oncol. Rep. 2004, 11, 559–595. [Google Scholar] [CrossRef]
- Kanai, M.; Hatano, E.; Kobayashi, S.; Fujiwara, Y.; Marubashi, S.; Miyamoto, A.; Shiomi, H.; Kubo, S.; Ikuta, S.; Yanagimoto, H.; et al. A multi-institution phase II study of gemcitabine/cisplatin/S-1 (GCS) combination chemotherapy for patients with advanced biliary tract cancer (KHBO 1002). Cancer Chemother. Pharmacol. 2015, 75, 293–300. [Google Scholar] [CrossRef]
- Chen, C.H.; Yang, H.J.; Shun, C.T.; Huang, C.Y.; Huang, K.H.; Yu, H.J.; Pu, Y.S. A cocktail regimen of intravesical mitomycin-C, doxorubicin, and cisplatin (MDP) for non-muscle-invasive bladder cancer. Urol. Oncol. 2012, 30, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The clinical role of the TME in solid cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Plamada, D.; Vodnar, D.C. Polyphenols-Gut Microbiota Interrelationship: A Transition to a New Generation of Prebiotics. Nutrients 2021, 14, 137. [Google Scholar] [CrossRef]
- Andrés, C.M.C.; Pérez de la Lastra, J.M.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. Polyphenols as Antioxidant/Pro-Oxidant Compounds and Donors of Reducing Species: Relationship with Human Antioxidant Metabolism. Processes 2023, 11, 2771. [Google Scholar] [CrossRef]
- Han, Y.; Jo, H.; Cho, J.H.; Dhanasekaran, D.N.; Song, Y.S. Resveratrol as a Tumor-Suppressive Nutraceutical Modulating Tumor Microenvironment and Malignant Behaviors of Cancer. Int. J. Mol. Sci. 2019, 20, 925. [Google Scholar] [CrossRef]
- Lightbourn, A.V.; Thomas, R.D. Crude Edible Fig (Ficus carica) Leaf Extract Prevents Diethylstilbestrol (DES)-Induced DNA Strand Breaks in Single-Cell Gel Electrophoresis (SCGE)/Comet Assay: Literature Review and Pilot Study. J. Bioequivalence Bioavailab. 2019, 11, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Pagliara, V.; Nasso, R.; Di Donato, P.; Finore, I.; Poli, A.; Masullo, M.; Arcone, R. Lemon Peel Polyphenol Extract Reduces Interleukin-6-Induced Cell Migration, Invasiveness, and Matrix Metalloproteinase-9/2 Expression in Human Gastric Adenocarcinoma MKN-28 and AGS Cell Lines. Biomolecules 2019, 9, 833. [Google Scholar] [CrossRef] [PubMed]
- Chaves, F.M.; Pavan, I.C.B.; da Silva, L.G.S.; de Freitas, L.B.; Rostagno, M.A.; Antunes, A.E.C.; Bezerra, R.M.N.; Simabuco, F.M. Pomegranate Juice and Peel Extracts are Able to Inhibit Proliferation, Migration and Colony Formation of Prostate Cancer Cell Lines and Modulate the Akt/mTOR/S6K Signaling Pathway. Plant Foods Hum. Nutr. 2020, 75, 54–62. [Google Scholar] [CrossRef]
- Abroodi, Z.; Sajedi, N.; Nikbakht, M.; Soleimani, M. Estrogen Receptor Beta (ERβ) May Act as Mediator in Apoptotic Induction of Grape Seed Extract (GSE). Asian Pac. J. Cancer Prev. 2019, 20, 3729–3734. [Google Scholar] [CrossRef]
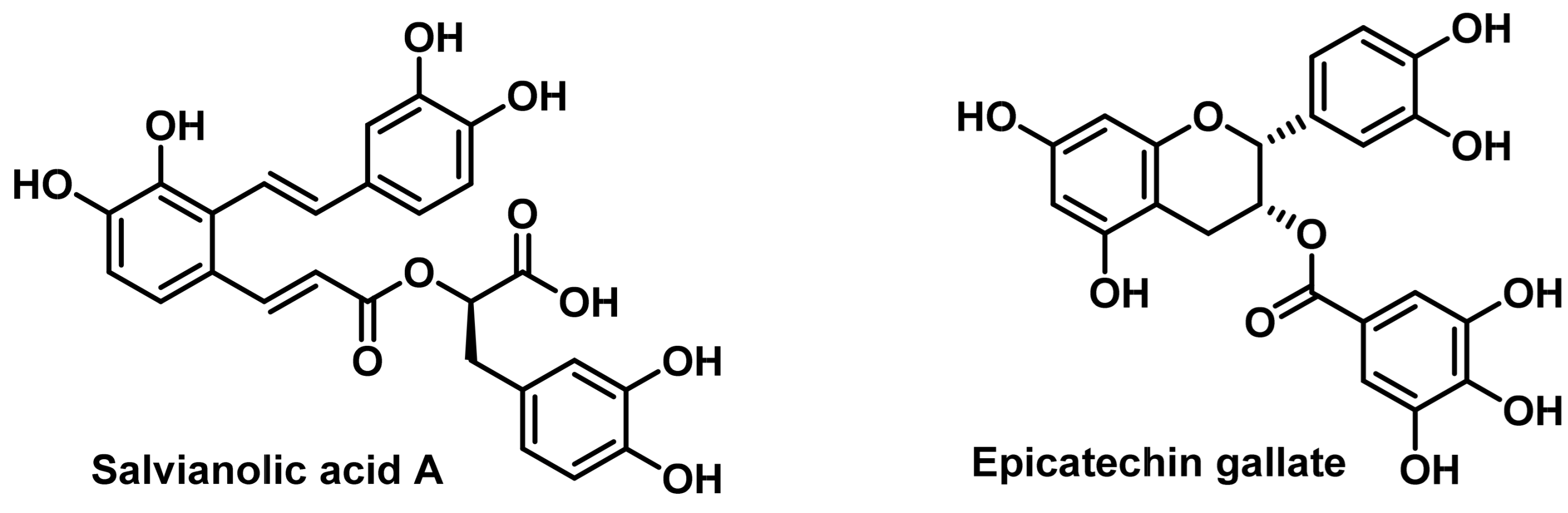
- Tang, X.L.; Yan, L.; Zhu, L.; Jiao, D.M.; Chen, J.; Chen, Q.Y. Salvianolic acid A reverses cisplatin resistance in lung cancer A549 cells by targeting c-met and attenuating Akt/mTOR pathway. J. Pharmacol. Sci. 2017, 135, 1–7. [Google Scholar] [CrossRef]
- Sharma, R.; Gatchie, L.; Williams, I.S.; Jain, S.K.; Vishwakarma, R.A.; Chaudhuri, B.; Bharate, S.B. Glycyrrhiza glabra extract and quercetin reverses cisplatin resistance in triple-negative MDA-MB-468 breast cancer cells via inhibition of cytochrome P450 1B1 enzyme. Bioorg. Med. Chem. Lett. 2017, 27, 5400–5403. [Google Scholar] [CrossRef] [PubMed]
- Domingo, I.K.; Latif, A.; Bhavsar, A.P. Pro-Inflammatory Signalling PRRopels Cisplatin-Induced Toxicity. Int. J. Mol. Sci. 2022, 23, 7227. [Google Scholar] [CrossRef]
- Desai, A.G.; Qazi, G.N.; Ganju, R.K.; El-Tamer, M.; Singh, J.; Saxena, A.K.; Bedi, Y.S.; Taneja, S.C.; Bhat, H.K. Medicinal plants and cancer chemoprevention. Curr. Drug Metab. 2008, 9, 581–591. [Google Scholar] [CrossRef]
- Zhou, J.; Nie, R.C.; Yin, Y.X.; Cai, X.X.; Xie, D.; Cai, M.Y. Protective Effect of Natural Antioxidants on Reducing Cisplatin-Induced Nephrotoxicity. Dis. Markers 2022, 2022, 1612348. [Google Scholar] [CrossRef]
- Mohamadi Yarijani, Z.; Godini, A.; Madani, S.H.; Najafi, H. Reduction of cisplatin-induced renal and hepatic side effects in rat through antioxidative and anti-inflammatory properties of Malva sylvestris L. extract. Biomed. Pharmacother. 2018, 106, 1767–1774. [Google Scholar] [CrossRef]
- Khalaf, A.A.; Hussein, S.; Tohamy, A.F.; Marouf, S.; Yassa, H.D.; Zaki, A.R.; Bishayee, A. Protective effect of Echinacea purpurea (Immulant) against cisplatin-induced immunotoxicity in rats. Daru 2019, 27, 233–241. [Google Scholar] [CrossRef]
- Rossi, B.M.; Palmero, E.I.; López-Kostner, F.; Sarroca, C.; Vaccaro, C.A.; Spirandelli, F.; Ashton-Prolla, P.; Rodriguez, Y.; de Campos Reis Galvão, H.; Reis, R.M.; et al. A survey of the clinicopathological and molecular characteristics of patients with suspected Lynch syndrome in Latin America. BMC Cancer 2017, 17, 623. [Google Scholar] [CrossRef]
- Almeer, R.S.; Abdel Moneim, A.E. Evaluation of the Protective Effect of Olive Leaf Extract on Cisplatin-Induced Testicular Damage in Rats. Oxid. Med. Cell Longev. 2018, 2018, 8487248. [Google Scholar] [CrossRef] [PubMed]
- Afsar, T.; Razak, S.; Almajwal, A.; Shabbir, M.; Khan, M.R. Evaluating the protective potency of Acacia hydaspica R. Parker on histological and biochemical changes induced by Cisplatin in the cardiac tissue of rats. BMC Complement. Altern. Med. 2019, 19, 182. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.; Bakhaat, G.A.; Tammam, H.G.; Mohamed, R.M.; El-Naggar, S.A. Cardioprotective effect of green tea extract and vitamin E on Cisplatin-induced cardiotoxicity in mice: Toxicological, histological and immunohistochemical studies. Biomed. Pharmacother. 2019, 113, 108731. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.P.; Peng, Y.X. Does the Oral Administration of Ginger Reduce Chemotherapy-Induced Nausea and Vomiting?: A Meta-analysis of 10 Randomized Controlled Trials. Cancer Nurs. 2019, 42, e14–e23. [Google Scholar] [CrossRef] [PubMed]
- Bossi, P.; Cortinovis, D.; Fatigoni, S.; Cossu Rocca, M.; Fabi, A.; Seminara, P.; Ripamonti, C.; Alfieri, S.; Granata, R.; Bergamini, C.; et al. A randomized, double-blind, placebo-controlled, multicenter study of a ginger extract in the management of chemotherapy-induced nausea and vomiting (CINV) in patients receiving high-dose cisplatin. Ann. Oncol. 2017, 28, 2547–2551. [Google Scholar] [CrossRef] [PubMed]
- Coffetti, G.; Moraschi, M.; Facchetti, G.; Rimoldi, I. The Challenging Treatment of Cisplatin-Resistant Tumors: State of the Art and Future Perspectives. Molecules 2023, 28, 3407. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.; Baskaran, K.; Pupulin, A.; Ruvinov, I.; Zaitoon, O.; Grewal, S.; Scaria, B.; Mehaidli, A.; Vegh, C.; Pandey, S. Hibiscus flower extract selectively induces apoptosis in breast cancer cells and positively interacts with common chemotherapeutics. BMC Complement. Altern. Med. 2019, 19, 98. [Google Scholar] [CrossRef] [PubMed]
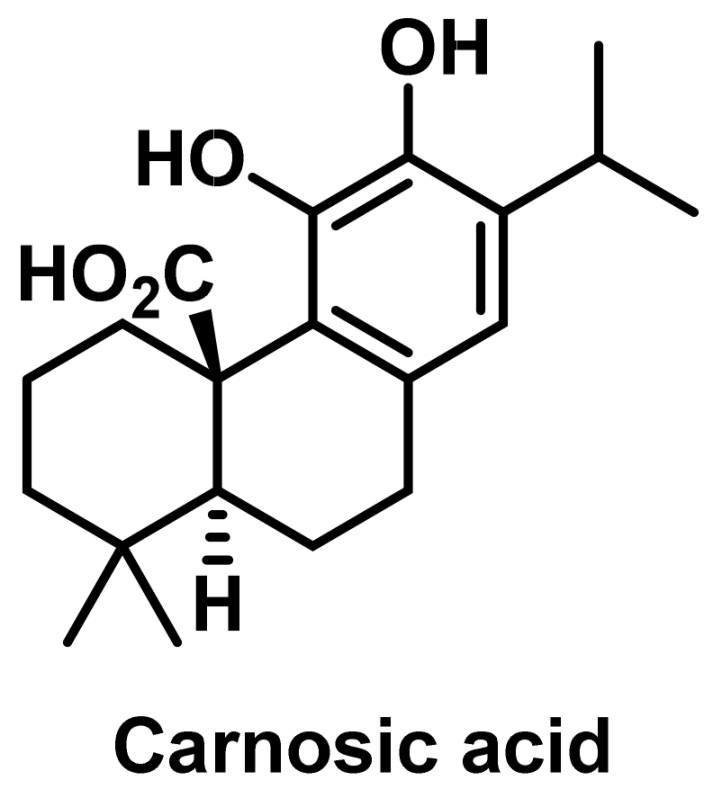
- Liu, W.; Wu, T.C.; Hong, D.M.; Hu, Y.; Fan, T.; Guo, W.J.; Xu, Q. Carnosic acid enhances the anti-lung cancer effect of cisplatin by inhibiting myeloid-derived suppressor cells. Chin. J. Nat. Med. 2018, 16, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Meier-Menches, S.M.; Casini, A. Design Strategies and Medicinal Applications of Metal-Peptidic Bioconjugates. Bioconjug. Chem. 2020, 31, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Soler, M.; Feliu, L.; Planas, M.; Ribas, X.; Costas, M. Peptide-mediated vectorization of metal complexes: Conjugation strategies and biomedical applications. Dalton Trans. 2016, 45, 12970–12982. [Google Scholar] [CrossRef]
- Hu, X.; Dong, Q.; Yang, J.; Zhang, Y. Recognizing metal and acid radical ion-binding sites by integrating ab initio modeling with template-based transferals. Bioinformatics 2016, 32, 3260–3269. [Google Scholar] [CrossRef]
- Truong, D.; Lam, N.Y.S.; Kamalov, M.; Riisom, M.; Jamieson, S.M.F.; Harris, P.W.R.; Brimble, M.A.; Metzler-Nolte, N.; Hartinger, C.G. A Solid Support-Based Synthetic Strategy for the Site-Selective Functionalization of Peptides with Organometallic Half-Sandwich Moieties. Chem. Eur. J. 2022, 28, e202104049. [Google Scholar] [CrossRef]
- Agbale, C.M.; Cardoso, M.H.; Galyuon, I.K.; Franco, O.L. Designing metallodrugs with nuclease and protease activity. Metallomics 2016, 8, 1159–1169. [Google Scholar] [CrossRef]
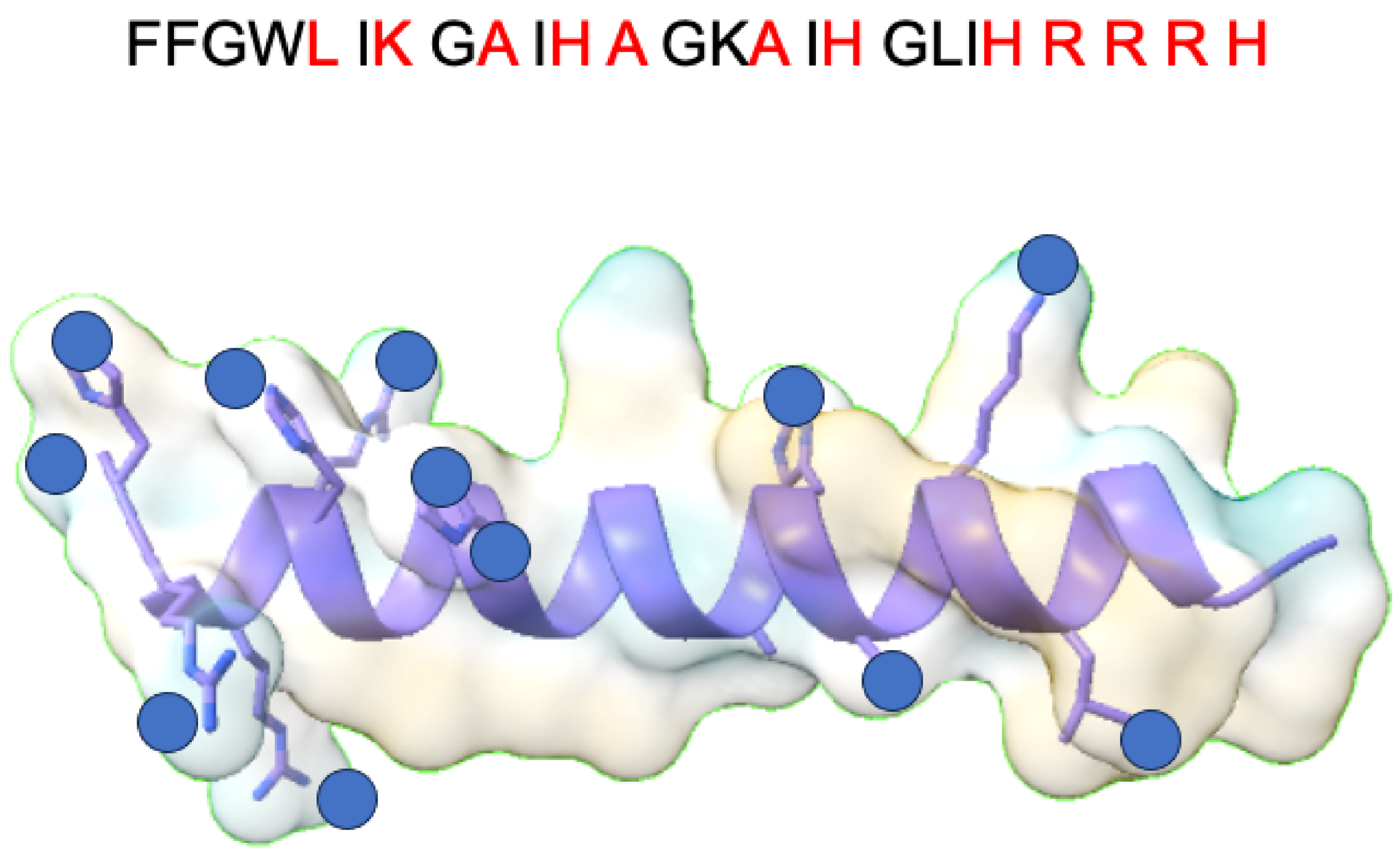
- Fu, R.; Rooney, M.T.; Zhang, R.; Cotten, M.L. Coordination of Redox Ions within a Membrane-Binding Peptide: A Tale of Aromatic Rings. J. Phys. Chem. Lett. 2021, 12, 4392–4399. [Google Scholar] [CrossRef]
- Paredes, S.D.; Kim, S.; Rooney, M.T.; Greenwood, A.I.; Hristova, K.; Cotten, M.L. Enhancing the membrane activity of Piscidin 1 through peptide metallation and the presence of oxidized lipid species: Implications for the unification of host defense mechanisms at lipid membranes. Biochim. Biophys. Acta (BBA) Biomembr. 2020, 1862, 183236. [Google Scholar] [CrossRef]
- Wu, J.; Chen, X.; Zhang, J.; Chen, J.; Wang, Y.; Wei, T.; Ma, J.; Li, Y.; Mo, T.; He, Z.; et al. Tachyplesin induces apoptosis in non-small cell lung cancer cells and enhances the chemosensitivity of A549/DDP cells to cisplatin by activating Fas and necroptosis pathway. Chem. Biol. Drug Des. 2021, 97, 809–820. [Google Scholar] [CrossRef]
- Kuzmin, D.V.; Emel’yanova, A.A.; Kalashnikova, M.B.; Panteleev, P.V.; Ovchinnikova, T.V. In Vitro Study of Antitumor Effect of Antimicrobial Peptide Tachyplesin I in Combination with Cisplatin. Bull. Exp. Biol. Med. 2018, 165, 220–224. [Google Scholar] [CrossRef]
- Meier, S.M.; Novak, M.; Kandioller, W.; Jakupec, M.A.; Arion, V.B.; Metzler-Nolte, N.; Keppler, B.K.; Hartinger, C.G. Identification of the structural determinants for anticancer activity of a ruthenium arene peptide conjugate. Chem. Eur. J. 2013, 19, 9297–9307. [Google Scholar] [CrossRef]
- Lucaciu, R.L.; Hangan, A.C.; Sevastre, B.; Oprean, L.S. Metallo-Drugs in Cancer Therapy: Past, Present and Future. Molecules 2022, 27, 6485. [Google Scholar] [CrossRef]
- Köpf, H.; Köpf-Maier, P. Titanocene dichloride—The first metallocene with cancerostatic activity. Angew. Chem. Int. Ed. Engl. 1979, 18, 477–478. [Google Scholar] [CrossRef]
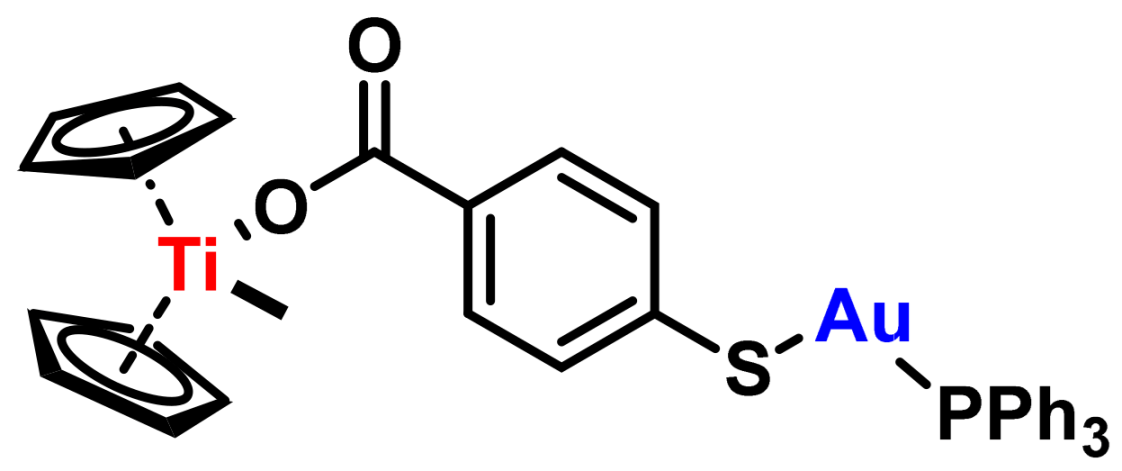
- Fernandez-Vega, L.; Ruiz Silva, V.A.; Domínguez-González, T.M.; Claudio-Betancourt, S.; Toro-Maldonado, R.E.; Capre Maso, L.C.; Ortiz, K.S.; Pérez-Verdejo, J.A.; González, J.R.; Rosado-Fraticelli, G.T.; et al. Evaluating Ligand Modifications of the Titanocene and Auranofin Moieties for the Development of More Potent Anticancer Drugs. Inorganics 2020, 8, 10. [Google Scholar] [CrossRef]
- Toney, J.H.; Marks, T.J. Hydrolysis chemistry of the metallocene dichlorides M(.eta.5-C5H5)2Cl2, M = titanium, vanadium, or zirconium. Aqueous kinetics, equilibria, and mechanistic implications for a new class of antitumor agents. J. Am. Chem. Soc. 1985, 107, 947–953. [Google Scholar] [CrossRef]
- Lümmen, G.; Sperling, H.; Luboldt, H.; Otto, T.; Rübben, H. Phase II trial of titanocene dichloride in advanced renal-cell carcinoma. Cancer Chemother. Pharmacol. 1998, 42, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Kröger, N.; Kleeberg, U.; Mross, K.; Edler, L.; Hossfeld, D. Phase II clinical trial of titanocene dichloride in patients with metastatic breast cancer. Oncol. Res. Treat. 2000, 23, 60–62. [Google Scholar] [CrossRef]
- Erxleben, A.; Claffey, J.; Tacke, M. Binding and hydrolysis studies of antitumoural titanocene dichloride and Titanocene Y with phosphate diesters. J. Inorg. Biochem. 2010, 104, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.; Keppler, K.B.; Heim, M.E.; Niebch, G.; Dietzfelbinger, H.; Rastetter, J.; Hanauske, A.R. Clinical phase I and pharmacokinetic trial of the new titanium complex budotitane. Investig. New Drugs 1996, 13, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.E.; Flechtner, H.; Keppler, B.K. Clinical studies with budotitane—A new non-platinum metal complex for cancer therapy. Ruthenium Other Non-Platin. Met. Complexes Cancer Chemother. 1989, 10, 217. [Google Scholar]
- Guo, Z.; Sadler, P.J. Metals in Medicine. Angew. Chem. Int. Ed. Engl. 1999, 38, 1512–1531. [Google Scholar] [CrossRef]
- Zeng, L.; Gupta, P.; Chen, Y.; Wang, E.; Ji, L.; Chao, H.; Chen, Z.S. The development of anticancer ruthenium(II) complexes: From single molecule compounds to nanomaterials. Chem. Soc. Rev. 2017, 46, 5771–5804. [Google Scholar] [CrossRef]
- Schluga, P.; Hartinger, C.G.; Egger, A.; Reisner, E.; Galanski, M.S.; Jakupec, M.A.; Keppler, B.K. Redox behavior of tumor-inhibiting ruthenium(iii) complexes and effects of physiological reductants on their binding to GMP. Dalton Trans. 2006, 1796–1802. [Google Scholar] [CrossRef]
- Groessl, M.; Tsybin, Y.O.; Hartinger, C.G.; Keppler, B.K.; Dyson, P.J. Ruthenium versus platinum: Interactions of anticancer metallodrugs with duplex oligonucleotides characterised by electrospray ionisation mass spectrometry. J. Biol. Inorg. Chem. 2010, 15, 677–688. [Google Scholar] [CrossRef]
- Liu, S.; Liang, A.; Wu, K.; Zeng, W.; Luo, Q.; Wang, F. Binding of Organometallic Ruthenium Anticancer Complexes to DNA: Thermodynamic Base and Sequence Selectivity. Int. J. Mol. Sci. 2018, 19, 2137. [Google Scholar] [CrossRef]
- van Rijt, S.H.; Sadler, P.J. Current applications and future potential for bioinorganic chemistry in the development of anticancer drugs. Drug Discov. Today 2009, 14, 1089–1097. [Google Scholar] [CrossRef]
- Keppler, B.K.; Rupp, W. Antitumor activity of imidazolium-bisimidazole-tetrachlororuthenate (III). A representative of a new class of inorganic antitumor agents. J. Cancer Res. Clin. Oncol. 1986, 111, 166–168. [Google Scholar] [CrossRef] [PubMed]
- Alessio, E.; Messori, L. NAMI-A and KP1019/1339, Two Iconic Ruthenium Anticancer Drug Candidates Face-to-Face: A Case Story in Medicinal Inorganic Chemistry. Molecules 2019, 24, 1995. [Google Scholar] [CrossRef] [PubMed]
- Sava, G.; Bergamo, A.; Zorzet, S.; Gava, B.; Casarsa, C.; Cocchietto, M.; Furlani, A.; Scarcia, V.; Serli, B.; Iengo, E.; et al. Influence of chemical stability on the activity of the antimetastasis ruthenium compound NAMI-A. Eur. J. Cancer 2002, 38, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Brindell, M.; Piotrowska, D.; Shoukry, A.A.; Stochel, G.; van Eldik, R. Kinetics and mechanism of the reduction of (ImH)[trans-RuCl4(dmso)(Im)] by ascorbic acid in acidic aqueous solution. J. Biol. Inorg. Chem. 2007, 12, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Sava, G. Linking the future of anticancer metal-complexes to the therapy of tumour metastases. Chem. Soc. Rev. 2015, 44, 8818–8835. [Google Scholar] [CrossRef] [PubMed]
- Galeano, A.; Berger, M.; Keppler, B. Antitumor activity of some ruthenium derivatives in human colon cancer cell lines in vitro. Arzneim. Forsch. 1992, 42, 821–824. [Google Scholar]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, a new redox-active anticancer agent–preclinical development and results of a clinical phase I study in tumor patients. Chem. Biodivers. 2008, 5, 2140–2155. [Google Scholar] [CrossRef] [PubMed]
- Wan, D.; Tang, B.; Wang, Y.-J.; Guo, B.-H.; Yin, H.; Yi, Q.-Y.; Liu, Y.-J. Synthesis and anticancer properties of ruthenium (II) complexes as potent apoptosis inducers through mitochondrial disruption. Eur. J. Med. Chem. 2017, 139, 180–190. [Google Scholar] [CrossRef]
- Burris, H.A.; Bakewell, S.; Bendell, J.C.; Infante, J.; Jones, S.F.; Spigel, D.R.; Weiss, G.J.; Ramanathan, R.K.; Ogden, A.; Von Hoff, D. Safety and activity of IT-139, a ruthenium-based compound, in patients with advanced solid tumours: A first-in-human, open-label, dose-escalation phase I study with expansion cohort. ESMO Open 2016, 1, e000154. [Google Scholar] [CrossRef]
- Kenny, R.G.; Marmion, C.J. Toward multi-targeted platinum and ruthenium drugs—A new paradigm in cancer drug treatment regimens? Chem. Rev. 2019, 119, 1058–1137. [Google Scholar] [CrossRef]
- Muralisankar, M.; Chen, J.-R.; Haribabu, J.; Ke, S.-C. Effective and Selective Ru(II)-Arene Complexes Containing 4,4′-Substituted 2,2′ Bipyridine Ligands Targeting Human Urinary Bladder Cancer Cells. Int. J. Mol. Sci. 2023, 24, 11896. [Google Scholar] [CrossRef]
- Renfrew, A.K.; Egger, A.E.; Scopelliti, R.; Hartinger, C.G.; Dyson, P.J. Synthesis and characterisation of the water soluble bis-phosphine complex [Ru (η6-cymene)(PPh2 (o-C6H4O)-κ2-P, O)(pta)]+ and an investigation of its cytotoxic effects. C. R. Chim. 2010, 13, 1144–1150. [Google Scholar] [CrossRef]
- Dorcier, A.; Hartinger, C.G.; Scopelliti, R.; Fish, R.H.; Keppler, B.K.; Dyson, P.J. Studies on the reactivity of organometallic Ru–, Rh–and Os–pta complexes with DNA model compounds. J. Inorg. Biochem. 2008, 102, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Scolaro, C.; Bergamo, A.; Brescacin, L.; Delfino, R.; Cocchietto, M.; Laurenczy, G.; Geldbach, T.J.; Sava, G.; Dyson, P.J. In vitro and in vivo evaluation of ruthenium (II)− arene PTA complexes. J. Med. Chem. 2005, 48, 4161–4171. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.S.; Menin, L.; Scopelliti, R.; Dyson, P.J. Conformational control of anticancer activity: The application of arene-linked dinuclear ruthenium (II) organometallics. Chem. Sci. 2014, 5, 2536–2545. [Google Scholar] [CrossRef]
- Dyson, P.J. Systematic Design of a Targeted Organometallic Antitumour Drug in Pre-clinical Development. CHIMIA Int. J. Chem. 2007, 61, 698. [Google Scholar] [CrossRef]
- Weiss, A.; Berndsen, R.H.; Dubois, M.; Müller, C.; Schibli, R.; Griffioen, A.W.; Dyson, P.J.; Nowak-Sliwinska, P. In vivo anti-tumor activity of the organometallic ruthenium (II)-arene complex [Ru (η 6-p-cymene) Cl 2 (pta)](RAPTA-C) in human ovarian and colorectal carcinomas. Chem. Sci. 2014, 5, 4742–4748. [Google Scholar] [CrossRef]
- Kilpin, K.J.; Cammack, S.M.; Clavel, C.M.; Dyson, P.J. Ruthenium (II) arene PTA (RAPTA) complexes: Impact of enantiomerically pure chiral ligands. Dalton Trans. 2013, 42, 2008–2014. [Google Scholar] [CrossRef]
- Renfrew, A.K.; Phillips, A.D.; Tapavicza, E.; Scopelliti, R.; Rothlisberger, U.; Dyson, P.J. Tuning the efficacy of ruthenium (II)-arene (RAPTA) antitumor compounds with fluorinated arene ligands. Organometallics 2009, 28, 5061–5071. [Google Scholar] [CrossRef]
- Blunden, B.M.; Lu, H.; Stenzel, M.H. Enhanced Delivery of the RAPTA-C Macromolecular Chemotherapeutic by Conjugation to Degradable Polymeric Micelles. Biomacromolecules 2013, 14, 4177–4188. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Kundu, S.; Bhattacharyya, A.; Hartinger, C.G.; Dyson, P.J. The ruthenium(II)-arene compound RAPTA-C induces apoptosis in EAC cells through mitochondrial and p53-JNK pathways. J. Biol. Inorg. Chem. 2008, 13, 1149–1155. [Google Scholar] [CrossRef]
- Swaminathan, S.; Haribabu, J.; Balakrishnan, N.; Vasanthakumar, P.; Karvembu, R. Piano stool Ru(II)-arene complexes having three monodentate legs: A comprehensive review on their development as anticancer therapeutics over the past decade. Coord. Chem. Rev. 2022, 459, 214403. [Google Scholar] [CrossRef]
- Fong, J.; Kasimova, K.; Arenas, Y.; Kaspler, P.; Lazic, S.; Mandel, A.; Lilge, L. A novel class of ruthenium-based photosensitizers effectively kills in vitro cancer cells and in vivo tumors. Photochem. Photobiol. Sci. 2015, 14, 2014–2023. [Google Scholar] [CrossRef] [PubMed]
- Poynton, F.E.; Bright, S.A.; Blasco, S.; Williams, D.C.; Kelly, J.M.; Gunnlaugsson, T. The development of ruthenium(ii) polypyridyl complexes and conjugates for in vitro cellular and in vivo applications. Chem. Soc. Rev. 2017, 46, 7706–7756. [Google Scholar] [CrossRef] [PubMed]
- Peña, B.; Saha, S.; Barhoumi, R.; Burghardt, R.C.; Dunbar, K.R. Ruthenium(II)-Polypyridyl Compounds with π-Extended Nitrogen Donor Ligands Induce Apoptosis in Human Lung Adenocarcinoma (A549) Cells by Triggering Caspase-3/7 Pathway. Inorg. Chem. 2018, 57, 12777–12786. [Google Scholar] [CrossRef] [PubMed]
- Notaro, A.; Jakubaszek, M.; Rotthowe, N.; Maschietto, F.; Vinck, R.; Felder, P.S.; Goud, B.; Tharaud, M.; Ciofini, I.; Bedioui, F.; et al. Increasing the Cytotoxicity of Ru(II) Polypyridyl Complexes by Tuning the Electronic Structure of Dioxo Ligands. J. Am. Chem. Soc. 2020, 142, 6066–6084. [Google Scholar] [CrossRef] [PubMed]
- Klajner, M.; Licona, C.; Fetzer, L.; Hebraud, P.; Mellitzer, G.; Pfeffer, M.; Harlepp, S.; Gaiddon, C. Subcellular Localization and Transport Kinetics of Ruthenium Organometallic Anticancer Compounds in Living Cells: A Dose-Dependent Role for Amino Acid and Iron Transporters. Inorg. Chem. 2014, 53, 5150–5158. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, F.; Karges, J.; Gasser, G. Critical Overview of the Use of Ru(II) Polypyridyl Complexes as Photosensitizers in One-Photon and Two-Photon Photodynamic Therapy. Acc. Chem. Res. 2017, 50, 2727–2736. [Google Scholar] [CrossRef]
- Correia, J.H.; Rodrigues, J.A.; Pimenta, S.; Dong, T.; Yang, Z. Photodynamic Therapy Review: Principles, Photosensitizers, Applications, and Future Directions. Pharmaceutics 2021, 13, 1332. [Google Scholar] [CrossRef]
- Tan, C.-P.; Zhong, Y.-M.; Ji, L.-N.; Mao, Z.-W. Phosphorescent metal complexes as theranostic anticancer agents: Combining imaging and therapy in a single molecule. Chem. Sci. 2021, 12, 2357–2367. [Google Scholar] [CrossRef] [PubMed]
- McFarland, S.A.; Mandel, A.; Dumoulin-White, R.; Gasser, G. Metal-based photosensitizers for photodynamic therapy: The future of multimodal oncology? Curr. Opin. Chem. Biol. 2020, 56, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, Y.; Li, G.; Zhang, P.; Jin, C.; Zeng, L.; Ji, L.; Chao, H. Ruthenium(II) polypyridyl complexes as mitochondria-targeted two-photon photodynamic anticancer agents. Biomaterials 2015, 56, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Karges, J.; Kuang, S.; Maschietto, F.; Blacque, O.; Ciofini, I.; Chao, H.; Gasser, G. Rationally designed ruthenium complexes for 1- and 2-photon photodynamic therapy. Nat. Commun. 2020, 11, 3262. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Wei, H.; Li, Q.; Su, X.; Liu, S.; Zhang, K.Y.; Lv, W.; Zhao, Q.; Li, X.; Huang, W. Achieving efficient photodynamic therapy under both normoxia and hypoxia using cyclometalated Ru(ii) photosensitizer through type I photochemical process. Chem. Sci. 2018, 9, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S. Ruthenium-Based Photoactivated Chemotherapy. J. Am. Chem. Soc. 2023, 145, 23397–23415. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Bai, L.; Zhang, P.; Zhao, H.; Zhou, Q. The Development of Ru(II)-Based Photoactivated Chemotherapy Agents. Molecules 2021, 26, 5679. [Google Scholar] [CrossRef] [PubMed]
- Karaoun, N.; Renfrew, A.K. A luminescent ruthenium(ii) complex for light-triggered drug release and live cell imaging. Chem. Commun. 2015, 51, 14038–14041. [Google Scholar] [CrossRef] [PubMed]
- Zamora, A.; Denning, C.A.; Heidary, D.K.; Wachter, E.; Nease, L.A.; Ruiz, J.; Glazer, E.C. Ruthenium-containing P450 inhibitors for dual enzyme inhibition and DNA damage. Dalton Trans. 2017, 46, 2165–2173. [Google Scholar] [CrossRef] [PubMed]
- van Rixel, V.H.S.; Ramu, V.; Auyeung, A.B.; Beztsinna, N.; Leger, D.Y.; Lameijer, L.N.; Hilt, S.T.; Le Dévédec, S.E.; Yildiz, T.; Betancourt, T.; et al. Photo-Uncaging of a Microtubule-Targeted Rigidin Analogue in Hypoxic Cancer Cells and in a Xenograft Mouse Model. J. Am. Chem. Soc. 2019, 141, 18444–18454. [Google Scholar] [CrossRef]
- D’Amato, A.; Mariconda, A.; Iacopetta, D.; Ceramella, J.; Catalano, A.; Sinicropi, M.S.; Longo, P. Complexes of Ruthenium(II) as Promising Dual-Active Agents against Cancer and Viral Infections. Pharmaceuticals 2023, 16, 1729. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, C.Y.; Nam, T.G. Ruthenium Complexes as Anticancer Agents: A Brief History and Perspectives. Drug Des. Dev. Ther. 2020, 14, 5375–5392. [Google Scholar] [CrossRef] [PubMed]
- Pierce, S.; Jennings, M.P.; Juliano, S.A.; Angeles-Boza, A.M. Peptide–Ruthenium Conjugate as an Efficient Photosensitizer for the Inactivation of Multidrug-Resistant Bacteria. Inorg. Chem. 2020, 59, 14866–14870. [Google Scholar] [CrossRef] [PubMed]
- Pricker, S.P. Medical uses of gold compounds: Past, present and future. Gold Bull. 1996, 29, 53–60. [Google Scholar] [CrossRef]
- Kim, J.H.; Reeder, E.; Parkin, S.; Awuah, S.G. Gold(I/III)-Phosphine Complexes as Potent Antiproliferative Agents. Sci. Rep. 2019, 9, 12335. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.I.; Ooi, K.K.; Tiekink, E.R.T. Gold-Based Medicine: A Paradigm Shift in Anti-Cancer Therapy? Molecules 2018, 23, 1410. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Yang, X.; Yan, M. Synthesis and Structure-Activity Relationship Study of Antimicrobial Auranofin against ESKAPE Pathogens. J. Med. Chem. 2019, 62, 7751–7768. [Google Scholar] [CrossRef] [PubMed]
- Fries, J.F.; Bloch, D.; Spitz, P.; Mitchell, D.M. Cancer in rheumatoid arthritis: A prospective long-term study of mortality. Am. J. Med. 1985, 78, 56–59. [Google Scholar] [CrossRef]
- Shin, D.W.; Kwon, Y.J.; Ye, D.J.; Baek, H.S.; Lee, J.E.; Chun, Y.J. Auranofin Suppresses Plasminogen Activator Inhibitor-2 Expression through Annexin A5 Induction in Human Prostate Cancer Cells. Biomol. Ther. 2017, 25, 177–185. [Google Scholar] [CrossRef]
- He, M.F.; Gao, X.P.; Li, S.C.; He, Z.H.; Chen, N.; Wang, Y.B.; She, J.X. Anti-angiogenic effect of auranofin on HUVECs in vitro and zebrafish in vivo. Eur. J. Pharmacol. 2014, 740, 240–247. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, H.J.; Huang, Q.; Lu, L.; Min, W. Novel action and mechanism of auranofin in inhibition of vascular endothelial growth factor receptor-3-dependent lymphangiogenesis. Anticancer Agents Med. Chem. 2014, 14, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Saba, N.; Shen, M.; Ghias, M.; Liu, J.; Gupta, S.D.; Chauhan, L.; Rao, R.; Gunewardena, S.; Schorno, K.; et al. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer Res. 2014, 74, 2520–2532. [Google Scholar] [CrossRef] [PubMed]
- Raninga, P.V.; Lee, A.C.; Sinha, D.; Shih, Y.Y.; Mittal, D.; Makhale, A.; Bain, A.L.; Nanayakarra, D.; Tonissen, K.F.; Kalimutho, M.; et al. Therapeutic cooperation between auranofin, a thioredoxin reductase inhibitor and anti-PD-L1 antibody for treatment of triple-negative breast cancer. Int. J. Cancer 2020, 146, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Selvaraju, K.; Saei, A.A.; D’Arcy, P.; Zubarev, R.A.; Arnér, E.S.; Linder, S. Repurposing of auranofin: Thioredoxin reductase remains a primary target of the drug. Biochimie 2019, 162, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Berners-Price, S.J. Gold-Based Therapeutic Agents: A New Perspective. In Bioinorganic Medicinal Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2011; pp. 197–222. [Google Scholar]
- Mármol, I.; Quero, J.; Rodríguez-Yoldi, M.J.; Cerrada, E. Gold as a Possible Alternative to Platinum-Based Chemotherapy for Colon Cancer Treatment. Cancers 2019, 11, 780. [Google Scholar] [CrossRef] [PubMed]
- Mirabelli, C.K.; Sung, C.M.; Zimmerman, J.P.; Hill, D.T.; Mong, S.; Crooke, S.T. Interactions of gold coordination complexes with DNA. Biochem. Pharmacol. 1986, 35, 1427–1433. [Google Scholar] [CrossRef] [PubMed]
- Datta, D. On Pearson’s HSAB principle. Inorg. Chem. 1992, 31, 2797–2800. [Google Scholar] [CrossRef]
- Onodera, T.; Momose, I.; Kawada, M. Potential Anticancer Activity of Auranofin. Chem. Pharm. Bull. 2019, 67, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.; Machado, R.C.; Grazul, R.M.; Lopes, M.T.; Corrêa, C.C.; Dos Santos, H.F.; de Almeida, M.V.; Silva, H. Novel antitumor adamantane-azole gold(I) complexes as potential inhibitors of thioredoxin reductase. J. Biol. Inorg. Chem. 2016, 21, 275–292. [Google Scholar] [CrossRef]
- Curran, D.; Dada, O.; Müller-Bunz, H.; Rothemund, M.; Sánchez-Sanz, G.; Schobert, R.; Zhu, X.; Tacke, M. Synthesis and Cytotoxicity Studies of Novel NHC*-Gold(I) Complexes Derived from Lepidiline A. Molecules 2018, 23, 2031. [Google Scholar] [CrossRef]
- Vergara, E.; Casini, A.; Sorrentino, F.; Zava, O.; Cerrada, E.; Rigobello, M.P.; Bindoli, A.; Laguna, M.; Dyson, P.J. Anticancer therapeutics that target selenoenzymes: Synthesis, characterization, in vitro cytotoxicity, and thioredoxin reductase inhibition of a series of gold(I) complexes containing hydrophilic phosphine ligands. ChemMedChem 2010, 5, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Vergara, E.; Cerrada, E.; Clavel, C.; Casini, A.; Laguna, M. Thiolato gold(i) complexes containing water-soluble phosphane ligands: A characterization of their chemical and biological properties. Dalton Trans. 2011, 40, 10927–10935. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, M.; Cao, R.; Zhang, W.; Yin, M.; Xiao, X.; Liu, Q.; Huang, N. A soluble bis-chelated gold(I) diphosphine compound with strong anticancer activity and low toxicity. J. Med. Chem. 2013, 56, 1455–1466. [Google Scholar] [CrossRef] [PubMed]
- Horvath, U.E.; Dobrzańska, L.; Strasser, C.E.; Bouwer Neé Potgieter, W.; Joone, G.; van Rensburg, C.E.; Cronje, S.; Raubenheimer, H.G. Amides of gold(I) diphosphines prepared from N-heterocyclic sources and their in vitro and in vivo screening for anticancer activity. J. Inorg. Biochem. 2012, 111, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Serebryanskaya, T.V.; Lyakhov, A.S.; Ivashkevich, L.S.; Schur, J.; Frias, C.; Prokop, A.; Ott, I. Gold(i) thiotetrazolates as thioredoxin reductase inhibitors and antiproliferative agents. Dalton Trans. 2015, 44, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Bagowski, C.P.; You, Y.; Scheffler, H.; Vlecken, D.H.; Schmitz, D.J.; Ott, I. Naphthalimide gold(i) phosphine complexes as anticancer metallodrugs. Dalton Trans. 2009, 10799–10805. [Google Scholar] [CrossRef] [PubMed]
- Tavares, T.T.; Azevedo, G.C.; Garcia, A.; Carpanez, A.G.; Lewer, P.M.; Paschoal, D.; Müller, B.L.; Dos Santos, H.F.; Matos, R.C.; Silva, H.; et al. Gold(I) complexes with aryl-thiosemicarbazones: Molecular modeling, synthesis, cytotoxicity and TrxR inhibition. Polyhedron 2017, 132, 95–104. [Google Scholar] [CrossRef]
- Seliman, A.A.A.; Altaf, M.; Odewunmi, N.A.; Kawde, A.-N.; Zierkiewicz, W.; Ahmad, S.; Altuwaijri, S.; Isab, A.A. Synthesis, X-ray structure, DFT calculations and anticancer activity of a selenourea coordinated gold(I)-carbene complex. Polyhedron 2017, 137, 197–206. [Google Scholar] [CrossRef]
- Seliman, A.A.A.; Altaf, M.; Onawole, A.T.; Ahmad, S.; Ahmed, M.Y.; Al-Saadi, A.A.; Altuwaijri, S.; Bhatia, G.; Singh, J.; Isab, A.A. Synthesis, X-ray structures and anticancer activity of gold(I)-carbene complexes with selenones as co-ligands and their molecular docking studies with thioredoxin reductase. J. Organomet. Chem. 2017, 848, 175–183. [Google Scholar] [CrossRef]
- Schuh, E.; Valiahdi, S.M.; Jakupec, M.A.; Keppler, B.K.; Chiba, P.; Mohr, F. Synthesis and biological studies of some gold(I) complexes containing functionalised alkynes. Dalton Trans. 2009, 10841–10845. [Google Scholar] [CrossRef]
- Tasan, S.; Licona, C.; Doulain, P.E.; Michelin, C.; Gros, C.P.; Le Gendre, P.; Harvey, P.D.; Paul, C.; Gaiddon, C.; Bodio, E. Gold-phosphine-porphyrin as potential metal-based theranostics. J. Biol. Inorg. Chem. 2015, 20, 143–154. [Google Scholar] [CrossRef] [PubMed]
- da Silva Maia, P.I.; Deflon, V.M.; Abram, U. Gold(III) Complexes in Medicinal Chemistry. Future Med. Chem. 2014, 6, 1515–1536. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, B.; Williams, M.R.M.; Bochmann, M. Gold(III) Complexes for Antitumor Applications: An Overview. Chem. Eur. J. 2018, 24, 11840–11851. [Google Scholar] [CrossRef] [PubMed]
- Nardon, C.; Fregona, D. Gold(III) Complexes in the Oncological Preclinical Arena: From Aminoderivatives to Peptidomimetics. Curr. Top. Med. Chem. 2016, 16, 360–380. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Hartinger, C.; Gabbiani, C.; Mini, E.; Dyson, P.J.; Keppler, B.K.; Messori, L. Gold(III) compounds as anticancer agents: Relevance of gold–protein interactions for their mechanism of action. J. Inorg. Biochem. 2008, 102, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Che, C.-M.; Sun, R.W.-Y. Therapeutic applications of gold complexes: Lipophilic gold(iii) cations and gold(i) complexes for anti-cancer treatment. Chem. Commun. 2011, 47, 9554–9560. [Google Scholar] [CrossRef] [PubMed]
- Cinellu, M.A.; Minghetti, G.; Pinna, M.V.; Stoccoro, S.; Zucca, A.; Manassero, M. Gold(III) derivatives with anionic oxygen ligands: Mononuclear hydroxo, alkoxo and acetato complexes. Crystal structure of [Au(bpy)(OMe)2][PF6]. J. Chem. Soc. Dalton Trans. 2000, 1261–1265. [Google Scholar] [CrossRef]
- Milacic, V.; Dou, Q.P. The tumor proteasome as a novel target for gold (III) complexes: Implications for breast cancer therapy. Coord. Chem. Rev. 2009, 253, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.W.-Y.; Che, C.-M. The anti-cancer properties of gold(III) compounds with dianionic porphyrin and tetradentate ligands. Coord. Chem. Rev. 2009, 253, 1682–1691. [Google Scholar] [CrossRef]
- Radisavljević, S.; Ćoćić, D.; Jovanović, S.; Šmit, B.; Petković, M.; Milivojević, N.; Planojević, N.; Marković, S.; Petrović, B. Synthesis, characterization, DFT study, DNA/BSA-binding affinity, and cytotoxicity of some dinuclear and trinuclear gold(III) complexes. J. Biol. Inorg. Chem. 2019, 24, 1057–1076. [Google Scholar] [CrossRef]
- Ronconi, L.; Marzano, C.; Zanello, P.; Corsini, M.; Miolo, G.; Maccà, C.; Trevisan, A.; Fregona, D. Gold(III) Dithiocarbamate Derivatives for the Treatment of Cancer: Solution Chemistry, DNA Binding, and Hemolytic Properties. J. Med. Chem. 2006, 49, 1648–1657. [Google Scholar] [CrossRef]
- Altaf, M.; Casagrande, N.; Mariotto, E.; Baig, N.; Kawde, A.N.; Corona, G.; Larcher, R.; Borghese, C.; Pavan, C.; Seliman, A.A.; et al. Potent In Vitro and In vivo Anticancer Activity of New Bipyridine and Bipyrimidine Gold (III) Dithiocarbamate Derivatives. Cancers 2019, 11, 474. [Google Scholar] [CrossRef] [PubMed]
- Serratrice, M.; Edafe, F.; Mendes, F.; Scopelliti, R.; Zakeeruddin, S.M.; Grätzel, M.; Santos, I.; Cinellu, M.A.; Casini, A. Cytotoxic gold compounds: Synthesis, biological characterization and investigation of their inhibition properties of the zinc finger protein PARP-1. Dalton Trans. 2012, 41, 3287–3293. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, M.N.; Bonsignore, R.; Thomas, S.R.; Bourissou, D.; Barone, G.; Casini, A. Cyclometalated Au(III) Complexes for Cysteine Arylation in Zinc Finger Protein Domains: Towards Controlled Reductive Elimination. Chemistry 2019, 25, 7628–7634. [Google Scholar] [CrossRef] [PubMed]
- Che, C.-M.; Sun, R.W.-Y.; Yu, W.-Y.; Ko, C.-B.; Zhu, N.; Sun, H. Gold(iii) porphyrins as a new class of anticancer drugs: Cytotoxicity, DNA binding and induction of apoptosis in human cervix epitheloid cancer cells. Chem. Commun. 2003, 1718–1719. [Google Scholar] [CrossRef] [PubMed]
- Lum, C.T.; Wai-Yin Sun, R.; Zou, T.; Che, C.-M. Gold(iii) complexes inhibit growth of cisplatin-resistant ovarian cancer in association with upregulation of proapoptotic PMS2 gene. Chem. Sci. 2014, 5, 1579–1584. [Google Scholar] [CrossRef]
- Fernández-Gallardo, J.; Elie, B.T.; Sadhukha, T.; Prabha, S.; Sanaú, M.; Rotenberg, S.A.; Ramos, J.W.; Contel, M. Heterometallic titanium-gold complexes inhibit renal cancer cells in vitro and in vivo. Chem. Sci. 2015, 6, 5269–5283. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Singh, S.; Sohi, H.; Dang, S. Advances in delivery of chemotherapeutic agents for cancer treatment. AAPS PharmSciTech 2022, 23, 25. [Google Scholar] [CrossRef]
- Rocha, M.; Chaves, N.; Báo, S. Nanobiotechnology for breast cancer treatment. In Breast Cancer—From Biology Medicine; IntechOpen: London, UK, 2017. [Google Scholar]
- Yan, L.; Shen, J.; Wang, J.; Yang, X.; Dong, S.; Lu, S. Nanoparticle-Based Drug Delivery System: A Patient-Friendly Chemotherapy for Oncology. Dose Response 2020, 18, 1559325820936161. [Google Scholar] [CrossRef]
- Gavas, S.; Quazi, S.; Karpiński, T.M. Nanoparticles for Cancer Therapy: Current Progress and Challenges. Nanoscale Res. Lett. 2021, 16, 173. [Google Scholar] [CrossRef]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef]
- Mao, X.; Calero-Pérez, P.; Montpeyó, D.; Bruna, J.; Yuste, V.J.; Candiota, A.P.; Lorenzo, J.; Novio, F.; Ruiz-Molina, D. Intranasal Administration of Catechol-Based Pt(IV) Coordination Polymer Nanoparticles for Glioblastoma Therapy. Nanomaterials 2022, 12, 1221. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Kim, H.C.; Wolfram, J.; Mu, C.; Zhang, W.; Liu, H.; Xie, Y.; Mai, J.; Zhang, H.; Li, Z.; et al. A Liposome Encapsulated Ruthenium Polypyridine Complex as a Theranostic Platform for Triple-Negative Breast Cancer. Nano Lett. 2017, 17, 2913–2920. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Wan, H.; Wang, W.; Zhang, X.; Uno, T.; Yang, Q.; Yue, J.; Gao, H.; Zhong, Y.; Tian, Y.; et al. A theranostic agent for cancer therapy and imaging in the second near-infrared window. Nano Res. 2019, 12, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Yücel, O.; Şengelen, A.; Emik, S.; Önay-Uçar, E.; Arda, N.; Gürdağ, G. Folic acid-modified methotrexate-conjugated gold nanoparticles as nano-sized trojans for drug delivery to folate receptor-positive cancer cells. Nanotechnology 2020, 31, 355101. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.Y.; Huan, M.L.; Wang, W.; Jia, Z.Y.; Wan, Y.H.; Zhou, S.Y.; Zhang, B.L. Tumor microenvironment and redox dual stimuli-responsive polymeric nanoparticles for the effective cisplatin-based cancer chemotherapy. Nanotechnology 2022, 34, 035101. [Google Scholar] [CrossRef] [PubMed]
- Irimie, A.I.; Sonea, L.; Jurj, A.; Mehterov, N.; Zimta, A.A.; Budisan, L.; Braicu, C.; Berindan-Neagoe, I. Future trends and emerging issues for nanodelivery systems in oral and oropharyngeal cancer. Int. J. Nanomed. 2017, 12, 4593–4606. [Google Scholar] [CrossRef] [PubMed]
- Drbohlavova, J.; Chomoucka, J.; Adam, V.; Ryvolova, M.; Eckschlager, T.; Hubalek, J.; Kizek, R. Nanocarriers for anticancer drugs—New trends in nanomedicine. Curr. Drug Metab. 2013, 14, 547–564. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Li, Z.; Tan, S.; Li, S.; Shen, Q.; Wang, K. Cancer drug delivery in the nano era: An overview and perspectives (Review). Oncol. Rep. 2017, 38, 611–624. [Google Scholar] [CrossRef]
- Yin, H.; Liao, L.; Fang, J. Enhanced permeability and retention (EPR) effect based tumor targeting: The concept, application and prospect. JSM Clin. Oncol. Res. 2014, 2, 1010. [Google Scholar]
- Nakamura, Y.; Mochida, A.; Choyke, P.L.; Kobayashi, H. Nanodrug Delivery: Is the Enhanced Permeability and Retention Effect Sufficient for Curing Cancer? Bioconjug. Chem. 2016, 27, 2225–2238. [Google Scholar] [CrossRef] [PubMed]
- Greish, K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Methods Mol. Biol. 2010, 624, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Dai, F.; Mao, Y.; Zhang, K.; Qin, Y.; Zheng, J. Metallodrugs in the battle against non-small cell lung cancer: Unlocking the potential for improved therapeutic outcomes. Front. Pharmacol. 2023, 14, 1242488. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrés, C.M.C.; Pérez de la Lastra, J.M.; Bustamante Munguira, E.; Andrés Juan, C.; Pérez-Lebeña, E. Anticancer Activity of Metallodrugs and Metallizing Host Defense Peptides—Current Developments in Structure-Activity Relationship. Int. J. Mol. Sci. 2024, 25, 7314. https://doi.org/10.3390/ijms25137314
Andrés CMC, Pérez de la Lastra JM, Bustamante Munguira E, Andrés Juan C, Pérez-Lebeña E. Anticancer Activity of Metallodrugs and Metallizing Host Defense Peptides—Current Developments in Structure-Activity Relationship. International Journal of Molecular Sciences. 2024; 25(13):7314. https://doi.org/10.3390/ijms25137314
Chicago/Turabian StyleAndrés, Celia María Curieses, José Manuel Pérez de la Lastra, Elena Bustamante Munguira, Celia Andrés Juan, and Eduardo Pérez-Lebeña. 2024. "Anticancer Activity of Metallodrugs and Metallizing Host Defense Peptides—Current Developments in Structure-Activity Relationship" International Journal of Molecular Sciences 25, no. 13: 7314. https://doi.org/10.3390/ijms25137314
APA StyleAndrés, C. M. C., Pérez de la Lastra, J. M., Bustamante Munguira, E., Andrés Juan, C., & Pérez-Lebeña, E. (2024). Anticancer Activity of Metallodrugs and Metallizing Host Defense Peptides—Current Developments in Structure-Activity Relationship. International Journal of Molecular Sciences, 25(13), 7314. https://doi.org/10.3390/ijms25137314