
Functional Genomics in Psoriasis


Abstract
1. Definition and General Understanding of Psoriasis

{kind=link}
| Section | Key Message | References |
|---|---|---|
| Definition and genetics of psoriasis | Psoriasis is a multifactorial, autoinflammatory, dermatological condition characterised by raised scaley lesions across the body. Whilst influenced by environmental and lifestyle factors, it has a large genetic component. Variants in PSORS1, located in the HLA-C gene, have been identified as the main genetic risk factor for psoriasis, accounting for 30–50% of disease heritability. | [2,3,4,5,9] |
| GWAS in psoriasis: benefits and limitations | The first large-scale GWAS in psoriasis by Cargill et al. confirmed the importance of IL12B and IL23R. Both are now used successfully as targets for treating psoriasis. More recent GWASs, such as the meta-analysis by Dand et al., have identified even more unique signatures. However, GWASs cannot assign a causal variant, gene, or cell type or elucidate the biological mechanism driving the SNP–phenotype relationship. | [10,11] |
| Post-GWAS analysis of psoriasis-associated SNPs using functional genomics | Techniques such as fine mapping, epigenetic analysis, chromatin conformation capture and eQTL analysis can reveal the structural context of genetic changes and identify physical interactions between genetic landmarks. Additionally, by identifying SNPs in cell type-specific enhancers, the causal cell types driving psoriasis phenotypes can be identified. | [12,13,14,15] |
| Examining phenotypic differences using advanced functional techniques | The revolutionary gene-editing technique CRISPR can be utilised to activate, repress, delete or alter a region of interest to assess the genetic and phenotypic consequences of suspected lead variants in psoriasis. This technique can be applied on the cellular level—both in cell lines and primary cells—as well as in organoid systems and whole organisms such as mouse models. | [16,17,18,19,20,21] |
| Towards novel psoriasis therapeutics | Drug repurposing utilises treatments that have already been used to treat other diseases. This method dramatically speeds up development time and reduces costs, as safety and pharmacodynamic profiles are already known for these drugs. Examples include the holistic treatment Esculetin and cancer drugs targeting POLI and IL-13. AI can also be used to speed up this process. While CRISPR-Cas9 has been used to treat sickle-cell disease and transfusion-dependent β-thalassemia, it has not yet been applied to psoriasis. Wan and colleagues suggest that the first RNP treatment for psoriasis could be on the horizon. | [22,23,24,25,26,27] |
2. The Significant Genetic Component in Psoriasis
| GWASs | GWASs determine associations between different genotypes and phenotypes to identify clusters of correlated SNPs associated with a particular trait [38]. Such screens can identify new genetic markers that increase susceptibility to psoriasis. |
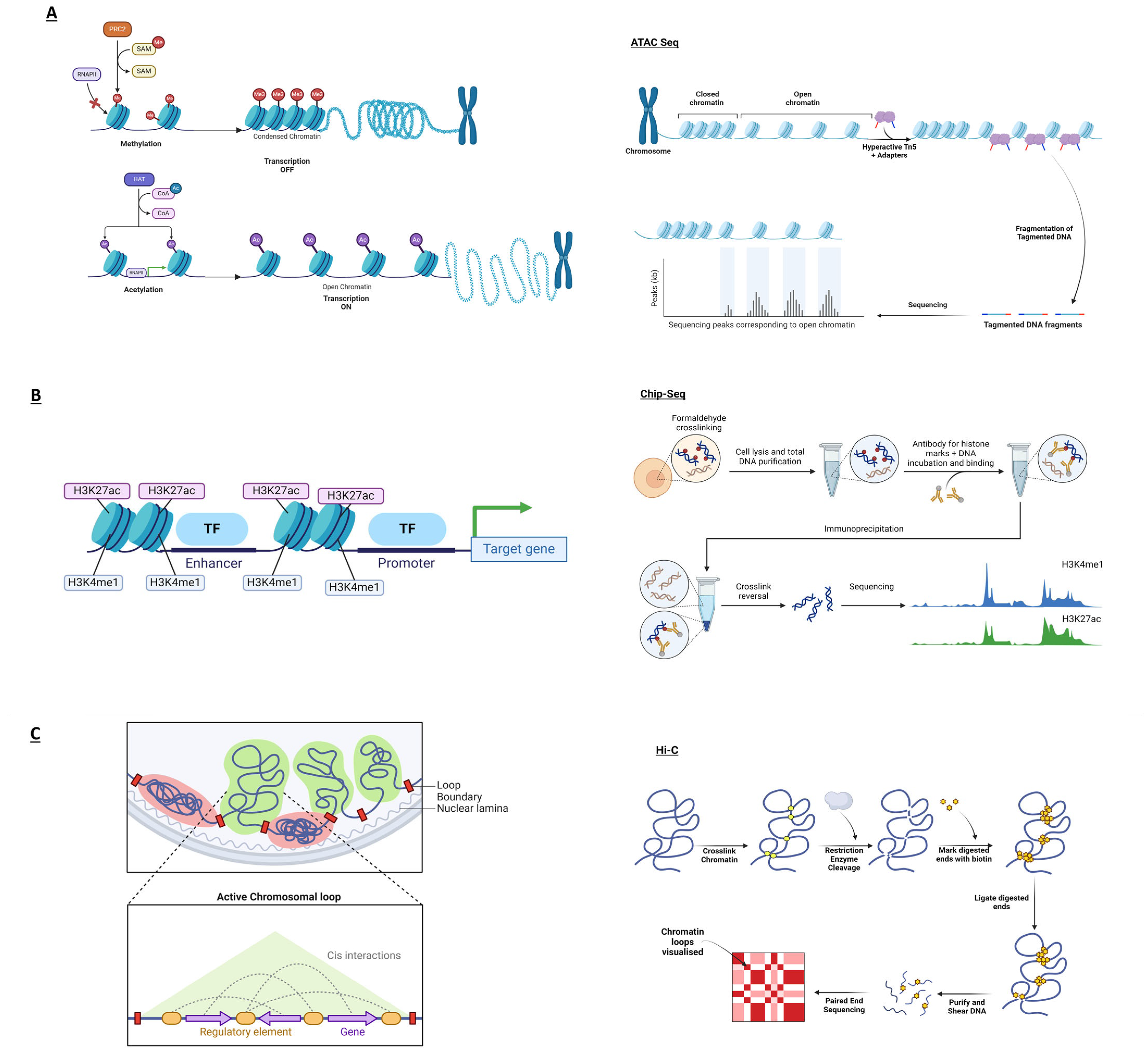
| ATAC-seq | ATAC-seq determines transcriptionally active, open chromatin regions [46]. Changes in chromatin accessibility in psoriasis offer clues into potentially critical genome spots in disease susceptibility. |
| ChIP-seq | ChIP-seq identifies histone modifications in targeted genomic regions, pinpointing the biological roles of epigenetic markers in different conditions or diseases [47]. In psoriasis, the ChIP-seq method can identify unknown gene regulatory mechanisms to identify novel therapeutic approaches. |
| Capture Hi-C (CHi-C) | CHi-C identifies specific regions—e.g., promoters or enhancers—through a hybridisation step, enriching and increasing the resolution of these areas of interest [48] compared to classical chromosome conformation capture techniques. Psoriasis-related long-range interactions can identify DNA rearrangements that could increase the risk of developing the disease. |
| CRISPR | The CRISPR system is a powerful tool that classically cuts targeted DNA sequences with breaks prone to alteration, deletion, or addition of genetic sequences [16]. This technique can help study genes or non-coding regions involved in psoriasis. |
3. GWASs in Psoriasis
4. Limitations and Potential Benefits of GWASs in Understanding Psoriasis
5. Post-GWAS Analysis of Psoriasis-Associated SNPs
6. The Use of Functional Genomics in Psoriasis Research
7. Chromosome Conformation Capture and eQTLs
8. Examining Phenotypic Differences Using Advanced Functional Techniques
9. Organoids
10. Epigenetic Studies in Psoriasis
11. Mouse Models
12. Towards Novel Psoriasis Therapeutics
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lebwohl, M. Psoriasis. Lancet 2003, 361, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Greb, J.E.; Goldminz, A.M.; Elder, J.T.; Lebwohl, M.G.; Gladman, D.D.; Wu, J.J.; Mehta, N.N.; Finlay, A.Y.; Gottlieb, A.B. Psoriasis. Nat. Rev. Dis. Primers 2016, 2, 16082. [Google Scholar] [CrossRef] [PubMed]
- Rendon, A.; Schäkel, K. Psoriasis Pathogenesis and Treatment. Int. J. Mol. Sci. 2019, 20, 1475. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.W. Psoriasis. JAMA Dermatol. 2017, 153, 956. [Google Scholar] [CrossRef] [PubMed]
- Parisi, R.; Symmons, D.P.M.; Griffiths, C.E.M.; Ashcroft, D.M. Global Epidemiology of Psoriasis: A Systematic Review of Incidence and Prevalence. J. Investig. Dermatol. 2013, 133, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Icen, M.; Crowson, C.S.; McEvoy, M.T.; Dann, F.J.; Gabriel, S.E.; Maradit Kremers, H. Trends in Incidence of Adult-Onset Psoriasis over Three Decades: A Population-Based Study. J. Am. Acad. Dermatol. 2009, 60, 394–401. [Google Scholar] [CrossRef]
- Rachakonda, T.D.; Schupp, C.W.; Armstrong, A.W. Psoriasis Prevalence among Adults in the United States. J. Am. Acad. Dermatol. 2014, 70, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Masson Regnault, M.; Shourick, J.; Jendoubi, F.; Tauber, M.; Paul, C. Time to Relapse After Discontinuing Systemic Treatment for Psoriasis: A Systematic Review. Am. J. Clin. Dermatol. 2022, 23, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.H.; Ameen, H.; Veal, C.; Evans, J.; Ramrakha-Jones, V.S.; Marsland, A.M.; David Burden, A.; Griffiths, C.E.M.; Trembath, R.C.; Barker, J.N.W.N. The Major Psoriasis Susceptibility Locus PSORS1 Is Not a Risk Factor for Late-Onset Psoriasis. J. Investig. Dermatol. 2005, 124, 103–106. [Google Scholar] [CrossRef]
- Cargill, M.; Schrodi, S.J.; Chang, M.; Garcia, V.E.; Brandon, R.; Callis, K.P.; Matsunami, N.; Ardlie, K.G.; Civello, D.; Catanese, J.J.; et al. A Large-Scale Genetic Association Study Confirms IL12B and Leads to the Identification of IL23R as Psoriasis-Risk Genes. Am. J. Hum. Genet. 2007, 80, 273–290. [Google Scholar] [CrossRef]
- Dand, N.; Stuart, P.E.; Bowes, J.; Ellinghaus, D.; Nititham, J.; Saklatvala, J.R.; Teder-Laving, M.; Thomas, L.F.; Traks, T.; Uebe, S.; et al. GWAS Meta-Analysis of Psoriasis Identifies New Susceptibility Alleles Impacting Disease Mechanisms and Therapeutic Targets. medRxiv 2023. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Stuart, P.E.; Tian, C.; Gudjonsson, J.E.; Das, S.; Zawistowski, M.; Ellinghaus, E.; Barker, J.N.; Chandran, V.; Dand, N.; et al. Large Scale Meta-Analysis Characterizes Genetic Architecture for Common Psoriasis Associated Variants. Nat. Commun. 2017, 8, 15382. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, A.; Asimit, J.; Wallace, C. Fine-Mapping Genetic Associations. Hum. Mol. Genet. 2020, 29, R81–R88. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, A.; Watson, H.; Wallace, C. Improving the Coverage of Credible Sets in Bayesian Genetic Fine-Mapping. PLoS Comput. Biol. 2020, 16, e1007829. [Google Scholar] [CrossRef] [PubMed]
- Gasperskaja, E.; Kučinskas, V. The Most Common Technologies and Tools for Functional Genome Analysis. Acta Medica Litu. 2017, 24, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Doudna, J.A. CRISPR Technology: A Decade of Genome Editing Is Only the Beginning. Science 2024, 379, eadd8643. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. Genome Editing. The New Frontier of Genome Engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Fellmann, C.; Gowen, B.G.; Lin, P.-C.; Doudna, J.A.; Corn, J.E. Cornerstones of CRISPR-Cas in Drug Discovery and Therapy. Nat. Rev. Drug Discov. 2017, 16, 89–100. [Google Scholar] [CrossRef]
- Boonekamp, K.E.; Kretzschmar, K.; Wiener, D.J.; Asra, P.; Derakhshan, S.; Puschhof, J.; López-Iglesias, C.; Peters, P.J.; Basak, O.; Clevers, H. Long-Term Expansion and Differentiation of Adult Murine Epidermal Stem Cells in 3D Organoid Cultures. Proc. Natl. Acad. Sci. USA 2019, 116, 14630–14638. [Google Scholar] [CrossRef]
- Lai Cheong, J.E.; Wessagowit, V.; McGrath, J.A. Molecular Abnormalities of the Desmosomal Protein Desmoplakin in Human Disease. Clin. Exp. Dermatol. 2005, 30, 261–266. [Google Scholar] [CrossRef]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A Genomic Sequencing Protocol That Yields a Positive Display of 5-Methylcytosine Residues in Individual DNA Strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef]
- Jeong, Y.; Song, J.; Lee, Y.; Choi, E.; Won, Y.; Kim, B.; Jang, W. A Transcriptome-Wide Analysis of Psoriasis: Identifying the Potential Causal Genes and Drug Candidates. Int. J. Mol. Sci. 2023, 24, 11717. [Google Scholar] [CrossRef]
- McInnes, G.; Yee, S.W.; Pershad, Y.; Altman, R.B. Genomewide Association Studies in Pharmacogenomics. Clin. Pharmacol. Ther. 2021, 110, 637–648. [Google Scholar] [CrossRef]
- Griffith, M.; Griffith, O.L.; Coffman, A.C.; Weible, J.V.; Mcmichael, J.F.; Spies, N.C.; Koval, J.; Das, I.; Callaway, M.B.; Eldred, J.M.; et al. DGIdb: Mining the Druggable Genome. Nat. Methods 2013, 10, 1209–1210. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, A.; Reeves, E.; Vollmer, S.; Arakawa, Y.; He, M.; Galinski, A.; Stöhr, J.; Dornmair, K.; James, E.; Prinz, J.C. ERAP1 Controls the Autoimmune Response against Melanocytes in Psoriasis by Generating the Melanocyte Autoantigen and Regulating Its Amount for HLA-C*06:02 Presentation. J. Immunol. 2021, 207, 2235–2244. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, Y.; Ekunwe, S.I.N.; Yi, X.; Liu, X.; Wang, H.; Pan, Y. Antioxidant Activity and Inhibition Effect on the Growth of Human Colon Carcinoma (HT-29) Cells of Esculetin from Cortex Fraxini. Med. Chem. Res. 2011, 20, 968–974. [Google Scholar] [CrossRef]
- Halling-Brown, M.D.; Bulusu, K.C.; Patel, M.; Tym, J.E.; Al-Lazikani, B. CanSAR: An Integrated Cancer Public Translational Research and Drug Discovery Resource. Nucleic Acids Res. 2012, 40, D947–D956. [Google Scholar] [CrossRef]
- Zeng, J.; Luo, S.; Huang, Y.; Lu, Q. Critical Role of Environmental Factors in the Pathogenesis of Psoriasis. J. Dermatol. 2017, 44, 863–872. [Google Scholar] [CrossRef]
- Rahman, P.; Elder, J.T. Genetic Epidemiology of Psoriasis and Psoriatic Arthritis. Ann. Rheum. Dis. 2005, 64 (Suppl. 2), ii37–ii39; discussion ii40–ii41. [Google Scholar] [CrossRef]
- Lønnberg, A.S.; Skov, L.; Skytthe, A.; Kyvik, K.O.; Pedersen, O.B.; Thomsen, S.F. Heritability of Psoriasis in a Large Twin Sample. Br. J. Dermatol. 2013, 169, 412–416. [Google Scholar] [CrossRef]
- Gudjonsson, J.E.; Elder, J.T. Psoriasis: Epidemiology. Clin. Dermatol. 2007, 25, 535–546. [Google Scholar] [CrossRef]
- Mahil, S.K.; Capon, F.; Barker, J.N. Genetics of Psoriasis. Dermatol. Clin. 2015, 33, 1–11. [Google Scholar] [CrossRef] [PubMed]
- International Psoriasis Genetics Consortium. The International Psoriasis Genetics Study: Assessing Linkage to 14 Candidate Susceptibility Loci in a Cohort of 942 Affected Sib Pairs. Am. J. Hum. Genet. 2003, 73, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Capon, F.; Trembath, R.C.; Barker, J.N. An Update on the Genetics of Psoriasis. Dermatol. Clin. 2004, 22, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Elder, J.T. PSORS1: Linking Genetics and Immunology. J. Investig. Dermatol. 2006, 126, 1205–1206. [Google Scholar] [CrossRef] [PubMed]
- Harden, J.L.; Krueger, J.G.; Bowcock, A.M. The Immunogenetics of Psoriasis: A Comprehensive Review. J. Autoimmun. 2015, 64, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Mateu-Arrom, L.; Puig, L. Genetic and Epigenetic Mechanisms of Psoriasis. Genes 2023, 14, 1619. [Google Scholar] [CrossRef] [PubMed]
- Uffelmann, E.; Huang, Q.Q.; Munung, N.S.; de Vries, J.; Okada, Y.; Martin, A.R.; Martin, H.C.; Lappalainen, T.; Posthuma, D. Genome-Wide Association Studies. Nat. Rev. Methods Primers 2021, 1, 59. [Google Scholar] [CrossRef]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.-Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement Factor H Polymorphism in Age-Related Macular Degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Sollis, E.; Mosaku, A.; Abid, A.; Buniello, A.; Cerezo, M.; Gil, L.; Groza, T.; Güneş, O.; Hall, P.; Hayhurst, J.; et al. The NHGRI-EBI GWAS Catalog: Knowledgebase and Deposition Resource. Nucleic Acids Res. 2023, 51, D977–D985. [Google Scholar] [CrossRef]
- Lee, J.J.; Wedow, R.; Okbay, A.; Kong, E.; Maghzian, O.; Zacher, M.; Nguyen-Viet, T.A.; Bowers, P.; Sidorenko, J.; Karlsson Linnér, R.; et al. Gene Discovery and Polygenic Prediction from a Genome-Wide Association Study of Educational Attainment in 1.1 Million Individuals. Nat. Genet. 2018, 50, 1112–1121. [Google Scholar] [CrossRef]
- Slatkin, M. Linkage Disequilibrium—Understanding the Evolutionary Past and Mapping the Medical Future. Nat. Rev. Genet. 2008, 9, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef]
- Cortes, A.; Brown, M.A. Promise and Pitfalls of the Immunochip. Arthritis Res. Ther. 2011, 13, 101. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.T.; Nijsten, T.; Elder, J.T. Recent Highlights in Psoriasis Research. J. Investig. Dermatol. 2017, 137, 550–556. [Google Scholar] [CrossRef]
- Grandi, F.C.; Modi, H.; Kampman, L.; Corces, M.R. Chromatin Accessibility Profiling by ATAC-Seq. Nat. Protoc. 2022, 17, 1518–1552. [Google Scholar] [CrossRef] [PubMed]
- Nakato, R.; Sakata, T. Methods for ChIP-Seq Analysis: A Practical Workflow and Advanced Applications. Methods 2021, 187, 44–53. [Google Scholar] [CrossRef]
- Aljogol, D.; Thompson, I.R.; Osborne, C.S.; Mifsud, B. Comparison of Capture Hi-C Analytical Pipelines. Front. Genet. 2022, 13, 786501. [Google Scholar] [CrossRef] [PubMed]
- Tsunemi, Y.; Saeki, H.; Nakamura, K.; Sekiya, T.; Hirai, K.; Fujita, H.; Asano, N.; Kishimoto, M.; Tanida, Y.; Kakinuma, T.; et al. Interleukin-12 P40 Gene (IL12B) 3′-Untranslated Region Polymorphism Is Associated with Susceptibility to Atopic Dermatitis and Psoriasis Vulgaris. J. Dermatol. Sci. 2002, 30, 161–166. [Google Scholar] [CrossRef]
- Han, B.; Eskin, E. Interpreting Meta-Analyses of Genome-Wide Association Studies. PLoS Genet. 2012, 8, e1002555. [Google Scholar] [CrossRef]
- Nishikawa, R.; Nagai, H.; Bito, T.; Ikeda, T.; Horikawa, T.; Adachi, A.; Matsubara, T.; Nishigori, C. Genetic Prediction of the Effectiveness of Biologics for Psoriasis Treatment. J. Dermatol. 2016, 43, 1273–1277. [Google Scholar] [CrossRef]
- Hirata, J.; Hirota, T.; Ozeki, T.; Kanai, M.; Sudo, T.; Tanaka, T.; Hizawa, N.; Nakagawa, H.; Sato, S.; Mushiroda, T.; et al. Variants at HLA-A, HLA-C, and HLA-DQB1 Confer Risk of Psoriasis Vulgaris in Japanese. J. Investig. Dermatol. 2018, 138, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, J.N. Genomewide Association Studies--Illuminating Biologic Pathways. N. Engl. J. Med. 2009, 360, 1699–1701. [Google Scholar] [CrossRef]
- Klein, R.J.; Xu, X.; Mukherjee, S.; Willis, J.; Hayes, J. Successes of Genome-Wide Association Studies. Cell 2010, 142, 350–351. [Google Scholar] [CrossRef]
- Boutet, M.-A.; Nerviani, A.; Gallo Afflitto, G.; Pitzalis, C. Role of the IL-23/IL-17 Axis in Psoriasis and Psoriatic Arthritis: The Clinical Importance of Its Divergence in Skin and Joints. Int. J. Mol. Sci. 2018, 19, 530. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Ellinghaus, E.; Nair, R.P.; Stuart, P.E.; Esko, T.; Metspalu, A.; Debrus, S.; Raelson, J.V.; Tejasvi, T.; Belouchi, M.; et al. Combined Analysis of Genome-Wide Association Studies for Crohn Disease and Psoriasis Identifies Seven Shared Susceptibility Loci. Am. J. Hum. Genet. 2012, 90, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Strange, A.; Capon, F.; Spencer, C.C.A.; Knight, J.; Weale, M.E.; Allen, M.H.; Barton, A.; Band, G.; Bellenguez, C.; Bergboer, J.G.M.; et al. A Genome-Wide Association Study Identifies New Psoriasis Susceptibility Loci and an Interaction between HLA-C and ERAP1. Nat. Genet. 2010, 42, 985–990. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the Missing Heritability of Complex Diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- McClellan, J.; King, M.-C. Genetic Heterogeneity in Human Disease. Cell 2010, 141, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef]
- Goldstein, D.B. Common Genetic Variation and Human Traits. N. Engl. J. Med. 2009, 360, 1696–1698. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Cortes, A.; Shipman, L.; Evans, H.G.; Attfield, K.E.; Jostins, L.; Barber, T.; Kaur, G.; Kuttikkatte, S.B.; Leach, O.A.; et al. Resolving TYK2 Locus Genotype-to-Phenotype Differences in Autoimmunity. Sci. Transl. Med. 2016, 8, 363ra149. [Google Scholar] [CrossRef] [PubMed]
- Ghoreschi, K.; Balato, A.; Enerbäck, C.; Sabat, R. Therapeutics Targeting the IL-23 and IL-17 Pathway in Psoriasis. Lancet 2021, 397, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Xu, Q.; Pu, C.; Zhu, K.; Lu, C.; Jiang, Y.; Xiao, L.; Han, Y.; Lu, L. Therapeutic Efficacy of Anti-CD19 CAR-T Cells in a Mouse Model of Systemic Lupus Erythematosus. Cell. Mol. Immunol. 2021, 18, 1896–1903. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Arany, Z.; Baur, J.A.; Epstein, J.A.; June, C.H. CAR T Therapy beyond Cancer: The Evolution of a Living Drug. Nature 2023, 619, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Owczarek, W. The Role of HLA-Cw6 in Psoriasis and Psoriatic Arthritis. Reumatologia 2022, 60, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Dand, N.; Mahil, S.K.; Capon, F.; Smith, C.H.; Simpson, M.A.; Barker, J.N. Psoriasis and Genetics. Acta Derm. Venereol. 2020, 100, adv00030. [Google Scholar] [CrossRef] [PubMed]
- Farh, K.K.H.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.H.; Shishkin, A.A.; et al. Genetic and Epigenetic Fine Mapping of Causal Autoimmune Disease Variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Terui, T.; Hayama, K.; Akiyama, M.; Ikeda, S.; Mabuchi, T.; Ozawa, A.; Kanekura, T.; Kurosawa, M.; Komine, M.; et al. Japanese Guidelines for the Management and Treatment of Generalized Pustular Psoriasis: The New Pathogenesis and Treatment of GPP. J. Dermatol. 2018, 45, 1235–1270. [Google Scholar] [CrossRef] [PubMed]
- Marrakchi, S.; Guigue, P.; Renshaw, B.R.; Puel, A.; Pei, X.-Y.; Fraitag, S.; Zribi, J.; Bal, E.; Cluzeau, C.; Chrabieh, M.; et al. Interleukin-36–Receptor Antagonist Deficiency and Generalized Pustular Psoriasis. N. Engl. J. Med. 2011, 365, 620–628. [Google Scholar] [CrossRef]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef]
- Heinz, S.; Romanoski, C.E.; Benner, C.; Glass, C.K. The Selection and Function of Cell Type-Specific Enhancers. Nat. Rev. Mol. Cell Biol. 2015, 16, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Bujold, D.; Morais, D.A.d.L.; Gauthier, C.; Côté, C.; Caron, M.; Kwan, T.; Chen, K.C.; Laperle, J.; Markovits, A.N.; Pastinen, T.; et al. The International Human Epigenome Consortium Data Portal. Cell Syst. 2016, 3, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.J.; Amode, M.R.; Aneja, A.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Becker, A.; Bennett, R.; Berry, A.; Bhai, J.; et al. Ensembl 2023. Nucleic Acids Res. 2023, 51, D933–D941. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) Project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of Functional Variation in Personal Genomes Using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, H. EQTL Studies: From Bulk Tissues to Single Cells. J. Genet. Genom. 2023, 50, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Belton, J.-M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi-C: A Comprehensive Technique to Capture the Conformation of Genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, Erasing and Reading Histone Lysine Methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef]
- Lin, Y.; Liu, L.; Sheng, Y.; Shen, C.; Zheng, X.; Zhou, F.; Yang, S.; Yin, X.; Zhang, X. A Catalog of Potential Putative Functional Variants in Psoriasis Genome-Wide Association Regions. PLoS ONE 2018, 13, e196635. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Stamatoyannopoulos, J.A.; Costello, J.F.; Ren, B.; Milosavljevic, A.; Meissner, A.; Kellis, M.; Marra, M.A.; Beaudet, A.L.; Ecker, J.R.; et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol. 2010, 28, 1045–1048. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Ray-Jones, H.; Duffus, K.; McGovern, A.; Martin, P.; Shi, C.; Hankinson, J.; Gough, O.; Yarwood, A.; Morris, A.P.; Adamson, A.; et al. Mapping DNA Interaction Landscapes in Psoriasis Susceptibility Loci Highlights KLF4 as a Target Gene in 9q31. BMC Biol. 2020, 18, 47. [Google Scholar] [CrossRef]
- Shi, C.; Ray-Jones, H.; Ding, J.; Duffus, K.; Fu, Y.; Gaddi, V.P.; Gough, O.; Hankinson, J.; Martin, P.; McGovern, A.; et al. Chromatin Looping Links Target Genes with Genetic Risk Loci for Dermatological Traits. J. Investig. Dermatol. 2021, 141, 1975–1984. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Lv, R.; Shi, X.; Yang, G.; Jin, J. CRISPR/DCas9 Tools: Epigenetic Mechanism and Application in Gene Transcriptional Regulation. Int. J. Mol. Sci. 2023, 24, 14865. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Yang, W.; Li, W.; He, L.; Lu, L.; Zhang, L.; Liu, Z.; Wang, Y.; Chao, T.; Huang, R.; et al. Zdhhc2 Is Essential for Plasmacytoid Dendritic Cells Mediated Inflammatory Response in Psoriasis. Front. Immunol. 2020, 11, 607442. [Google Scholar] [CrossRef] [PubMed]
- Wan, T.; Pan, Q.; Ping, Y. Microneedle-Assisted Genome Editing: A Transdermal Strategy of Targeting NLRP3 by CRISPR-Cas9 for Synergistic Therapy of Inflammatory Skin Disorders. Sci. Adv. 2021, 7, eabe2888. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Chen, X.; Dowbaj, A.M.; Sljukic, A.; Bratlie, K.; Lin, L.; Fong, E.L.S.; Balachander, G.M.; Chen, Z.; Soragni, A.; et al. Organoids. Nat. Rev. Methods Primers 2022, 2, 94. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef]
- Rusiñol, L.; Puig, L. Multi-Omics Approach to Improved Diagnosis and Treatment of Atopic Dermatitis and Psoriasis. Int. J. Mol. Sci. 2024, 25, 1042. [Google Scholar] [CrossRef]
- Dopytalska, K.; Ciechanowicz, P.; Wiszniewski, K.; Szymańska, E.; Walecka, I. The Role of Epigenetic Factors in Psoriasis. Int. J. Mol. Sci. 2021, 22, 9294. [Google Scholar] [CrossRef]
- Tost, J. DNA Methylation: An Introduction to the Biology and the Disease-Associated Changes of a Promising Biomarker. Mol. Biotechnol. 2010, 44, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.O.; Liu, Y.; Ryan, C.; Joyce, C.E.; Duan, S.; Cao, L.; Martin, A.; Liao, W.; Menter, A.; Bowcock, A.M. A Subset of Methylated CpG Sites Differentiate Psoriatic from Normal Skin. J. Investig. Dermatol. 2012, 132, 583–592. [Google Scholar] [CrossRef]
- Zhang, P.; Su, Y.; Chen, H.; Zhao, M.; Lu, Q. Abnormal DNA Methylation in Skin Lesions and PBMCs of Patients with Psoriasis Vulgaris. J. Dermatol. Sci. 2010, 60, 40–42. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Shen, C.; Xu, J.; Gao, J.; Zheng, X.; Ko, R.; Dou, J.; Cheng, Y.; Zhu, C.; Xu, S.; et al. Epigenome-Wide Association Data Implicates DNA Methylation-Mediated Genetic Risk in Psoriasis. Clin. Epigenet. 2016, 8, 131. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yao, Q.; Mariscal, A.G.; Wu, X.; Hülse, J.; Pedersen, E.; Helin, K.; Waisman, A.; Vinkel, C.; Thomsen, S.F.; et al. Epigenetic Control of IL-23 Expression in Keratinocytes Is Important for Chronic Skin Inflammation. Nat. Commun. 2018, 9, 1420. [Google Scholar] [CrossRef] [PubMed]
- Ovejero-Benito, M.C.; Reolid, A.; Sánchez-Jiménez, P.; Saiz-Rodríguez, M.; Muñoz-Aceituno, E.; Llamas-Velasco, M.; Martín-Vilchez, S.; Cabaleiro, T.; Román, M.; Ochoa, D.; et al. Histone Modifications Associated with Biological Drug Response in Moderate-to-Severe Psoriasis. Exp. Dermatol. 2018, 27, 1361–1371. [Google Scholar] [CrossRef]
- Cohen-Karni, D.; Xu, D.; Apone, L.; Fomenkov, A.; Sun, Z.; Davis, P.J.; Kinney, S.R.M.; Yamada-Mabuchi, M.; Xu, S.; Davis, T.; et al. The MspJI Family of Modification-Dependent Restriction Endonucleases for Epigenetic Studies. Proc. Natl. Acad. Sci. USA 2011, 108, 11040–11045. [Google Scholar] [CrossRef]
- Chandra, A.; Senapati, S.; Roy, S.; Chatterjee, G.; Chatterjee, R. Epigenome-Wide DNA Methylation Regulates Cardinal Pathological Features of Psoriasis. Clin. Epigenet. 2018, 10, 108. [Google Scholar] [CrossRef]
- Gudjonsson, J.E.; Johnston, A.; Dyson, M.; Valdimarsson, H.; Elder, J.T. Mouse Models of Psoriasis. J. Investig. Dermatol. 2007, 127, 1292–1308. [Google Scholar] [CrossRef]
- Gates, A.H.; Karasek, M. Hereditary Absence of Sebaceous Glands in the Mouse. Science 1965, 148, 1471–1473. [Google Scholar] [CrossRef] [PubMed]
- Gangwar, R.S.; Gudjonsson, J.E.; Ward, N.L. Mouse Models of Psoriasis: A Comprehensive Review. J. Investig. Dermatol. 2022, 142, 884–897. [Google Scholar] [CrossRef] [PubMed]
- Visel, A.; Zhu, Y.; May, D.; Afzal, V.; Gong, E.; Attanasio, C.; Blow, M.J.; Cohen, J.C.; Rubin, E.M.; Pennacchio, L.A. Targeted Deletion of the 9p21 Non-Coding Coronary Artery Disease Risk Interval in Mice. Nature 2010, 464, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Hou, G.; Harley, I.T.W.; Lu, X.; Zhou, T.; Xu, N.; Yao, C.; Qin, Y.; Ouyang, Y.; Ma, J.; Zhu, X.; et al. SLE Non-Coding Genetic Risk Variant Determines the Epigenetic Dysfunction of an Immune Cell Specific Enhancer That Controls Disease-Critical MicroRNA Expression. Nat. Commun. 2021, 12, 135. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S. Humanized Mice: A Brief Overview on Their Diverse Applications in Biomedical Research. J. Cell. Physiol. 2018, 233, 2889–2901. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, J.J. Pathophysiology of Psoriasis. Annu. Rev. Med. 1977, 28. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Oak, A.S.W.; Elewski, B.E. Use of IL-23 Inhibitors for the Treatment of Plaque Psoriasis and Psoriatic Arthritis: A Comprehensive Review. Am. J. Clin. Dermatol. 2021, 22, 173–192. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, Q.; Liu, H.; Lu, C.; Liang, C.L.; Qiu, F.; Han, L.; Dai, Z. Esculetin Ameliorates Psoriasis-like Skin Disease in Mice by Inducing CD4+Foxp3+ Regulatory T Cells. Front. Immunol. 2018, 9, 2092. [Google Scholar] [CrossRef] [PubMed]
- Nanda, H.; Ponnusamy, N.; Odumpatta, R.; Jeyakanthan, J.; Mohanapriya, A. Exploring Genetic Targets of Psoriasis Using Genome Wide Association Studies (GWAS) for Drug Repurposing. 3 Biotech 2020, 10, 43. [Google Scholar] [CrossRef]
- Eder, L.; Chandran, V.; Pellett, F.; Pollock, R.; Shanmugarajah, S.; Rosen, C.F.; Rahman, P.; Gladman, D.D. IL13 Gene Polymorphism Is a Marker for Psoriatic Arthritis among Psoriasis Patients. Ann. Rheum. Dis. 2011, 70, 1594–1598. [Google Scholar] [CrossRef]
- Yan, C.; Grabowska, M.E.; Dickson, A.L.; Li, B.; Wen, Z.; Roden, D.M.; Michael Stein, C.; Embí, P.J.; Peterson, J.F.; Feng, Q.; et al. Leveraging Generative AI to Prioritize Drug Repurposing Candidates for Alzheimer’s Disease with Real-World Clinical Validation. NPJ Digit. Med. 2024, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Song, X.; Ma, T.; Pan, X.; Zhou, Y.; Hou, Y.; Zhang, Z.; Li, K.; Karypis, G.; Cheng, F. Repurpose Open Data to Discover Therapeutics for COVID-19 Using Deep Learning. J. Proteome Res. 2020, 19, 4624–4636. [Google Scholar] [CrossRef] [PubMed]
- Hofmarcher, M.; Mayr, A.; Rumetshofer, E.; Ruch, P.; Renz, P.; Schimunek, J.; Seidl, P.; Vall, A.; Widrich, M.; Hochreiter, S.; et al. Large-Scale Ligand-Based Virtual Screening for SARS-CoV-2 Inhibitors Using Deep Neural Networks. arXiv 2020, arXiv:2004.00979. [Google Scholar] [CrossRef]
- Patrick, M.T.; Bardhi, R.; Raja, K.; He, K.; Tsoi, L.C. Advancement in Predicting Interactions between Drugs Used to Treat Psoriasis and Its Comorbidities by Integrating Molecular and Clinical Resources. J. Am. Med. Inform. Assoc. 2021, 28, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Caputo, V.; Strafella, C.; Cosio, T.; Lanna, C.; Campione, E.; Novelli, G.; Giardina, E.; Cascella, R. Pharmacogenomics: An Update on Biologics and Small-Molecule Drugs in the Treatment of Psoriasis. Genes 2021, 12, 1398. [Google Scholar] [CrossRef] [PubMed]
- Wong, C. UK First to Approve CRISPR Treatment for Diseases: What You Need to Know. Nature 2023, 623, 676–677. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Hao, S.; Zheng, H.; Zhao, X.; Li, Y. Association of NLRP1 and NLRP3 Polymorphisms with Psoriasis Vulgaris Risk in the Chinese Han Population. BioMed Res. Int. 2018, 2018, 4714836. [Google Scholar] [CrossRef]
- Carlström, M.; Ekman, A.K.; Petersson, S.; Söderkvist, P.; Enerbäck, C. Genetic Support for the Role of the NLRP3 Inflammasome in Psoriasis Susceptibility. Exp. Dermatol. 2012, 21, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Ekman, A.K.; Verma, D.; Fredrikson, M.; Bivik, C.; Enerbäck, C. Genetic Variations of NLRP1: Susceptibility in Psoriasis. Br. J. Dermatol. 2014, 171, 1517–1520. [Google Scholar] [CrossRef]
- Ran, D.; Cai, M.; Zhang, X. Genetics of Psoriasis: A Basis for Precision Medicine. Precis. Clin. Med. 2019, 2, 120–130. [Google Scholar] [CrossRef]
- Shen, C.; Gao, J.; Yin, X.; Sheng, Y.; Sun, L.; Cui, Y.; Zhang, X. Association of the Late Cornified Envelope-3 Genes with Psoriasis and Psoriatic Arthritis: A Systematic Review. J. Genet. Genom. 2015, 42, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Jin, X.; Xu, J.; Gao, J.; Du, X.; Duan, D.; Li, B.; Zhao, J.; Zhan, W.; Tang, H.; et al. Sequencing-Based Approach Identified Three New Susceptibility Loci for Psoriasis. Nat. Commun. 2014, 5, 4331. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Cao, L.; Roberson, E.D.O.; Pierson, K.C.; Yang, C.-F.; Joyce, C.E.; Ryan, C.; Duan, S.; Helms, C.A.; Liu, Y.; et al. PSORS2 Is Due to Mutations in CARD14. Am. J. Hum. Genet. 2012, 90, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, L.C.; Spain, S.L.; Knight, J.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Ding, J.; Li, Y.; Tejasvi, T.; Gudjonsson, J.E.; et al. Identification of 15 New Psoriasis Susceptibility Loci Highlights the Role of Innate Immunity. Nat. Genet. 2012, 44, 1341–1348. [Google Scholar] [CrossRef]
- Piganis, R.A.R.; De Weerd, N.A.; Gould, J.A.; Schindler, C.W.; Mansell, A.; Nicholson, S.E.; Hertzog, P.J. Suppressor of Cytokine Signaling (SOCS) 1 Inhibits Type I Interferon (IFN) Signaling via the Interferon α Receptor (IFNAR1)-Associated Tyrosine Kinase Tyk2 *. J. Biol. Chem. 2011, 286, 33811–33818. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, S.; Richards, E.L.; Orozco, G.; Eyre, S. Functional Genomics in Psoriasis. Int. J. Mol. Sci. 2024, 25, 7349. https://doi.org/10.3390/ijms25137349
Rossi S, Richards EL, Orozco G, Eyre S. Functional Genomics in Psoriasis. International Journal of Molecular Sciences. 2024; 25(13):7349. https://doi.org/10.3390/ijms25137349
Chicago/Turabian StyleRossi, Stefano, Ellie Louise Richards, Gisela Orozco, and Stephen Eyre. 2024. "Functional Genomics in Psoriasis" International Journal of Molecular Sciences 25, no. 13: 7349. https://doi.org/10.3390/ijms25137349
APA StyleRossi, S., Richards, E. L., Orozco, G., & Eyre, S. (2024). Functional Genomics in Psoriasis. International Journal of Molecular Sciences, 25(13), 7349. https://doi.org/10.3390/ijms25137349