
Detection and Characterization of Methylated Circulating Tumor DNA in Gastric Cancer

 , ,
, , 
Abstract
1. Introduction
1.1. Gastric Cancer and Liquid Biopsy
1.2. Methylation
1.3. Fragmentomics (Size and 5′End-Motif)
2. Results
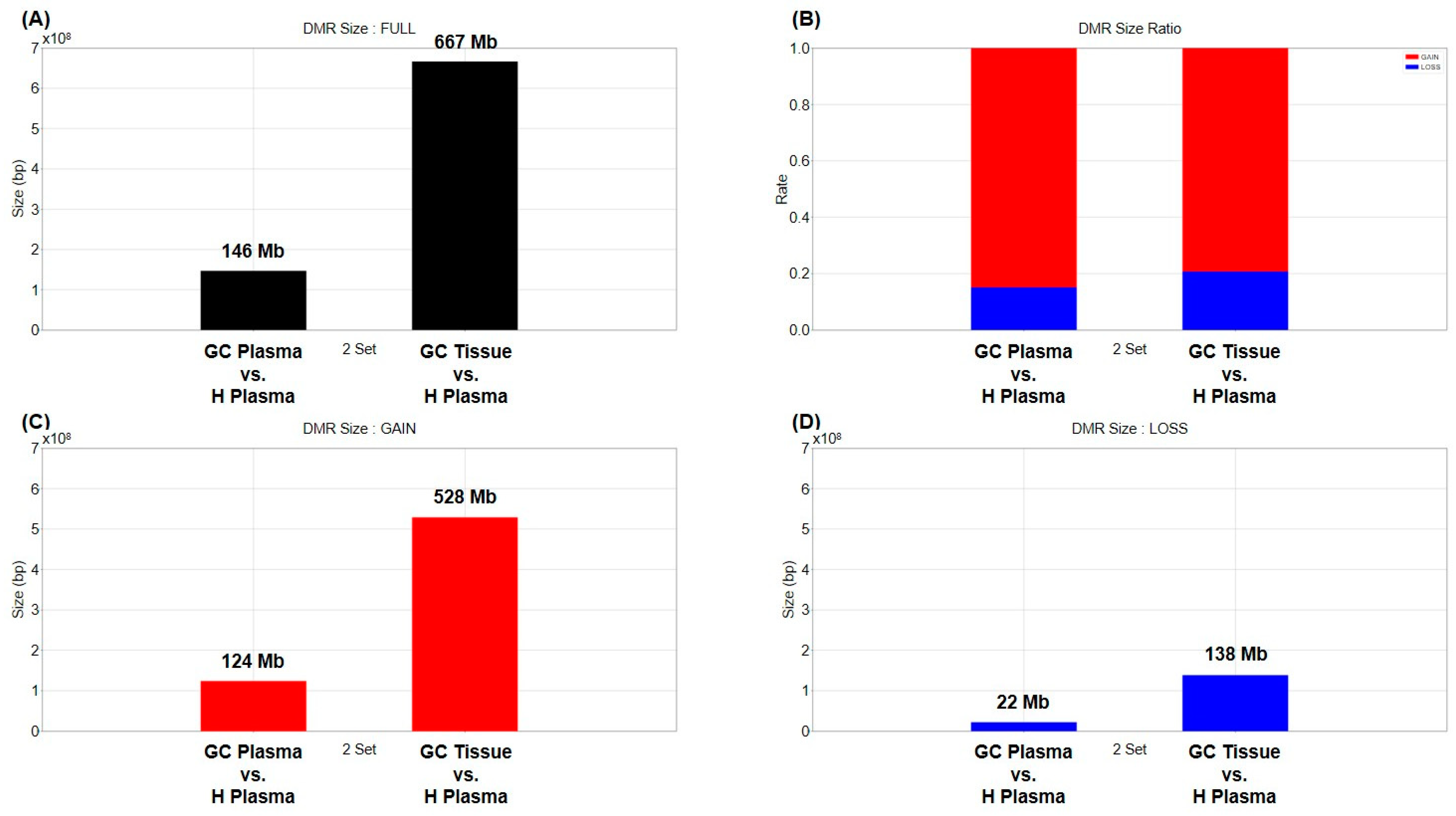
2.1. Differentially Methylated Region (DMR)
2.2. Commonly Methylated Region (CMR)
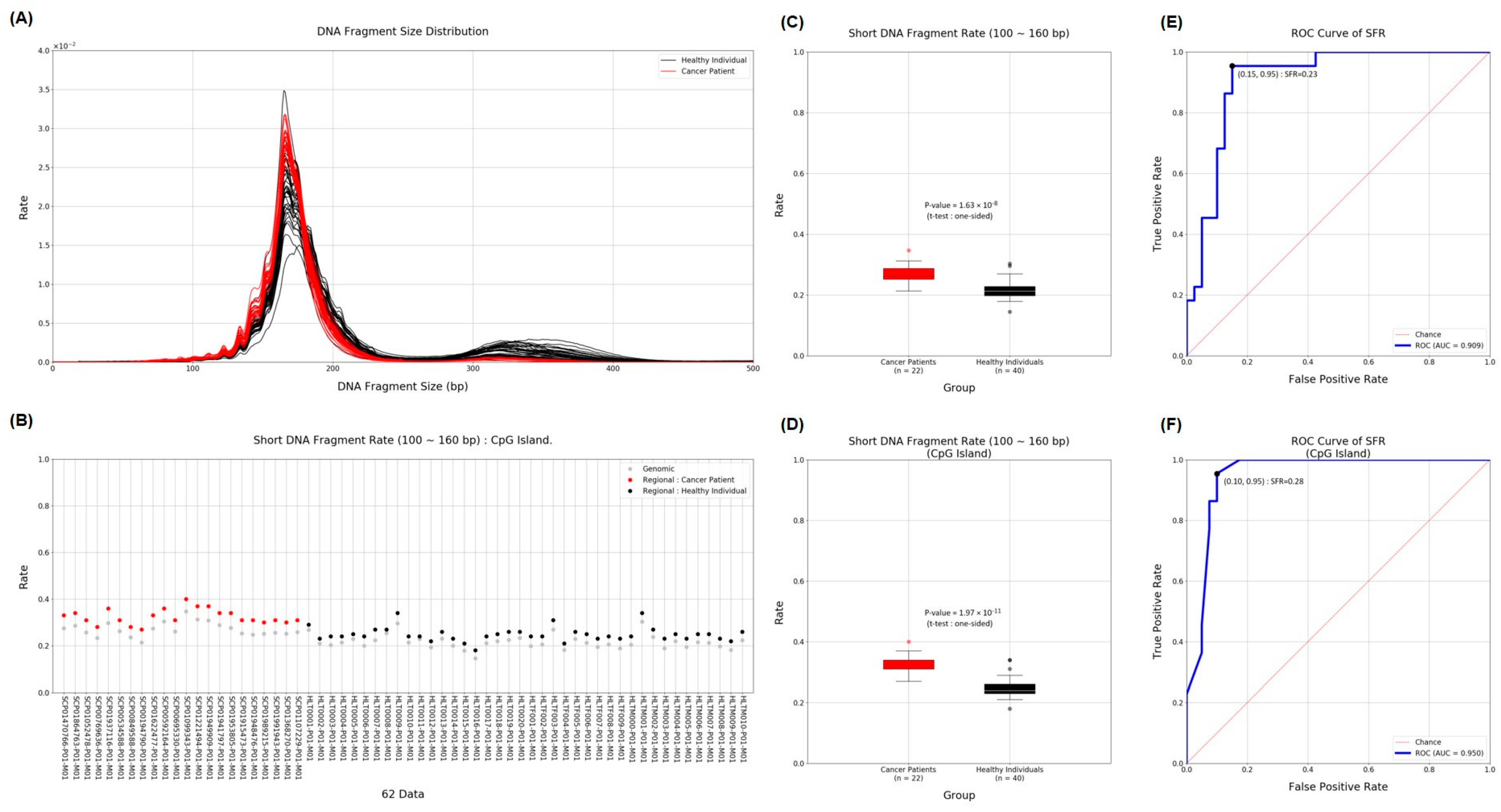
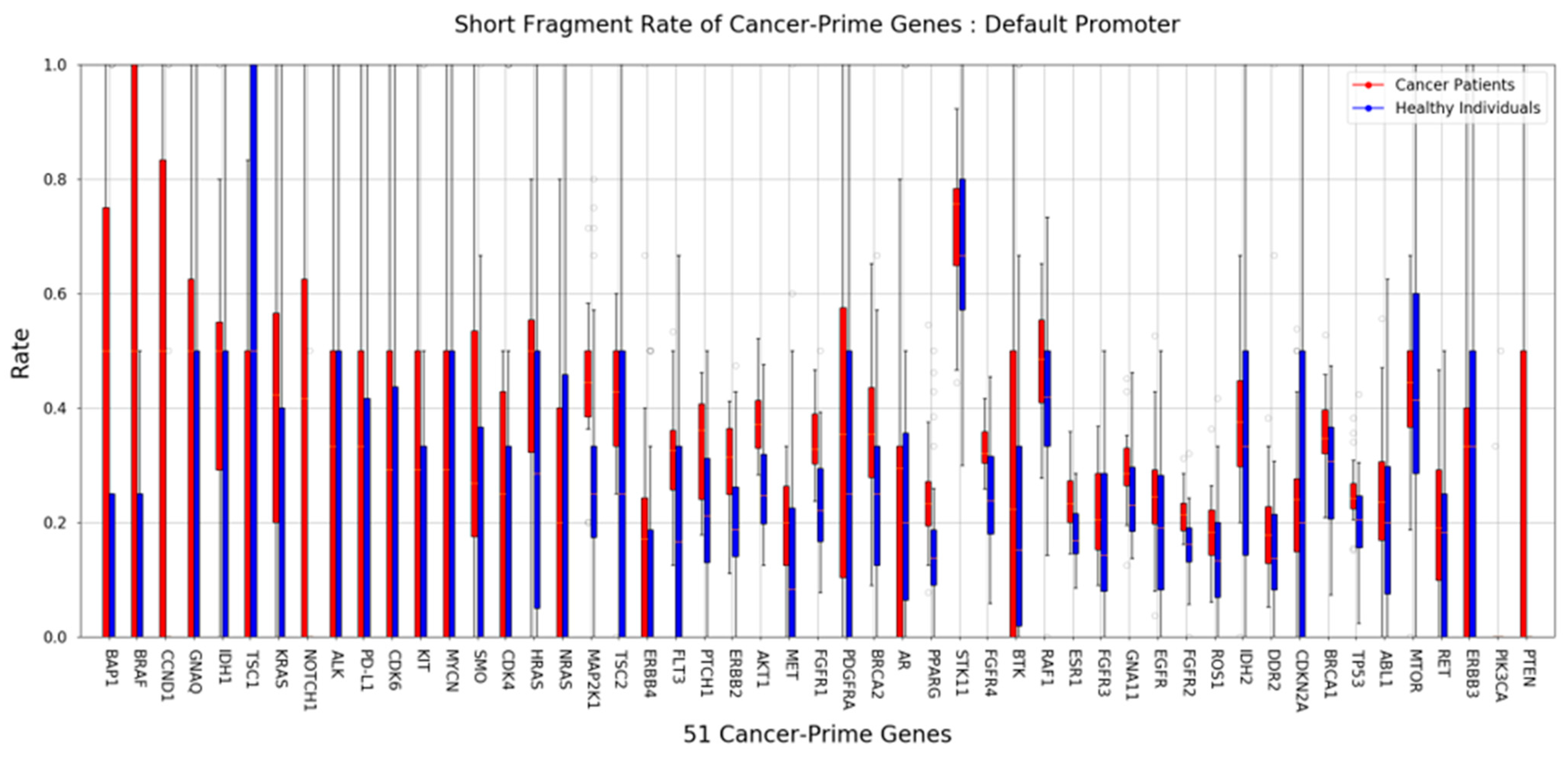
2.3. Short Fragment Ratio (SFR)
2.4. 5′End-Motif
3. Discussion
4. Materials and Methods
4.1. 40 Healthy Donor Plasmas, 22 Pairs of GC Tissue and Blood, DNA Extraction
4.2. MeDIP Sequencing
4.3. Data Processing and Mapping
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nagai, H.; Kim, Y.H. Cancer prevention from the perspective of global cancer burden patterns. J. Thorac. Dis. 2017, 9, 448. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Rutherford, M.J.; Bardot, A.; Ferlay, J.; Andersson, T.M.; Myklebust, T.Å.; Tervonen, H.; Thursfield, V.; Ransom, D.; Shack, L. Progress in cancer survival, mortality, and incidence in seven high-income countries 1995–2014 (ICBP SURVMARK-2): A population-based study. Lancet Oncol. 2019, 20, 1493–1505. [Google Scholar] [CrossRef]
- Park, S.H.; Kang, M.J.; Yun, E.H.; Jung, K.-W. Epidemiology of gastric cancer in Korea: Trends in incidence and survival based on Korea Central Cancer Registry Data (1999–2019). J. Gastric Cancer 2022, 22, 160. [Google Scholar] [CrossRef]
- Hong, S.; Won, Y.-J.; Lee, J.J.; Jung, K.-W.; Kong, H.-J.; Im, J.-S.; Seo, H.G. Cancer statistics in Korea: Incidence, mortality, survival, and prevalence in 2018. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2021, 53, 301. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zhou, M.; Xu, Y.; Gu, X.; Zou, M.; Abudushalamu, G.; Yao, Y.; Fan, X.; Wu, G. Clinical application and detection techniques of liquid biopsy in gastric cancer. Mol. Cancer 2023, 22, 7. [Google Scholar] [CrossRef] [PubMed]
- Lone, S.N.; Nisar, S.; Masoodi, T.; Singh, M.; Rizwan, A.; Hashem, S.; El-Rifai, W.; Bedognetti, D.; Batra, S.K.; Haris, M. Liquid biopsy: A step closer to transform diagnosis, prognosis and future of cancer treatments. Mol. Cancer 2022, 21, 79. [Google Scholar] [CrossRef] [PubMed]
- Soda, N.; Clack, K.; Shiddiky, M.J. Recent advances in liquid biopsy technologies for cancer biomarker detection. Sens. Diagn. 2022, 1, 343–375. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Pantel, K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Fujii, H.; Nagakura, H.; Kobayashi, N.; Kubo, S.; Tanaka, K.; Watanabe, K.; Horita, N.; Hara, Y.; Nishikawa, M.; Miura, K. Liquid biopsy for detecting epidermal growth factor receptor mutation among patients with non-small cell lung cancer treated with afatinib: A multicenter prospective study. BMC Cancer 2022, 22, 1035. [Google Scholar] [CrossRef] [PubMed]
- Nigro, M.C.; Marchese, P.V.; Deiana, C.; Casadio, C.; Galvani, L.; Di Federico, A.; De Giglio, A. Clinical utility and application of liquid biopsy genotyping in lung cancer: A comprehensive review. Lung Cancer Targets Ther. 2023, 14, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Nacchio, M.; Sgariglia, R.; Gristina, V.; Pisapia, P.; Pepe, F.; De Luca, C.; Migliatico, I.; Clery, E.; Greco, L.; Vigliar, E. KRAS mutations testing in non-small cell lung cancer: The role of Liquid biopsy in the basal setting. J. Thorac. Dis. 2020, 12, 3836. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, G.; Rodrigues, A.; Matos, C.; Barata, F.; Cirnes, L.; Ferreira, L.; Lopes, J.A.; Felizardo, M.; Fidalgo, P.; Brito, U. Liquid biopsy in the management of advanced lung cancer: Implementation and practical aspects. Cancer Treat. Res. Commun. 2023, 36, 100725. [Google Scholar] [CrossRef] [PubMed]
- Di Sario, G.; Rossella, V.; Famulari, E.S.; Maurizio, A.; Lazarevic, D.; Giannese, F.; Felici, C. Enhancing clinical potential of liquid biopsy through a multi-omic approach: A systematic review. Front. Genet. 2023, 14, 1152470. [Google Scholar] [CrossRef]
- Amelio, I.; Bertolo, R.; Bove, P.; Buonomo, O.C.; Candi, E.; Chiocchi, M.; Cipriani, C.; Di Daniele, N.; Ganini, C.; Juhl, H. Liquid biopsies and cancer omics. Cell Death Discov. 2020, 6, 131. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, J.; Fu, Q.; Taly, V.; Tan, F. Integrative analysis of multi-omics data for liquid biopsy. Br. J. Cancer 2023, 128, 505–518. [Google Scholar] [CrossRef]
- Dulbecco, R. A turning point in cancer research: Sequencing the human genome. Science 1986, 231, 1055–1056. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef]
- Zhou, J.; Sears, R.L.; Xing, X.; Zhang, B.; Li, D.; Rockweiler, N.B.; Jang, H.S.; Choudhary, M.N.; Lee, H.J.; Lowdon, R.F. Tissue-specific DNA methylation is conserved across human, mouse, and rat, and driven by primary sequence conservation. BMC Genom. 2017, 18, 1–17. [Google Scholar] [CrossRef]
- Ghosh, S.; Yates, A.J.; Frühwald, M.C.; Miecznikowski, J.C.; Plass, C.; Smiraglia, D. Tissue specific DNA methylation of CpG islands in normal human adult somatic tissues distinguishes neural from non-neural tissues. Epigenetics 2010, 5, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Jiang, P.; Chan, K.A.; Wong, J.; Cheng, Y.K.; Liang, R.H.; Chan, W.-K.; Ma, E.S.; Chan, S.L.; Cheng, S.H. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc. Natl. Acad. Sci. USA 2015, 112, E5503–E5512. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, Q.; Kang, S.; Same, M.; Zhou, Y.; Sun, C.; Liu, C.-C.; Matsuoka, L.; Sher, L.; Wong, W.H. CancerDetector: Ultrasensitive and non-invasive cancer detection at the resolution of individual reads using cell-free DNA methylation sequencing data. Nucleic Acids Res. 2018, 46, e89. [Google Scholar] [CrossRef] [PubMed]
- Mouliere, F.; Rosenfeld, N. Circulating tumor-derived DNA is shorter than somatic DNA in plasma. Proc. Natl. Acad. Sci. USA 2015, 112, 3178–3179. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Cario, C.L.; Leong, L.; Lopez, K.; Márquez, C.P.; Chu, C.; Li, P.S.; Oropeza, E.; Tenggara, I.; Cowan, J. Cell-free DNA concentration and fragment size as a biomarker for prostate cancer. Sci. Rep. 2021, 11, 5040. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.D.; Han, D.S.; Jiang, P.; Chiu, R.W. Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science 2021, 372, eaaw3616. [Google Scholar] [CrossRef] [PubMed]
- Cristiano, S.; Leal, A.; Phallen, J.; Fiksel, J.; Adleff, V.; Bruhm, D.C.; Jensen, S.Ø.; Medina, J.E.; Hruban, C.; White, J.R. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019, 570, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.A.; Shankar, V. Single-strand-specific nucleases. FEMS Microbiol. Rev. 2003, 26, 457–491. [Google Scholar] [CrossRef]
- Balian, A.; Hernandez, F.J. Nucleases as molecular targets for cancer diagnosis. Biomark. Res. 2021, 9, 1–16. [Google Scholar] [CrossRef]
- Yang, W. Nucleases: Diversity of structure, function and mechanism. Q. Rev. Biophys. 2011, 44, 1–93. [Google Scholar] [CrossRef]
- Zhou, Z.; Ma, M.-J.L.; Chan, R.W.; Lam, W.J.; Peng, W.; Gai, W.; Hu, X.; Ding, S.C.; Ji, L.; Zhou, Q. Fragmentation landscape of cell-free DNA revealed by deconvolutional analysis of end motifs. Proc. Natl. Acad. Sci. USA 2023, 120, e2220982120. [Google Scholar] [CrossRef]
- Lienhard, M.; Grimm, C.; Morkel, M.; Herwig, R.; Chavez, L. MEDIPS: Genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics 2014, 30, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Vrba, L.; Futscher, B.W. A suite of DNA methylation markers that can detect most common human cancers. Epigenetics 2018, 13, 61–72. [Google Scholar] [CrossRef]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, 1–9. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, C.K.; Lee, S.Y.; Choi, C.M. Treatment outcomes and safety of afatinib in advanced squamous cell lung cancer progressed after platinum-based doublet chemotherapy and immunotherapy (SPACE study). Thorac. Cancer 2021, 12, 1264–1268. [Google Scholar] [CrossRef] [PubMed]
- Boks, M.P.; Derks, E.M.; Weisenberger, D.J.; Strengman, E.; Janson, E.; Sommer, I.E.; Kahn, R.S.; Ophoff, R.A. The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PLoS ONE 2009, 4, e6767. [Google Scholar] [CrossRef]
- Lakshminarasimhan, R.; Liang, G. The role of DNA methylation in cancer. Adv. Exp. Med. Biol. 2016, 945, 151–172. [Google Scholar] [CrossRef]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef]
- Stejskal, P.; Goodarzi, H.; Srovnal, J.; Hajdúch, M.; van’t Veer, L.J.; Magbanua, M.J.M. Circulating tumor nucleic acids: Biology, release mechanisms, and clinical relevance. Mol. Cancer 2023, 22, 15. [Google Scholar] [CrossRef]
- Neumann, M.H.; Bender, S.; Krahn, T.; Schlange, T. ctDNA and CTCs in liquid biopsy–current status and where we need to progress. Comput. Struct. Biotechnol. J. 2018, 16, 190–195. [Google Scholar] [CrossRef]
- Shen, S.Y.; Burgener, J.M.; Bratman, S.V.; De Carvalho, D.D. Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nat. Protoc. 2019, 14, 2749–2780. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Ethnicity | Tissue | Sample | Capture | # Samples | # Individual |
|---|---|---|---|---|---|---|
| Gastric Cancer | Korean | Plasma | cfDNA | MeDIP | 22 | 22 |
| Cancer | gDNA | MeDIP | 22 | |||
| Healthy | Hispanic | Plasma | cfDNA | MeDIP | 40 | 40 |
| Motif | Average Frequency (Rate) | p-Value | |
|---|---|---|---|
| Cancer Patients | Healthy Individuals | ||
| CCAC | 0.012198 | 0.010076 | 5.03 × 10−11 |
| AGCT | 0.003254 | 0.004846 | 6.11 × 10−11 |
| TCCT | 0.004531 | 0.003161 | 6.11 × 10−11 |
| TCAG | 0.002890 | 0.001785 | 8.16 × 10−11 |
| TACG | 0.000666 | 0.000449 | 9.85 × 10−11 |
| AGTC | 0.002373 | 0.003192 | 9.89 × 10−11 |
| AGTT | 0.002356 | 0.004342 | 1.20 × 10−10 |
| TCAA | 0.002575 | 0.001606 | 1.20 × 10−10 |
| AGAT | 0.002691 | 0.003770 | 1.32 × 10−10 |
| AGCC | 0.003252 | 0.003959 | 1.45 × 10−10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, S.Y.; Youn, S.H.; Bae, J.-H.; Lee, S.-H.; Lee, S.Y. Detection and Characterization of Methylated Circulating Tumor DNA in Gastric Cancer. Int. J. Mol. Sci. 2024, 25, 7377. https://doi.org/10.3390/ijms25137377
Seo SY, Youn SH, Bae J-H, Lee S-H, Lee SY. Detection and Characterization of Methylated Circulating Tumor DNA in Gastric Cancer. International Journal of Molecular Sciences. 2024; 25(13):7377. https://doi.org/10.3390/ijms25137377
Chicago/Turabian StyleSeo, Seung Young, Sang Hee Youn, Jin-Han Bae, Sung-Hun Lee, and Sun Young Lee. 2024. "Detection and Characterization of Methylated Circulating Tumor DNA in Gastric Cancer" International Journal of Molecular Sciences 25, no. 13: 7377. https://doi.org/10.3390/ijms25137377
APA StyleSeo, S. Y., Youn, S. H., Bae, J.-H., Lee, S.-H., & Lee, S. Y. (2024). Detection and Characterization of Methylated Circulating Tumor DNA in Gastric Cancer. International Journal of Molecular Sciences, 25(13), 7377. https://doi.org/10.3390/ijms25137377