
The Response of the Gut Physiological Function and Microbiome of a Wild Freshwater Fish (Megalobrama terminalis) to Alterations in Reproductive Behavior

Abstract
:1. Introduction
2. Results
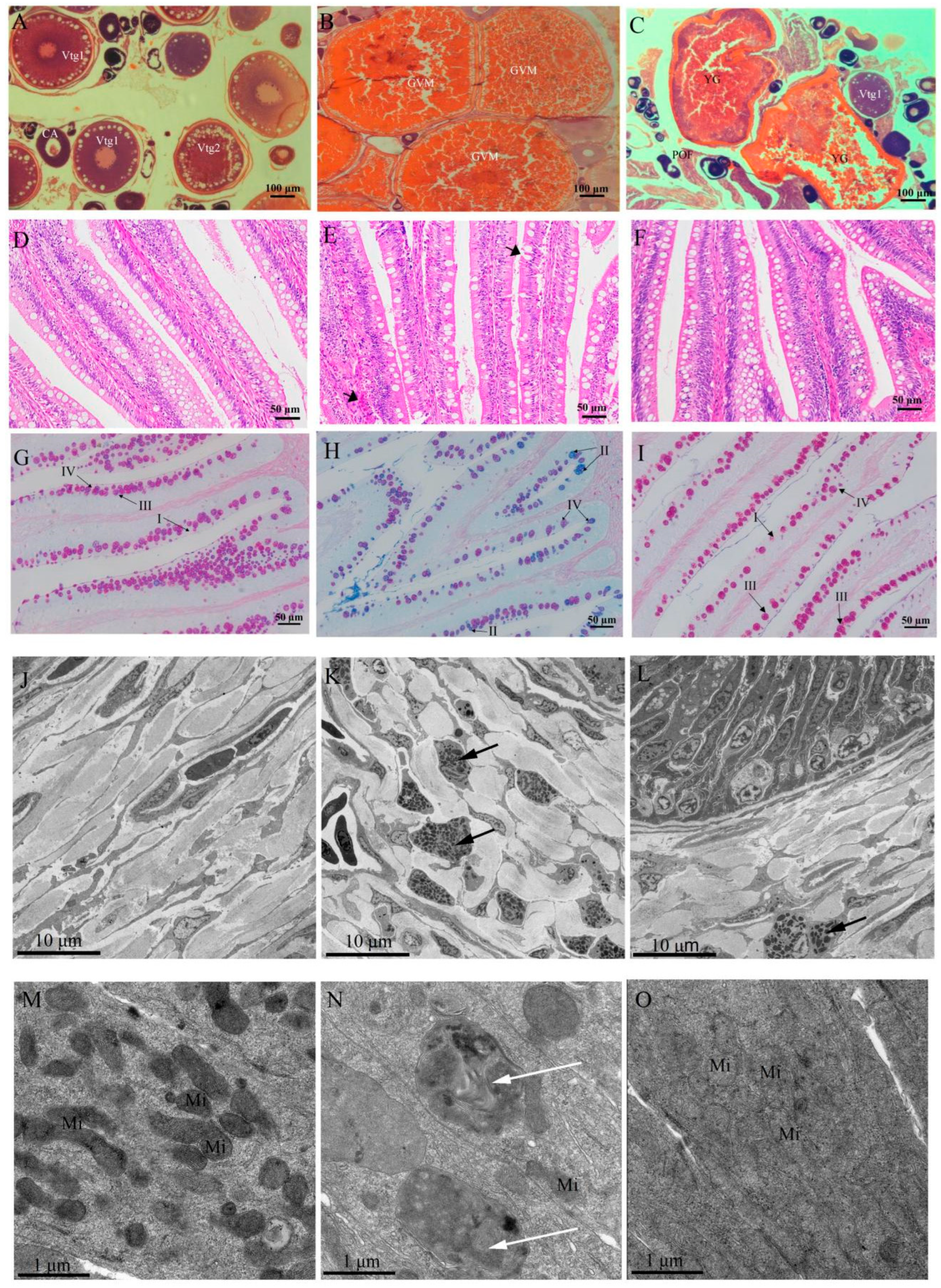
2.1. Biological, Gut Morphological and Physiological Responses in Different Reproductive Stages
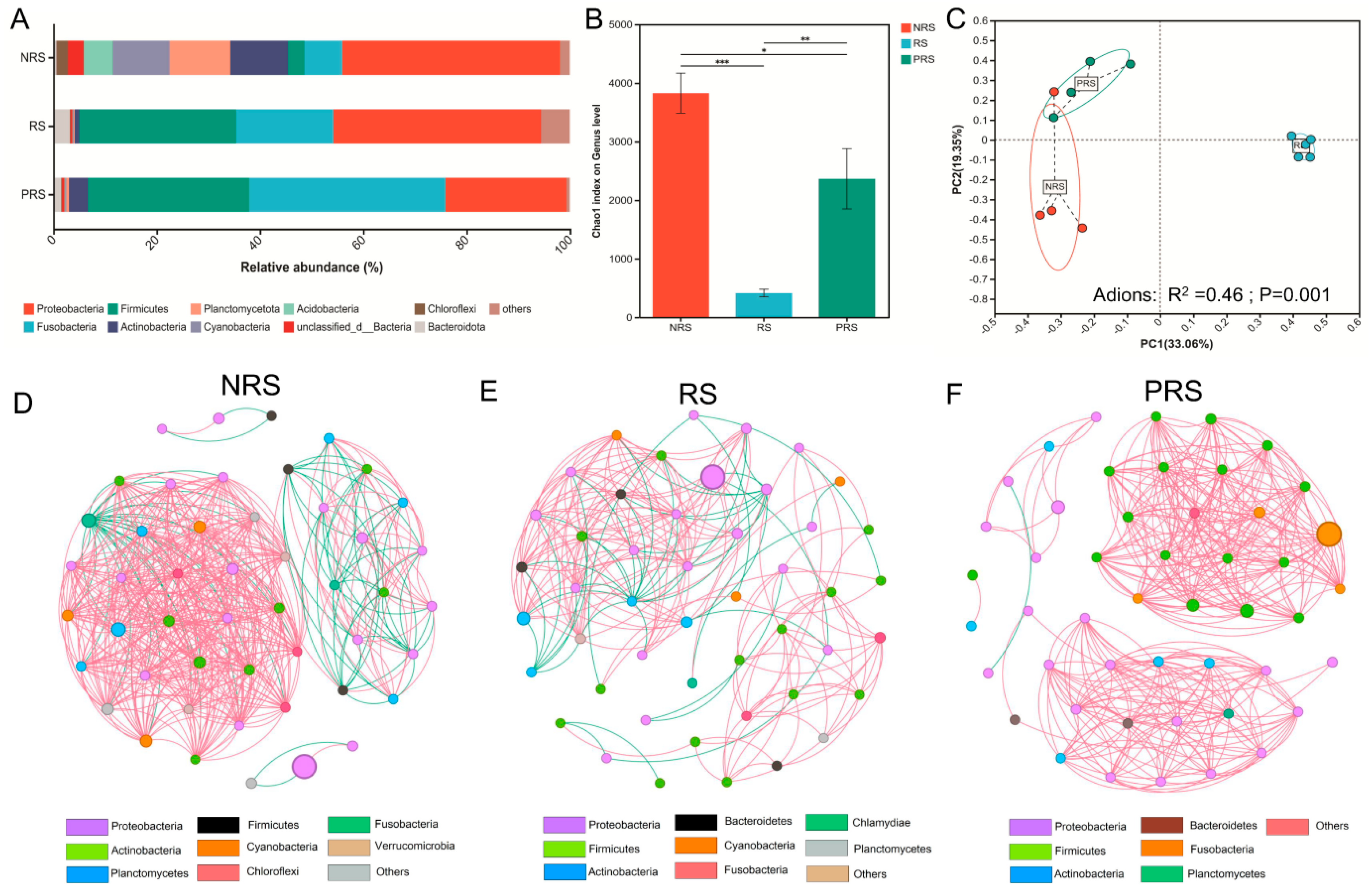
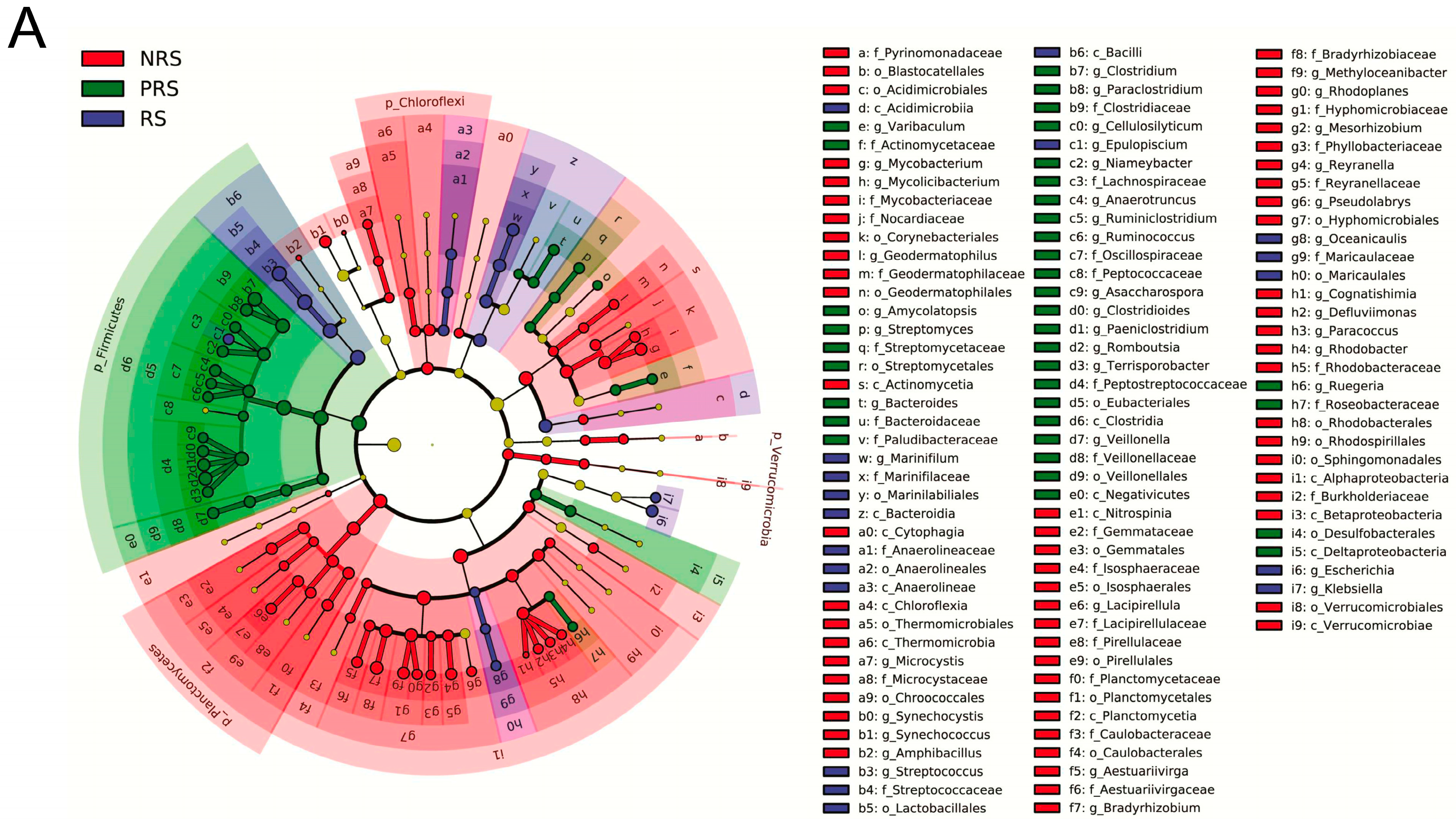
2.2. Gut Microbiota Diversity and Ecological Networks among the Non-Reproductive, Reproductive and Post-Reproductive Groups
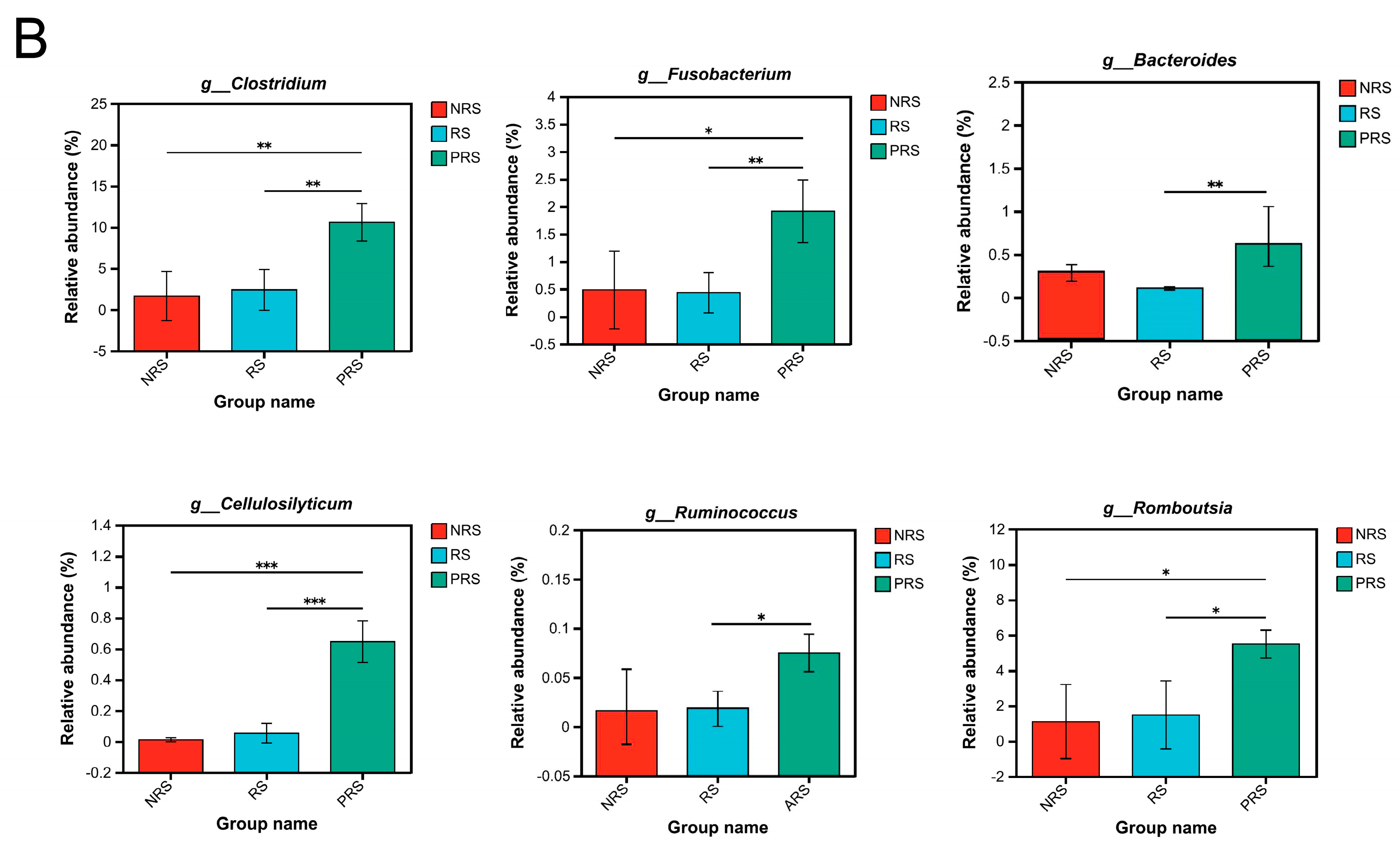
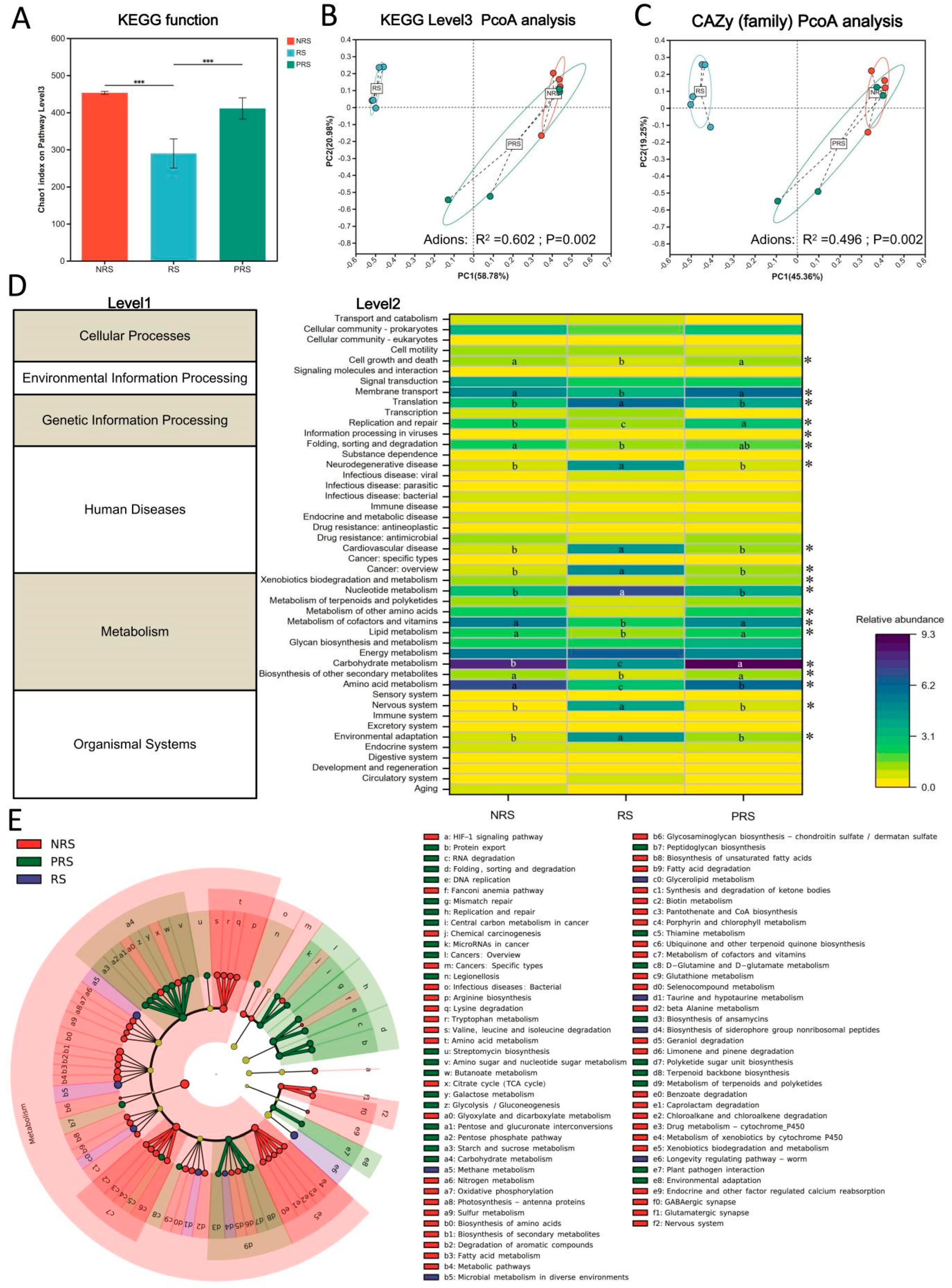
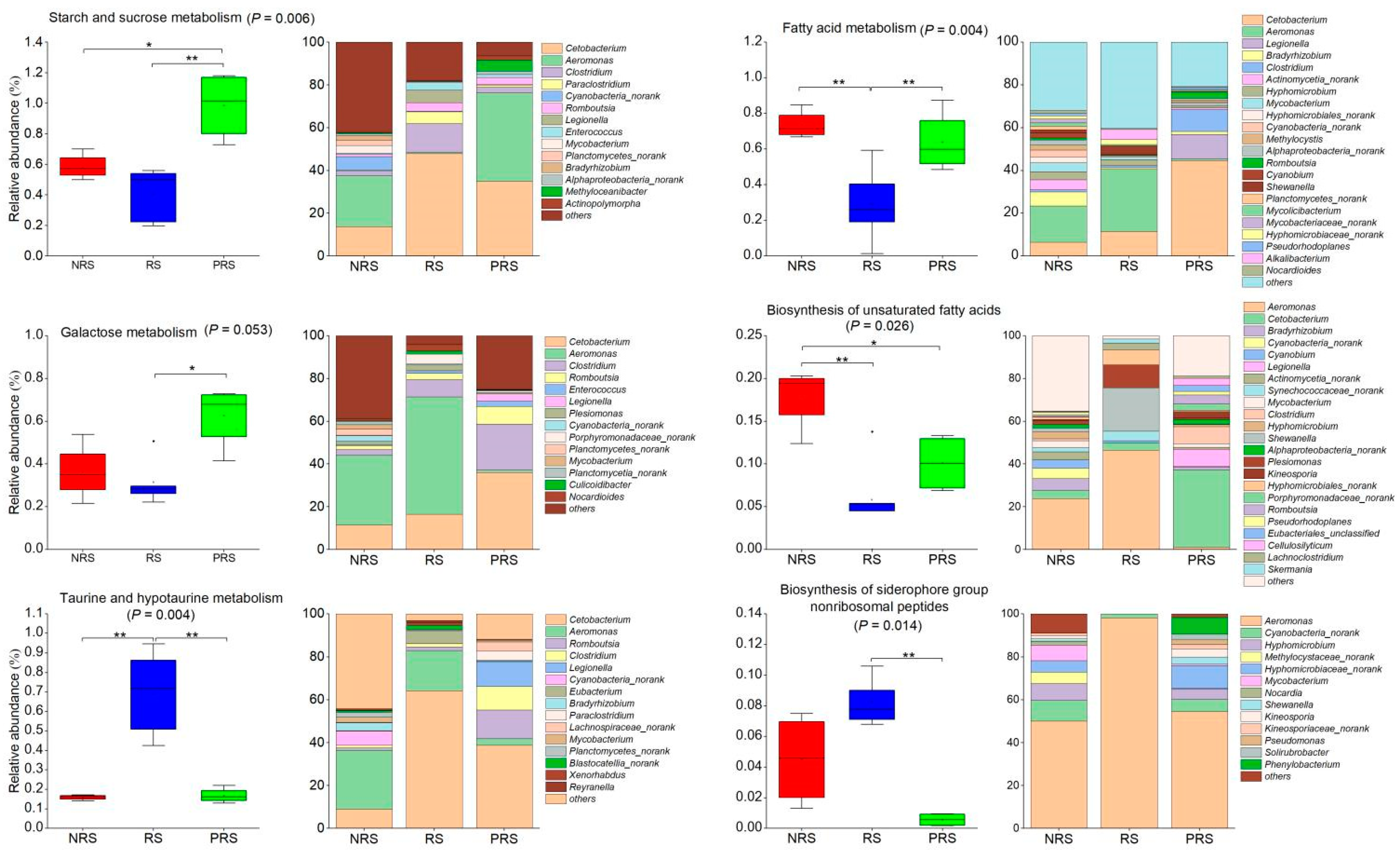
2.3. Gut Microbiota Functional Differences among Non-Reproductive, Reproductive and Post-Reproductive Groups
3. Discussion
4. Materials and Methods
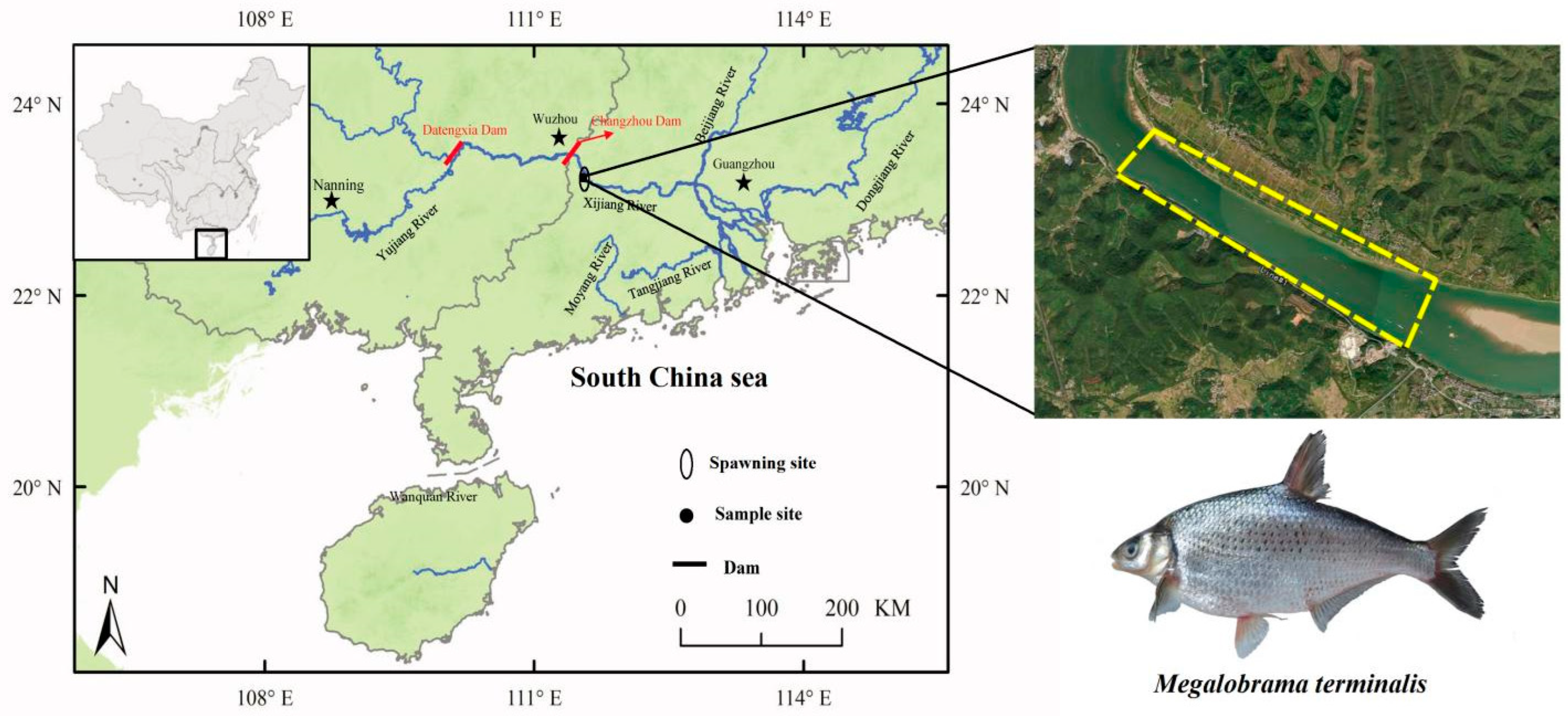
4.1. Fish Sampling
4.2. Biochemical Assays
4.3. Histological and Ultrastructural Investigations
4.3.1. Light Microscopy
4.3.2. Transmission Electron Microscopy
4.3.3. Morphometric Measurements
4.4. Enzyme Assays
4.5. Metagenomic Analysis
4.5.1. Fish Gut Contents Collection
4.5.2. DNA Extraction, Library Construction, and Metagenomic Sequencing
4.5.3. Sequence Quality Control and Genome Assembly
4.5.4. Gene Prediction, Taxonomy, and Functional Annotation
4.5.5. Bioinformatic Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation | Full title |
| AMY | Amylase |
| AP | Alkaline Phosphatase |
| CAT | Catalase |
| FD | Fullness degree |
| GR | Glutathione reductase |
| GSI | Gonadosomatic index |
| GVM | Germinal vesicle migration |
| K | Fatness |
| LAP | Leucine–alanine peptidase |
| LIP | Lipase |
| Lip | Lipid content |
| MAL | Maltase |
| NRS | Non-reproductive stage |
| POF | Post-ovulatory follicle |
| Pro | Protein content |
| PRS | Post-reproductive stage |
| RGL | Relative gut length |
| RS | Reproductive stage |
| SL | Standard length |
| SOD | Superoxide dismutase |
| TRY | Trypsin |
| Vtg1 | Primary vitellogenic oocyte |
| Vtg2 | Secondary vitellogenic oocyte |
| Wat | Water content |
| Wt | Body weight |
| YG | Yolk |
References
- Comizzoli, P.; Power, M. Reproductive microbiomes in wild animal species: A new dimension in conservation biology. Adv. Exp. Med. Biol. 2019, 1200, 225–240. [Google Scholar]
- Mallick, H.; Franzosa, E.; McLver, L.; Banerjee, S.; Sirota-Madi, A.; Kostic, A.; Clish, C.; Vlamakis, H.; Xavier, R.; Huttenhower, C. Predictive metabolomic profiling of microbial communities using amplicon or metagenomic sequences. Nat. Commun. 2019, 10, 3136. [Google Scholar] [CrossRef]
- Han, H.; Yi, B.; Zhong, R.; Wang, M.; Zhang, S.; Ma, J.; Yin, Y.; Yin, J.; Chen, L.; Zhang, H. From gut microbiota to host appetite: Gut microbiota-derived metabolites as key regulators. Microbiome 2021, 9, 162. [Google Scholar] [CrossRef]
- Huang, G.; Qu, Q.; Wang, M.; Huang, M.; Zhou, W.; Wei, F. Global landscape of gut microbiome diversity and antibiotic resistomes across vertebrates. Sci. Total Environ. 2022, 838, 156178. [Google Scholar] [CrossRef]
- Levin, D.; Raab, N.; Pinto, Y.; Rothschild, D.; Zanir, G.; Godneva, A.; Mellul, N.; Futorian, D.; Gal, D.; Leviatan, S.; et al. Diversity and functional landscapes in the microbiota of animals in the wild. Science 2021, 372, eabb5352. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Liang, X.; Lu, H.; Li, X.; Zhang, Z.; Lu, X.; Wang, H.; Meng, Y.; Roy, A.; Luo, W.; et al. Adaptation of gut microbiome and host metabolic systems to lignocellulosic degradation in bamboo rats. ISME J. 2022, 16, 1980–1992. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kou, C.; Li, Y.; Li, J.; Zhu, S. Fish gut microbiome analysis provides insight into differences in physiology and behavior of invasive Nile tilapia and indigenous fish in a large subtropical river in China. Animals 2023, 13, 2413. [Google Scholar] [CrossRef]
- Qi, X.; Yun, C.; Pang, Y.; Qiao, J. The impact of the gut microbiota on the reproductive and metabolic endocrine system. Gut Microbes 2021, 13, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.C.; Cable, J.; Lima-Maximino, M.G.; Maximino, C.; Xavier, R. Using fish models toinvestigate the links between microbiome and social behaviour: The next step for translational microbiome research? Fish Fishes 2019, 20, 640–652. [Google Scholar] [CrossRef]
- Comizzoli, P.; Power, M.L.; Bornbusch, S.L.; Muletz-Wolz, C.R. Interactions between reproductive biology and microbiomes in wild animal species. Anim. Microbiome 2021, 3, 87. [Google Scholar] [CrossRef]
- Antwis, R.E.; Edwards, K.L.; Unwin, B.; Walker, S.L.; Shultz, S. Rare gut microbiota associated with breeding success, hormone metabolites and ovarian cycle phase in the critically endangered eastern black rhino. Microbiome 2019, 7, 27. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.; Li, J.; Chen, W. The gut microbiome composition and degradation enzymes activity of black Amur bream (Megalobrama terminalis) in response to breeding migratory behavior. Ecol. Evol. 2021, 11, 5150–5163. [Google Scholar] [CrossRef] [PubMed]
- Korry, B.; Belenky, P. Trophic level and proteobacteria abundance drive antibiotic resistance levels in fish from coastal New England. Anim. Microbiome 2023, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Yukgehnaish, K.; Kumar, P.; Sivachandran, P.; Marimuthu, K.; Arshad, A.; Paray, B.A.; Arockiaraj, J. Gut microbiota metagenomics in aquaculture: Factors influencing gut microbiome and its physiological role in fish. Rev. Aquac. 2020, 12, 1903–1927. [Google Scholar] [CrossRef]
- Escalas, A.; Auguet, J.-C.; Avouac, A.; Seguin, R.; Gradel, A.; Borrossi, L.; Villéger, S. Ecological Specialization Within a Carnivorous Fish Family Is Supported by a Herbivorous Microbiome Shaped by a Combination of Gut Traits and Specific Diet. Front. Mar. Sci. 2021, 8, 622883. [Google Scholar] [CrossRef]
- Ghilardi, M.; Schiettekatte, N.M.D.; Casey, J.M.; Brandl, S.J.; Degregori, S.; Mercière, A.; Morat, F.; Letourneur, Y.; Bejarano, S.; Parravicini, V. Phylogeny, body morphology, and trophic level shape intestinal traits in coral reef fishes. Ecol. Evol. 2021, 11, 13218–13231. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.; Li, Y.; Li, J.; Zhu, S. Gut microbiomes of Cyprinid fishes exhibit host-species symbiosis along gut trait and diet. Front. Microbiol. 2022, 13, 936601. [Google Scholar]
- Liu, Y.; Li, Y.; Li, J.; Zhou, Q.; Li, X. Gut microbiomes analyses of wild migratory freshwater fish (Megalobrama terminalis) through geographic isolation. Front. Microbiol. 2022, 13, 858454. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, W.; Li, Y.; Li, J.; Li, X. Growth and ontogenetic development of digestive functionality in black Amur bream (Megalobrama terminalis). Aquaclture Res. 2020, 51, 3593–3601. [Google Scholar] [CrossRef]
- Fu, S.J.; Dong, Y.W.; Killen, S.S. Aerobic scope in fishes with different lifestyles and across habitats: Trade-offs among hypoxia tolerance, swimming performance and digestion. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2022, 272, 111277. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.; Li, Y.; Chen, W.; Li, J.; Zhu, S. Reproductive biology and strategy of Megalobrama terminalis of Xijiang River. J. Lake Sci. 2021, 33, 232–241. [Google Scholar]
- Liu, Y.; Li, X.; Li, J.; Li, Y. Insights into energy accumulation and allocation strategy of reproductive migration of black Amur bream (Megalobrama terminalis) in the Pearl River basin, China. Front. Ecol. Evol. 2022, 10, 848228. [Google Scholar] [CrossRef]
- Burns, M.D.; Bloom, D.D. Migratory lineages rapidly evolve larger body sizes than non- migratory relatives in ray- finned fishes. Proc. Royal Soc. B 2020, 287, 20192615. [Google Scholar] [CrossRef] [PubMed]
- Zaboukas, N.; Miliou, H.; Megalofonou, P.; Moraitou–Apostolopoulou, M. Biochemical composition of the Atlantic bonito Sarda sarda from the Aegean Sea (eastern Mediterranean Sea) in different stages of sexual maturity. J. Fish. Biol 2006, 69, 347–362. [Google Scholar] [CrossRef]
- Sieiro, P.; Otero, J.; Aubourg, S.P. Biochemical Composition and Energy Strategy Along the Reproductive Cycle of Female Octopus vulgaris in Galician Waters (NW Spain). Front. Physiol. 2020, 11, 760. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, N.; Jonsson, B.; Hansen, L. Energetic cost of spawning in male and female Atlantic salmon (Salmo salar L.). J. Fish Biol. 1997, 39, 739–744. [Google Scholar] [CrossRef]
- Hinch, S.; Rand, P. Optimal swim speeds and forward-assisted propulsion: Energy-conserving behavious of upriver-migrating adult salmon. Can. J. Fish. Aquat. Sci. 2000, 57, 2470–2478. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.; Wang, Z. Study on distribution characteristics of intestinal mucous cells and digestive enzyme activities in Paramisgurnus dabryanus. Acta Hydrobiol. Sin. 2017, 41, 1048–1053. [Google Scholar]
- Fassarella, M.; Blaak, E.E.; Penders, J.; Nauta, A.; Smidt, H.; Zoetendal, E.G. Gut microbiome stability and resilience: Elucidating the response to perturbations in order to modulate gut health. Gut 2021, 70, 595–605. [Google Scholar] [CrossRef]
- Reifel, C.; Travill, A. Structure and carbohydrate histochemistry of the intestine in ten teleostean species. J. Morphol. 1979, 162, 343–359. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-González, C.A.; Moyano-López, F.J.; Civera-Cerecedo, R.; Carrasco-Chávez, V.; Ortiz-Galindo, J.L.; Dumas, S. Development of digestive enzyme activity in larvae of spotted sand bass Paralabrax maculatofasciatus. Fish Physiol. Biochem. 2008, 34, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.K.; Williams, C.V.; Junge, R.; Mahefarisoa, K.L.; Rajaonarivelo, T.; Rakotondrainibe, H.; O’connell, T.M.; Drea, C.M. A role for gut microbiota in host niche differentiation. ISME J. 2020, 14, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Burnham, C.M.; McKenney, E.A.; van Heugten, K.A.; Minter, L.J.; Trivedi, S. Effects of age, seasonality, and reproductive status on the gut microbiome of Southern White Rhinoceros (Ceratotherium simum simum) at the North Carolina zoo. Anim. Microbiome 2023, 5, 27. [Google Scholar] [CrossRef]
- Mallott, E.K.; Borries, C.; Koenig, A.; Amato, K.R.; Lu, A. Reproductive hormones mediate changes in the gut microbiome during pregnancy and lactation in Phayre’s leaf monkeys. Sci. Rep. 2020, 10, 9961. [Google Scholar] [CrossRef]
- Kokou, F.; Sasson, G.; Friedman, J.; Eyal, S.; Ovadia, O.; Harpaz, S.; Cnaani, A.; Mizrahi, I. Core gut microbial communities are maintained by beneficial interactions and strain variability in fish. Nat. Microbiol. 2019, 4, 2456–2465. [Google Scholar] [CrossRef] [PubMed]
- Riera, J.; Baldo, L. Microbial co-occurrence networks of gut microbiota reveal community conservation and diet-associated shifts in cichlid fishes. Anim. Microbiome 2020, 2, 36. [Google Scholar] [CrossRef]
- Fan, C.; Zhang, L.; Jia, S.; Tang, X.; Fu, H.; Li, W.; Liu, C.; Zhang, H.; Cheng, Q.; Zhang, Y. Seasonal variations in the composition and functional profi les of gut microbiota refl ect dietary changes in plateau pikas. Integr. Zool. 2022, 17, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Porter, N.T.; Luis, A.S.; Martens, E.C. Bacteroides thetaiotaomicron. Trends Microbiol. 2018, 26, 966–967. [Google Scholar] [CrossRef]
- Johansson, M.E.; Jakobsson, H.E.; Holmén-Larsson, J.; Schütte, A.; Ermund, A.; Rodríguez-Piñeiro, A.M.; Arike, L.; Wising, C.; Svensson, F.; Bäckhed, F.; et al. Normalization of Host Intestinal Mucus Layers Requires Long-Term Microbial Colonization. Cell Host Microbe 2015, 18, 582–592. [Google Scholar] [CrossRef]
- Melo-Gonzalez, F.; Fenton, T.M.; Forss, C.; Smedley, C.; Goenka, A.; Macdonald, A.S.; Thornton, D.J.; Travis, M.A. Intestinal mucin activates human dendritic cells and IL-8 production in a glycan-specific manner. J. Biol. Chem. 2018, 293, 8543–8553. [Google Scholar] [CrossRef] [PubMed]
- Byndloss, M.X.; Pernitzsch, S.R.; Bäumler, A.J. Healthy hosts rule within: Ecological forces shaping the gut microbiota. Mucosal Immunol. 2018, 11, 1299–1305. [Google Scholar] [CrossRef]
- Bae, M.; Ahmed, K.; Yim, J.E. Beneficial Effects of Taurine on Metabolic Parameters in Animals and Humans. J. Obes. Metab. Syndr. 2022, 31, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Gollapalli, K.; Mangiola, S.; Schranner, D.; Yusuf, M.A.; Chamoli, M.; Shi, S.L.; Bastos, B.L.; Nair, T.; Riermeier, A.; et al. Taurine deficiency as a driver of aging. Science 2023, 380, eabn9257. [Google Scholar] [CrossRef] [PubMed]
- Silva, H.V.R.; Silva, A.R.; da Silvada, L.D.M.; Comizzoli, P. Semen Cryopreser-vation and Banking for the Conservation of Neotropical Carnivores. Biopreserv. Biobank. 2019, 17, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Zainuddin, Z.Z.; Mohamed Tarmizi, M.R.; Yap, K.C.; Comizzoli, P.; Sipangkui, S. First evaluations and cryopreservation of semen samples from sundaclouded leopards (Neofelis diardi). Animals 2020, 10, 1072. [Google Scholar] [CrossRef]
- Yin, M. Fish Ecology; China Agriculture Press: Beijing, China, 1993. [Google Scholar]
- Hach, C.; Scott, V.; Kopelove, A. A powerful Kjeldahl nitrogen method using peroxymonosulfuric acid. J. Agric. Food Chem. 1985, 33, 1117–1123. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane–Stanley, G. A simple method for the isolation and purification of total lipid from animal tissue. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Z. A Study on Structural Characteristics of Intestinal Tract of the Air-Breathing Loach, Paramisgurnus dabryanus (Sauvage, 1878). Pak. J. Zool. 2017, 49, 1223–1230. [Google Scholar] [CrossRef]
- Bradford, M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Allen, G.; Flores-Vergara, M.; Krasynanski, S.; Kumar, S.; Thompson, W.F. A modified protocol for rapid DNAisolation from plant tissues using cetyltrimethylammoniumbromide. Nat. Protoc. 2006, 1, 2320–2325. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, 60. [Google Scholar] [CrossRef]
- Clarke, K. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | NRS | RS | PRS | |
|---|---|---|---|---|
| biological information | SL (mm) | 254 ± 31 (20) | 263 ± 21(17) | 257 ± 26 (18) |
| Wt (g) | 371.6 ± 26.9 (20) b | 420.6 ± 35.4 (17) a | 350.6 ± 33.7 (18) b | |
| GSI (%) | 2.35 ± 0.38 (20) b | 10.59 ± 2.14 (17) a | 4.31 ± 1.97(18) b | |
| K | 2.41 ± 0.35 (20) b | 2.62 ± 0.28 (17) a | 2.35 ± 0.31 (18) b | |
| RGL | 3.51 ± 0.23 (20) a | 3.21 ± 0.54 (17) b | 3.52 ± 0.42 (18) a | |
| FD | 3.75 ± 0.15(20) b | 0.65 ± 0.12 (17) c | 4.25 ± 0.25 (18) a | |
| biochemical constituent proportion of ovary | Pro (%) | 15.31 ± 2.14 (10) | 13.08 ± 3.01(10) | 11.35 ± 1.96 (10) |
| Lip (%) | 11.93 ± 1.84 (10) ab | 15.88 ± 2.94 (10) a | 9.58 ± 1.47 (10) b | |
| Wat (%) | 68.25 ± 3.65 (10) ab | 63.56 ± 4.15 (10) b | 73.57 ± 5.11 (10) a |
| Item | NRS | RS | PRS | |
|---|---|---|---|---|
| Mucous cell | Type I | 1.27 ± 0.21 a | 0.51 ± 0.23 b | 1.16 ± 0.31 a |
| Type II | 0.89 ± 0.14 b | 2.18 ± 0.37 a | 0.61 ± 0.11 c | |
| Type III | 8.75 ± 1.19 a | 2.02 ± 0.34 c | 5.75 ± 0.75 b | |
| Type IV | 3.68 ± 0.75 b | 5.82 ± 1.05 a | 3.95 ± 0.93 b | |
| Total | 14.59 ± 2.27 a | 10.53 ± 1.94 b | 11.47 ± 1.87 ab | |
| Mitochondria in intestinal epithelial cells | 5.71 ± 1.07 a | 2.14 ± 0.83 b | 6.92 ± 1.32 a | |
| Eosinophilic granulocyte in intestinal submucosa | 0.45 ± 0.33 b | 3.86 ± 0.93 a | 0.79 ± 0.56 b |
| Fullness | Description |
|---|---|
| 0 | Jejunum or a very small amount of food was observed in the intestinal tract |
| 1 | Only part of the bowel has a small amount of food or a quarter of the bowel is food |
| 2 | There is only a small amount of food or half of the food in the entire bowel |
| 3 | More food, medium filling degree, food accounted for 3/4 of the bowel |
| 4 | It is a lot of food. It is filling up the intestines |
| 5 | There was too much food, and the intestines swelled |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Kou, C.; Chen, J.; Li, Y.; Li, J. The Response of the Gut Physiological Function and Microbiome of a Wild Freshwater Fish (Megalobrama terminalis) to Alterations in Reproductive Behavior. Int. J. Mol. Sci. 2024, 25, 7425. https://doi.org/10.3390/ijms25137425
Liu Y, Kou C, Chen J, Li Y, Li J. The Response of the Gut Physiological Function and Microbiome of a Wild Freshwater Fish (Megalobrama terminalis) to Alterations in Reproductive Behavior. International Journal of Molecular Sciences. 2024; 25(13):7425. https://doi.org/10.3390/ijms25137425
Chicago/Turabian StyleLiu, Yaqiu, Chunni Kou, Jiayue Chen, Yuefei Li, and Jie Li. 2024. "The Response of the Gut Physiological Function and Microbiome of a Wild Freshwater Fish (Megalobrama terminalis) to Alterations in Reproductive Behavior" International Journal of Molecular Sciences 25, no. 13: 7425. https://doi.org/10.3390/ijms25137425