
The Gut and Skin Microbiome and Its Association with Aging Clocks



Abstract
1. Introduction
2. Methods
3. Results and Discussion
3.1. Literature Search
3.2. Gut Microbiome Aging Clocks
3.2.1. Taxonomic Clocks
3.2.2. Biodiversity Clocks
3.2.3. Functional Clocks
3.2.4. Meta-Metabolomic and Proteomic Clocks
3.2.5. Gut Microbiome Modulation of Other Aging Clocks
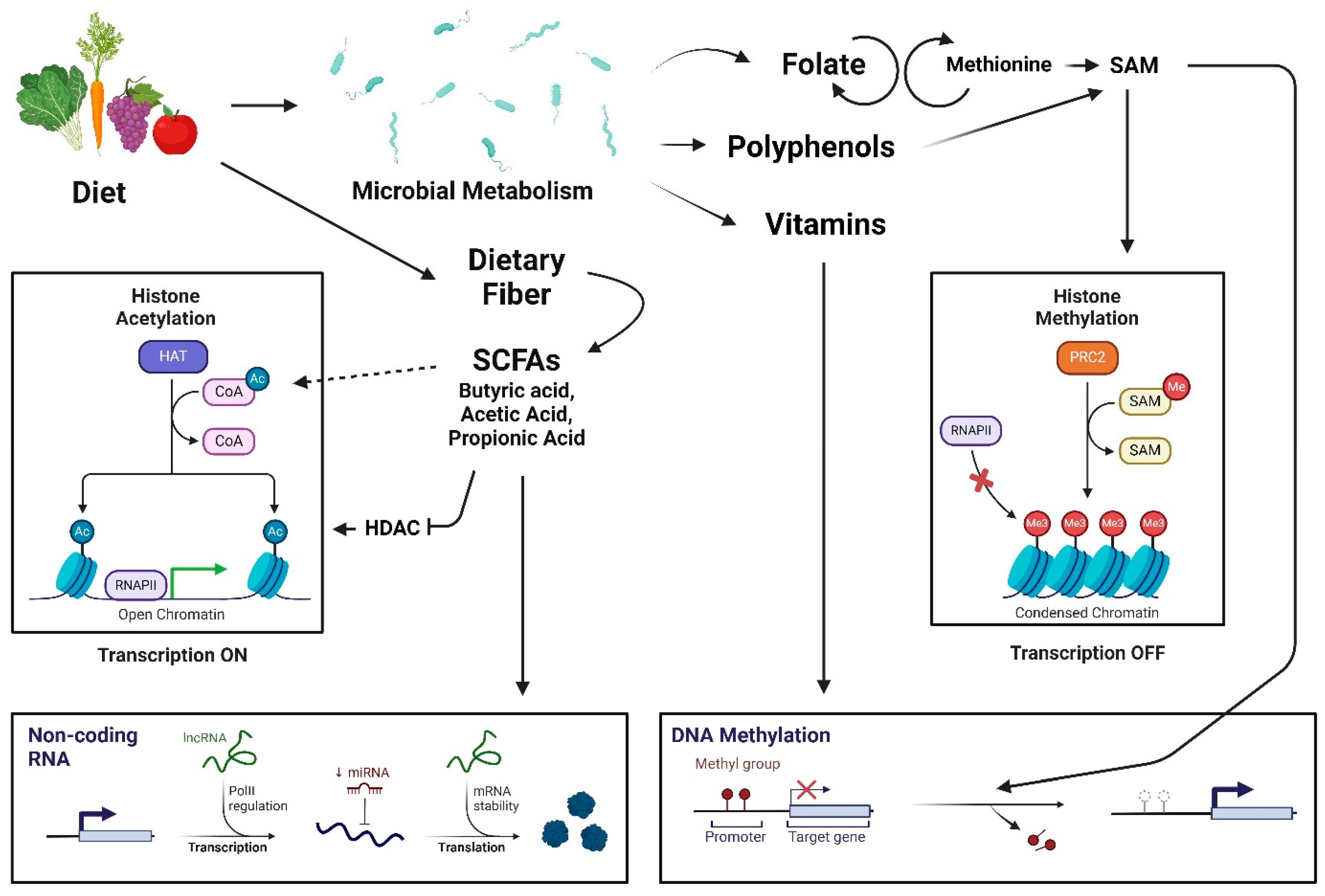
3.2.6. Gut Microbiome-Derived Secondary Metabolites
3.3. Skin Microbiome Aging Clocks
3.3.1. Skin Microbiome Modulation of Other Aging Clocks
3.3.2. Skin Microbiome-Derived Secondary Metabolites
3.4. Technological Advances and Measurement Techniques
3.5. Challenges and Limitations
3.5.1. Interindividual Variability
3.5.2. Standardization and Validation
3.5.3. Ethical Considerations
3.6. Clinical Implications and Future Directions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dodig, S.; Cepelak, I.; Pavic, I. Hallmarks of senescence and aging. Biochem. Med. 2019, 29, 030501. [Google Scholar] [CrossRef] [PubMed]
- Salih, A.; Nichols, T.; Szabo, L.; Petersen, S.E.; Raisi-Estabragh, Z. Conceptual Overview of Biological Age Estimation. Aging Dis. 2023, 14, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Maltoni, R.; Ravaioli, S.; Bronte, G.; Mazza, M.; Cerchione, C.; Massa, I.; Balzi, W.; Cortesi, M.; Zanoni, M.; Bravaccini, S. Chronological age or biological age: What drives the choice of adjuvant treatment in elderly breast cancer patients? Transl. Oncol. 2022, 15, 101300. [Google Scholar] [CrossRef] [PubMed]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Howard, B.; Bascom, C.C.; Hu, P.; Binder, R.L.; Fadayel, G.; Huggins, T.G.; Jarrold, B.B.; Osborne, R.; Rocchetta, H.L.; Swift, D.; et al. Aging-Associated Changes in the Adult Human Skin Microbiome and the Host Factors that Affect Skin Microbiome Composition. J. Investig. Dermatol. 2022, 142, 1934–1946.e21. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Oh, H.N.; Park, T.; Kim, H.; Lee, H.G.; An, S.; Sul, W.J. Aged related human skin microbiome and mycobiome in Korean women. Sci. Rep. 2022, 12, 2351. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Segre, J.A. Dialogue between skin microbiota and immunity. Science 2014, 346, 954–959. [Google Scholar] [CrossRef]
- Etchegaray, J.P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef]
- Krautkramer, K.A.; Kreznar, J.H.; Romano, K.A.; Vivas, E.I.; Barrett-Wilt, G.A.; Rabaglia, M.E.; Keller, M.P.; Attie, A.D.; Rey, F.E.; Denu, J.M. Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues. Mol. Cell 2016, 64, 982–992. [Google Scholar] [CrossRef]
- Ratanapokasatit, Y.; Laisuan, W.; Rattananukrom, T.; Petchlorlian, A.; Thaipisuttikul, I.; Sompornrattanaphan, M. How Microbiomes Affect Skin Aging: The Updated Evidence and Current Perspectives. Life 2022, 12, 936. [Google Scholar] [CrossRef]
- Lan, Y.; Kriete, A.; Rosen, G.L. Selecting age-related functional characteristics in the human gut microbiome. Microbiome 2013, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, P.W.; Jeffery, I.B. Gut microbiota and aging. Science 2015, 350, 1214–1215. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Hua, Y.; Zeng, B.; Ning, R.; Li, Y.; Zhao, J. Gut microbiota signatures of longevity. Curr. Biol. 2016, 26, R832–R833. [Google Scholar] [CrossRef]
- Shibagaki, N.; Suda, W.; Clavaud, C.; Bastien, P.; Takayasu, L.; Iioka, E.; Kurokawa, R.; Yamashita, N.; Hattori, Y.; Shindo, C.; et al. Aging-related changes in the diversity of women’s skin microbiomes associated with oral bacteria. Sci. Rep. 2017, 7, 10567. [Google Scholar] [CrossRef]
- Juge, R.; Rouaud-Tinguely, P.; Breugnot, J.; Servaes, K.; Grimaldi, C.; Roth, M.P.; Coppin, H.; Closs, B. Shift in skin microbiota of Western European women across aging. J. Appl. Microbiol. 2018, 125, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Mitra, R.; Maitra, A.; Gupta, S.; Kumaran, S.; Chakrabortty, A.; Majumder, P.P. Sebum and Hydration Levels in Specific Regions of Human Face Significantly Predict the Nature and Diversity of Facial Skin Microbiome. Sci. Rep. 2016, 6, 36062. [Google Scholar] [CrossRef]
- Galkin, F.; Mamoshina, P.; Aliper, A.; Putin, E.; Moskalev, V.; Gladyshev, V.N.; Zhavoronkov, A. Human Gut Microbiome Aging Clock Based on Taxonomic Profiling and Deep Learning. iScience 2020, 23, 101199. [Google Scholar] [CrossRef]
- Huang, S.; Haiminen, N.; Carrieri, A.P.; Hu, R.; Jiang, L.; Parida, L.; Russell, B.; Allaband, C.; Zarrinpar, A.; Vazquez-Baeza, Y.; et al. Human Skin, Oral, and Gut Microbiomes Predict Chronological Age. mSystems 2020, 5, e00630-19. [Google Scholar] [CrossRef]
- Sala, C.; Giampieri, E.; Vitali, S.; Garagnani, P.; Remondini, D.; Bazzani, A.; Franceschi, C.; Castellani, G.C. Gut microbiota ecology: Biodiversity estimated from hybrid neutral-niche model increases with health status and aging. PLoS ONE 2020, 15, e0237207. [Google Scholar] [CrossRef]
- Wilmanski, T.; Diener, C.; Rappaport, N.; Patwardhan, S.; Wiedrick, J.; Lapidus, J.; Earls, J.C.; Zimmer, A.; Glusman, G.; Robinson, M.; et al. Gut microbiome pattern reflects healthy ageing and predicts survival in humans. Nat. Metab. 2021, 3, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, H.; Lu, W.; Wu, T.; Yuan, W.; Zhu, J.; Lee, Y.K.; Zhao, J.; Zhang, H.; Chen, W. Human gut microbiome aging clocks based on taxonomic and functional signatures through multi-view learning. Gut Microbes 2022, 14, 2025016. [Google Scholar] [CrossRef] [PubMed]
- Gopu, V.; Camacho, F.R.; Toma, R.; Torres, P.J.; Cai, Y.; Krishnan, S.; Rajagopal, S.; Tily, H.; Vuyisich, M.; Banavar, G. An accurate aging clock developed from large-scale gut microbiome and human gene expression data. iScience 2024, 27, 108538. [Google Scholar] [CrossRef] [PubMed]
- Nogal, A.; Valdes, A.M.; Menni, C. The role of short-chain fatty acids in the interplay between gut microbiota and diet in cardio-metabolic health. Gut Microbes 2021, 13, 1897212. [Google Scholar] [CrossRef] [PubMed]
- Phongsisay, V. The immunobiology of Campylobacter jejuni: Innate immunity and autoimmune diseases. Immunobiology 2016, 221, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, C.B.; Lichatz, R.; Pich, A.; Muhlfeld, C.; Woltemate, S.; Vital, M.; Brandenberger, C. Short-chain fatty acids improve inflamm-aging and acute lung injury in old mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2023, 324, L480–L492. [Google Scholar] [CrossRef] [PubMed]
- Jang, L.G.; Choi, G.; Kim, S.W.; Kim, B.Y.; Lee, S.; Park, H. The combination of sport and sport-specific diet is associated with characteristics of gut microbiota: An observational study. J. Int. Soc. Sports Nutr. 2019, 16, 21. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liang, H.; Hu, Y.; Lu, L.; Zheng, C.; Fan, Y.; Wu, B.; Zou, T.; Luo, X.; Zhang, X.; et al. Gut bacterial profiles in Parkinson’s disease: A systematic review. CNS Neurosci. Ther. 2023, 29, 140–157. [Google Scholar] [CrossRef] [PubMed]
- Shelest, I.I.; Soroka, N.A. Characteristics of the inhibitory processes in the neurons of the posterior lateral thalamic nucleus when exposed to different afferent stimulations. Fiziol. Zhurnal 1986, 32, 741–748. [Google Scholar]
- Biagi, E.; Franceschi, C.; Rampelli, S.; Severgnini, M.; Ostan, R.; Turroni, S.; Consolandi, C.; Quercia, S.; Scurti, M.; Monti, D.; et al. Gut Microbiota and Extreme Longevity. Curr. Biol. 2016, 26, 1480–1485. [Google Scholar] [CrossRef]
- Hung, C.C.; Chang, C.C.; Huang, C.W.; Nouchi, R.; Cheng, C.H. Gut microbiota in patients with Alzheimer’s disease spectrum: A systematic review and meta-analysis. Aging 2022, 14, 477–496. [Google Scholar] [CrossRef] [PubMed]
- Mariat, D.; Firmesse, O.; Levenez, F.; Guimaraes, V.; Sokol, H.; Dore, J.; Corthier, G.; Furet, J.P. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009, 9, 123. [Google Scholar] [CrossRef]
- Nemet, I.; Saha, P.P.; Gupta, N.; Zhu, W.; Romano, K.A.; Skye, S.M.; Cajka, T.; Mohan, M.L.; Li, L.; Wu, Y.; et al. A Cardiovascular Disease-Linked Gut Microbial Metabolite Acts via Adrenergic Receptors. Cell 2020, 180, 862–877.e822. [Google Scholar] [CrossRef]
- Levy, P.Y.; Fenollar, F.; Stein, A.; Borrione, F.; Raoult, D. Finegoldia magna: A forgotten pathogen in prosthetic joint infection rediscovered by molecular biology. Clin. Infect. Dis. 2009, 49, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.Y.; Nam, Y.D. Gut microbiome in healthy aging versus those associated with frailty. Gut Microbes 2023, 15, 2278225. [Google Scholar] [CrossRef] [PubMed]
- White, P.J.; Newgard, C.B. Branched-chain amino acids in disease. Science 2019, 363, 582–583. [Google Scholar] [CrossRef]
- Johnson, L.C.; Parker, K.; Aguirre, B.F.; Nemkov, T.G.; D’Alessandro, A.; Johnson, S.A.; Seals, D.R.; Martens, C.R. The plasma metabolome as a predictor of biological aging in humans. Geroscience 2019, 41, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Belsky, D.W.; Caspi, A.; Corcoran, D.L.; Sugden, K.; Poulton, R.; Arseneault, L.; Baccarelli, A.; Chamarti, K.; Gao, X.; Hannon, E.; et al. DunedinPACE, a DNA methylation biomarker of the pace of aging. eLife 2022, 11, e73420. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef]
- Ansari, I.; Raddatz, G.; Gutekunst, J.; Ridnik, M.; Cohen, D.; Abu-Remaileh, M.; Tuganbaev, T.; Shapiro, H.; Pikarsky, E.; Elinav, E.; et al. The microbiota programs DNA methylation to control intestinal homeostasis and inflammation. Nat. Microbiol. 2020, 5, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Tahara, T.; Hirata, I.; Nakano, N.; Tahara, S.; Horiguchi, N.; Kawamura, T.; Okubo, M.; Ishizuka, T.; Yamada, H.; Yoshida, D.; et al. Potential link between Fusobacterium enrichment and DNA methylation accumulation in the inflammatory colonic mucosa in ulcerative colitis. Oncotarget 2017, 8, 61917–61926. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.H.; Sommer, F.; Falk-Paulsen, M.; Ulas, T.; Best, P.; Fazio, A.; Kachroo, P.; Luzius, A.; Jentzsch, M.; Rehman, A.; et al. Exposure to the gut microbiota drives distinct methylome and transcriptome changes in intestinal epithelial cells during postnatal development. Genome Med. 2018, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Gong, D.; Nguyen, D.N.; Zhang, X.; Hu, Q.; Lu, H.; Fredholm, M.; Sangild, P.T.; Gao, F. Early microbial colonization affects DNA methylation of genes related to intestinal immunity and metabolism in preterm pigs. DNA Res. 2018, 25, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Amaretti, A.; Raimondi, S. Folate production by probiotic bacteria. Nutrients 2011, 3, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, Z.; Cai, H.; Zhou, C. Progress in the microbial production of S-adenosyl-L-methionine. World J. Microbiol. Biotechnol. 2016, 32, 153. [Google Scholar] [CrossRef] [PubMed]
- Waldecker, M.; Kautenburger, T.; Daumann, H.; Busch, C.; Schrenk, D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J. Nutr. Biochem. 2008, 19, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhao, S.; Xiang, T.; Feng, H.; Ma, L.; Fu, P. Epigenetic connection between gut microbiota-derived short-chain fatty acids and chromatin histone modification in kidney diseases. Chin. Med. J. 2022, 135, 1692–1694. [Google Scholar] [CrossRef] [PubMed]
- Boopathi, S.; Kumar, R.M.S.; Priya, P.S.; Haridevamuthu, B.; Nayak, S.; Chulenbayeva, L.; Almagul, K.; Arockiaraj, J. Gut Enterobacteriaceae and uraemic toxins—Perpetrators for ageing. Exp. Gerontol. 2023, 173, 112088. [Google Scholar] [CrossRef]
- Ebersole, J.L.; Nagarajan, R.; Kirakodu, S.; Gonzalez, O.A. Oral Microbiome and Gingival Gene Expression of Inflammatory Biomolecules With Aging and Periodontitis. Front. Oral Health 2021, 2, 725115. [Google Scholar] [CrossRef]
- Xu, K.; Guo, Y.; Wang, Y.; Ren, Y.; Low, V.; Cho, S.; Ping, L.; Peng, K.; Li, X.; Qiu, Y.; et al. Decreased Enterobacteriaceae translocation due to gut microbiota remodeling mediates the alleviation of premature aging by a high-fat diet. Aging Cell 2023, 22, e13760. [Google Scholar] [CrossRef] [PubMed]
- Sayed, N.; Huang, Y.; Nguyen, K.; Krejciova-Rajaniemi, Z.; Grawe, A.P.; Gao, T.; Tibshirani, R.; Hastie, T.; Alpert, A.; Cui, L.; et al. An inflammatory aging clock (iAge) based on deep learning tracks multimorbidity, immunosenescence, frailty and cardiovascular aging. Nat. Aging 2021, 1, 598–615. [Google Scholar] [CrossRef] [PubMed]
- Mylonas, A.; Hawerkamp, H.C.; Wang, Y.; Chen, J.; Messina, F.; Demaria, O.; Meller, S.; Homey, B.; Di Domizio, J.; Mazzolai, L.; et al. Type I IFNs link skin-associated dysbiotic commensal bacteria to pathogenic inflammation and angiogenesis in rosacea. JCI Insight 2023, 8, e151846. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; He, J.; Li, J.; Zou, Q.; Si, J.; Guo, Y.; Yu, J.; Li, C.; Wang, F.; Chan, T.; et al. Microbiome and Metabolome Analyses Reveal Novel Interplay Between the Skin Microbiota and Plasma Metabolites in Psoriasis. Front. Microbiol. 2021, 12, 643449. [Google Scholar] [CrossRef] [PubMed]
- Trompette, A.; Pernot, J.; Perdijk, O.; Alqahtani, R.A.A.; Domingo, J.S.; Camacho-Munoz, D.; Wong, N.C.; Kendall, A.C.; Wiederkehr, A.; Nicod, L.P.; et al. Gut-derived short-chain fatty acids modulate skin barrier integrity by promoting keratinocyte metabolism and differentiation. Mucosal Immunol. 2022, 15, 908–926. [Google Scholar] [CrossRef] [PubMed]
- Racine, P.J.; Janvier, X.; Clabaut, M.; Catovic, C.; Souak, D.; Boukerb, A.M.; Groboillot, A.; Konto-Ghiorghi, Y.; Duclairoir-Poc, C.; Lesouhaitier, O.; et al. Dialog between skin and its microbiota: Emergence of “Cutaneous Bacterial Endocrinology”. Exp. Dermatol. 2020, 29, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Arellano, K.; Lee, Y.; Yeo, S.; Ji, Y.; Ko, J.; Holzapfel, W. Pilot Study on the Forehead Skin Microbiome and Short Chain Fatty Acids Depending on the SC Functional Index in Korean Cohorts. Microorganisms 2021, 9, 2216. [Google Scholar] [CrossRef] [PubMed]
- Janda, J.M.; Abbott, S.L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses, perils, and pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef] [PubMed]
- Jylhava, J.; Pedersen, N.L.; Hagg, S. Biological Age Predictors. eBioMedicine 2017, 21, 29–36. [Google Scholar] [CrossRef]
- Field, A.E.; Robertson, N.A.; Wang, T.; Havas, A.; Ideker, T.; Adams, P.D. DNA Methylation Clocks in Aging: Categories, Causes, and Consequences. Mol. Cell 2018, 71, 882–895. [Google Scholar] [CrossRef]
- Yang, Z.; Wong, A.; Kuh, D.; Paul, D.S.; Rakyan, V.K.; Leslie, R.D.; Zheng, S.C.; Widschwendter, M.; Beck, S.; Teschendorff, A.E. Correlation of an epigenetic mitotic clock with cancer risk. Genome Biol. 2016, 17, 205. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Christensen, B.C.; Gladyshev, V.N.; Heijmans, B.T.; Horvath, S.; et al. DNA methylation aging clocks: Challenges and recommendations. Genome Biol. 2019, 20, 249. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Oshima, J.; Martin, G.M.; Lu, A.T.; Quach, A.; Cohen, H.; Felton, S.; Matsuyama, M.; Lowe, D.; Kabacik, S.; et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging 2018, 10, 1758–1775. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Quach, A.; Levine, M.E.; Tanaka, T.; Lu, A.T.; Chen, B.H.; Ferrucci, L.; Ritz, B.; Bandinelli, S.; Neuhouser, M.L.; Beasley, J.M.; et al. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging 2017, 9, 419–446. [Google Scholar] [CrossRef] [PubMed]
- Gensous, N.; Garagnani, P.; Santoro, A.; Giuliani, C.; Ostan, R.; Fabbri, C.; Milazzo, M.; Gentilini, D.; di Blasio, A.M.; Pietruszka, B.; et al. One-year Mediterranean diet promotes epigenetic rejuvenation with country- and sex-specific effects: A pilot study from the NU-AGE project. Geroscience 2020, 42, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Hindorff, L.A.; Bonham, V.L.; Brody, L.C.; Ginoza, M.E.C.; Hutter, C.M.; Manolio, T.A.; Green, E.D. Prioritizing diversity in human genomics research. Nat. Rev. Genet. 2018, 19, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Mackey, T. An ethical assessment of anti-aging medicine. J. Anti Aging Med. 2003, 6, 187–204. [Google Scholar] [CrossRef]
- Dupras, C.; Beauchamp, E.; Joly, Y. Selling direct-to-consumer epigenetic tests: Are we ready? Nat. Rev. Genet. 2020, 21, 335–336. [Google Scholar] [CrossRef]
- De Oliveira, N.F.P.; de Souza, B.F.; de Castro Coelho, M. UV Radiation and Its Relation to DNA Methylation in Epidermal Cells: A Review. Epigenomes 2020, 4, 23. [Google Scholar] [CrossRef] [PubMed]
- Boroni, M.; Zonari, A.; Reis de Oliveira, C.; Alkatib, K.; Ochoa Cruz, E.A.; Brace, L.E.; Lott de Carvalho, J. Highly accurate skin-specific methylome analysis algorithm as a platform to screen and validate therapeutics for healthy aging. Clin. Epigenetics 2020, 12, 105. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mao, K.; Zhai, H.; Jackie Han, J.D. Clinical application of facial aging clocks. Lancet Reg. Health West. Pac. 2023, 37, 100858. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Author (Year) | Type of Aging Clock | Gut or Skin Microbiome | Key Findings |
|---|---|---|---|
| Galkin et al. (2020) [18] | Taxonomic | Gut | Bifidobacterium spp., Akkermansia muciniphila, and Bacteroides spp. or pathogenic bacteria, such as Escheria coli and Campylobacter jejuni, were most predictive. |
| Huang et al. (2020) [19] | Taxonomic | Gut | Bifidobacterium and Blautia or the families Lachnospiraceae, Ruminococcaceae, and Clostridiaceae had the highest feature importance scores. |
| Sala et al. (2020) [20] | Biodiversity | Gut | There was a decrease in microbial diversity with unhealthy phenotypes at older ages. |
| Wilmanski et al. (2021) [21] | Biodiversity | Gut | The β-diversity of gut microbiomes becomes more unique with age and is related to metabolomic changes. |
| Chen et al. (2022) [22] | Functional | Gut | Acetyl-CoA biosynthesis and nicotinate degradation were the most predictive. Finegoldia magna, Bifidobacterium dentium, and Clostridium clostridioforme increased, while Prevotella copri and Burkholderialse bacterium decreased with age. |
| Gopu et al. (2024) [23] | Functional | Gut | Ruminococcaceae, Bifidobacteriaceae, Lachnospiraceae, and Clostridiaceae families were the most predictive. |
| Huang et al. (2020) [19] | Taxonomic | Skin | Mycoplasma, Enterobacteriaceae, and Pasteurellaceae were negatively correlated with age. |
| Author (Year) | Type of Aging Clock | Gut or Skin Microbiome | Type of Sequencing | R2 | Mean Absolute Error (Years) |
|---|---|---|---|---|---|
| Galkin et al. (2020) [18] | Taxonomic | Gut | Metagenomic | 0.20 | 10.6 |
| Huang et al. (2020) [19] | Taxonomic | Gut | 16S rRNA | 0.17 | 11.5 |
| Sala et al. (2020) [20] | Biodiversity | Gut | 16S rRNA | * | * |
| Wilmanski et al. (2021) [21] | Biodiversity | Gut | 16S rRNA | * | * |
| Chen et al. (2022) [22] | Functional | Gut | Metagenomic | 0.6 | 8.3 |
| Gopu et al. (2024) [23] | Functional | Gut | Metagenomic | 0.42 | 9.5 |
| Huang et al. (2020) [19] | Taxonomic | Skin | 16S rRNA | 0.74 | 3.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Min, M.; Egli, C.; Sivamani, R.K. The Gut and Skin Microbiome and Its Association with Aging Clocks. Int. J. Mol. Sci. 2024, 25, 7471. https://doi.org/10.3390/ijms25137471
Min M, Egli C, Sivamani RK. The Gut and Skin Microbiome and Its Association with Aging Clocks. International Journal of Molecular Sciences. 2024; 25(13):7471. https://doi.org/10.3390/ijms25137471
Chicago/Turabian StyleMin, Mildred, Caitlin Egli, and Raja K. Sivamani. 2024. "The Gut and Skin Microbiome and Its Association with Aging Clocks" International Journal of Molecular Sciences 25, no. 13: 7471. https://doi.org/10.3390/ijms25137471
APA StyleMin, M., Egli, C., & Sivamani, R. K. (2024). The Gut and Skin Microbiome and Its Association with Aging Clocks. International Journal of Molecular Sciences, 25(13), 7471. https://doi.org/10.3390/ijms25137471