
Resveratrol and Vitamin D: Eclectic Molecules Promoting Mitochondrial Health in Sarcopenia

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Molecular Mechanisms of Myogenesis in Adulthood
3. Resveratrol and Skeletal Muscle Function
4. Vitamin D and Skeletal Muscle Function
5. Role of Mitochondria of Skeletal Muscle Fibers in Sarcopenia Physiopathology
5.1. Mitochondrial Biogenesis in Sarcopenia
5.2. Mitophagy in Sarcopenia
5.3. Role of Neuromuscular Junction Mitochondria in Sarcopenia Pathophysiology
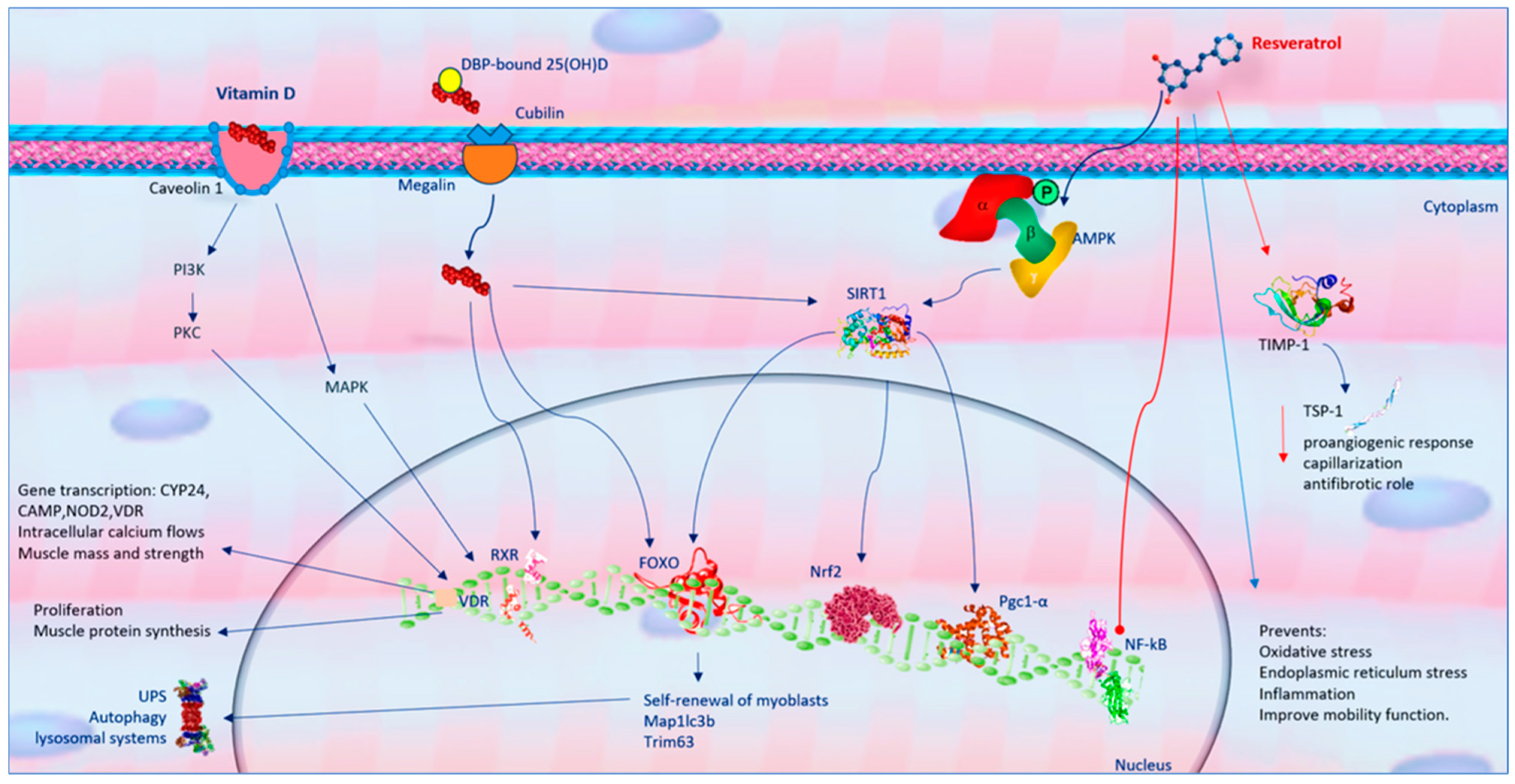
6. Resveratrol Molecular Mechanisms in Mitochondria Health
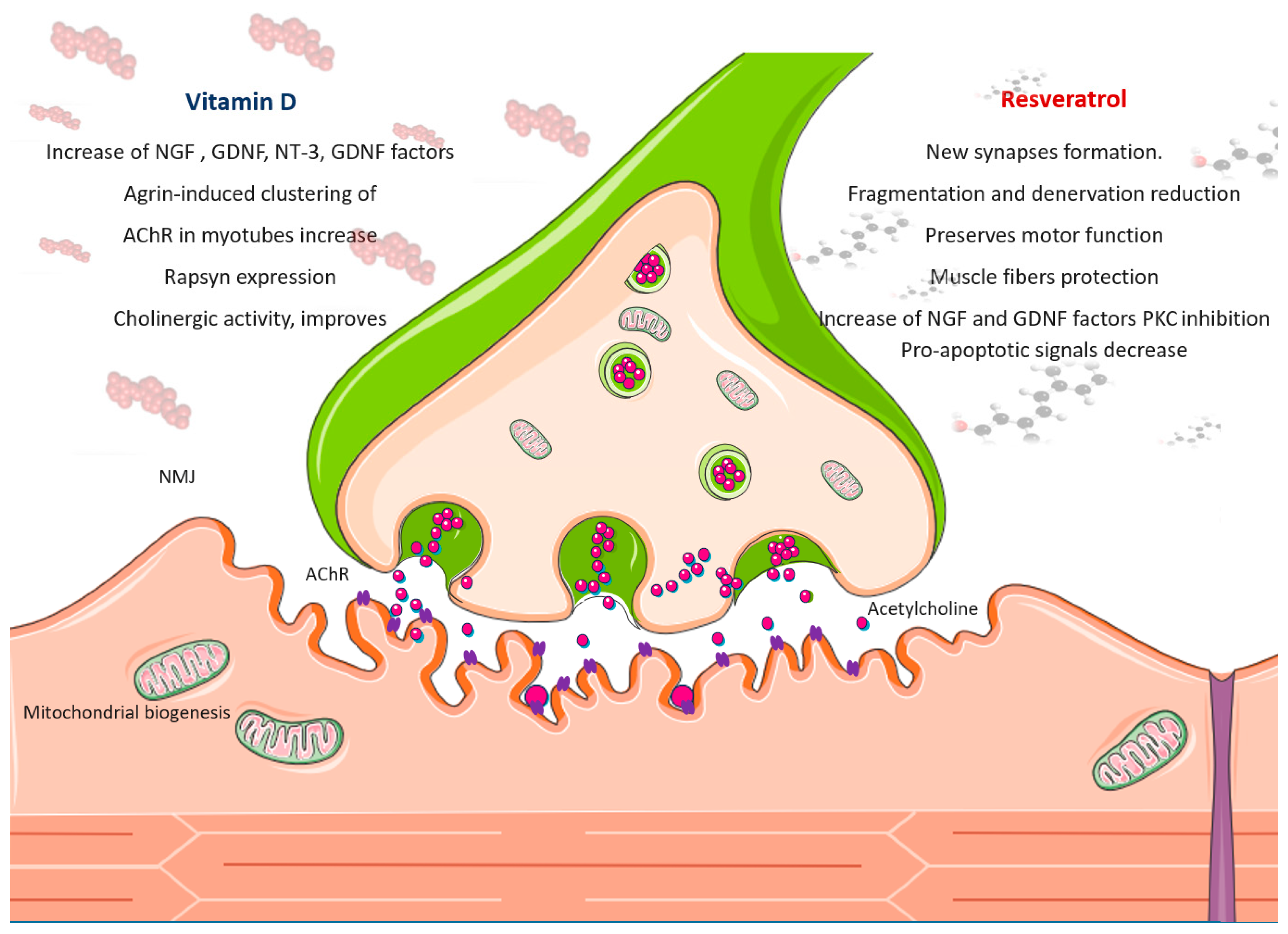
The Effect of Resveratrol on NMJ Function
7. Vitamin D Molecular Mechanisms in Mitochondria Health of Skeletal Muscle
The Effect of Vitamin D on NMJ Function
8. Concluding Remarks and Future Direction
Author Contributions
Funding
Conflicts of Interest
References
- Cruz-Jentoft, A.J.; Sayer, A.A. Sarcopenia. Lancet 2019, 393, 2636–2646. [Google Scholar] [CrossRef] [PubMed]
- Beaudart, C.; Demonceau, C.; Reginster, J.Y.; Locquet, M.; Cesari, M.; Cruz Jentoft, A.J.; Bruyere, O. Sarcopenia and health-related quality of life: A systematic review and meta-analysis. J. Cachexia Sarcopenia Muscle 2023, 14, 1228–1243. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, T.; Ulfhake, B. Sarcopenia: What Is the Origin of This Aging-Induced Disorder? Front. Genet. 2021, 12, 688526. [Google Scholar] [CrossRef] [PubMed]
- Westbury, L.D.; Beaudart, C.; Bruyere, O.; Cauley, J.A.; Cawthon, P.; Cruz-Jentoft, A.J.; Curtis, E.M.; Ensrud, K.; Fielding, R.A.; Johansson, H.; et al. Recent sarcopenia definitions-prevalence, agreement and mortality associations among men: Findings from population-based cohorts. J. Cachexia Sarcopenia Muscle 2023, 14, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Russo, C.; Valle, M.S.; Malaguarnera, L. Antioxidative effects of vitamin D in muscle dysfunction. Redox Exp. Med. 2023, 2023, e230013. [Google Scholar] [CrossRef]
- Prado, C.M.; Batsis, J.A.; Donini, L.M.; Gonzalez, M.C.; Siervo, M. Sarcopenic obesity in older adults: A clinical overview. Nat. Rev. Endocrinol. 2024, 20, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Valle, M.S.; Russo, C.; Casabona, A.; Crimi, N.; Crimi, C.; Colaianni, V.; Cioni, M.; Malaguarnera, L. Anti-inflammatory role of vitamin D in muscle dysfunctions of patients with chronic obstructive pulmonary disease: A comprehensive review. Minerva Medica 2023, 114, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Russo, C.; Valle, M.S.; Casabona, A.; Spicuzza, L.; Sambataro, G.; Malaguarnera, L. Vitamin D Impacts on Skeletal Muscle Dysfunction in Patients with COPD Promoting Mitochondrial Health. Biomedicines 2022, 10, 898. [Google Scholar] [CrossRef]
- Genovese, C.; D’Angeli, F.; Attanasio, F.; Caserta, G.; Scarpaci, K.S.; Nicolosi, D. Phytochemical composition and biological activities of Orobanche crenata Forssk.: A review. Nat. Prod. Res. 2021, 35, 4579–4595. [Google Scholar] [CrossRef]
- Roman, W.; Pinheiro, H.; Pimentel, M.R.; Segales, J.; Oliveira, L.M.; Garcia-Dominguez, E.; Gomez-Cabrera, M.C.; Serrano, A.L.; Gomes, E.R.; Munoz-Canoves, P. Muscle repair after physiological damage relies on nuclear migration for cellular reconstruction. Science 2021, 374, 355–359. [Google Scholar] [CrossRef]
- Lepper, C.; Partridge, T.A.; Fan, C.M. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 2011, 138, 3639–3646. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, V.D.; Punch, V.G.; Kawabe, Y.; Jones, A.E.; Palidwor, G.A.; Porter, C.J.; Cross, J.W.; Carvajal, J.J.; Kockx, C.E.; van Ijcken, W.F.; et al. Transcriptional dominance of Pax7 in adult myogenesis is due to high-affinity recognition of homeodomain motifs. Dev. Cell 2012, 22, 1208–1220. [Google Scholar] [CrossRef] [PubMed]
- Olguin, H.C.; Olwin, B.B. Pax-7 up-regulation inhibits myogenesis and cell cycle progression in satellite cells: A potential mechanism for self-renewal. Dev. Biol. 2004, 275, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Musarò, A. The basis of muscle regeneration. Adv. Biol. 2014, 2014, 612471. [Google Scholar] [CrossRef]
- Hernandez-Hernandez, J.M.; Garcia-Gonzalez, E.G.; Brun, C.E.; Rudnicki, M.A. The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin. Cell Dev. Biol. 2017, 72, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Li, Y.L. Inflammation balance in skeletal muscle damage and repair. Front. Immunol. 2023, 14, 1133355. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, M.; De Gregorio, C.; Malaguarnera, G.; Tuttobene, M.; Biazzo, F.; Malaguarnera, L. Evaluation of AMCase and CHIT-1 expression in monocyte macrophages lineage. Mol. Cell. Biochem. 2013, 374, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, A.; Kawashima, M.; Nagata, I.; Miyoshi, M.; Miyakawa, M.; Sugiyama, M.; Sakuraya, T.; Sonomura, T.; Arakawa, T. Icing after skeletal muscle injury decreases M1 macrophage accumulation and TNF-alpha expression during the early phase of muscle regeneration in rats. Histochem. Cell Biol. 2023, 159, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.M.; Bakkar, N.; Guttridge, D.C. NF-kappaB signaling in skeletal muscle health and disease. Curr. Top. Dev. Biol. 2011, 96, 85–119. [Google Scholar] [CrossRef]
- Guttridge, D.C.; Mayo, M.W.; Madrid, L.V.; Wang, C.Y.; Baldwin, A.S., Jr. NF-kappaB-induced loss of MyoD messenger RNA: Possible role in muscle decay and cachexia. Science 2000, 289, 2363–2366. [Google Scholar] [CrossRef]
- He, W.A.; Berardi, E.; Cardillo, V.M.; Acharyya, S.; Aulino, P.; Thomas-Ahner, J.; Wang, J.; Bloomston, M.; Muscarella, P.; Nau, P.; et al. NF-kappaB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Investig. 2013, 123, 4821–4835. [Google Scholar] [CrossRef] [PubMed]
- Careccia, G.; Mangiavini, L.; Cirillo, F. Regulation of Satellite Cells Functions during Skeletal Muscle Regeneration: A Critical Step in Physiological and Pathological Conditions. Int. J. Mol. Sci. 2023, 25, 512. [Google Scholar] [CrossRef] [PubMed]
- Villalta, S.A.; Rinaldi, C.; Deng, B.; Liu, G.; Fedor, B.; Tidball, J.G. Interleukin-10 reduces the pathology of mdx muscular dystrophy by deactivating M1 macrophages and modulating macrophage phenotype. Hum. Mol. Genet. 2011, 20, 790–805. [Google Scholar] [CrossRef] [PubMed]
- Jejurikar, S.S.; Kuzon, W.M., Jr. Satellite cell depletion in degenerative skeletal muscle. Apoptosis 2003, 8, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, D.; Mourkioti, F.; Gambardella, A.; Kirstetter, P.; Lopez, R.G.; Rosenthal, N.; Nerlov, C. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc. Natl. Acad. Sci. USA 2009, 106, 17475–17480. [Google Scholar] [CrossRef]
- Fu, X.; Xiao, J.; Wei, Y.; Li, S.; Liu, Y.; Yin, J.; Sun, K.; Sun, H.; Wang, H.; Zhang, Z.; et al. Combination of inflammation-related cytokines promotes long-term muscle stem cell expansion. Cell Res. 2015, 25, 655–673. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, S.; Wang, C.; Kuang, S. The hypoxia-inducible factors HIF1alpha and HIF2alpha are dispensable for embryonic muscle development but essential for postnatal muscle regeneration. J. Biol. Chem. 2017, 292, 5981–5991. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, F.; Resmini, G.; Angelino, E.; Ferrara, M.; Tarantino, A.; Piccoli, M.; Rota, P.; Ghiroldi, A.; Monasky, M.M.; Ciconte, G.; et al. HIF-1alpha Directly Controls WNT7A Expression During Myogenesis. Front. Cell Dev. Biol. 2020, 8, 593508. [Google Scholar] [CrossRef]
- Gustafsson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.; Ruas, J.L.; Poellinger, L.; Lendahl, U.; Bondesson, M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell 2005, 9, 617–628. [Google Scholar] [CrossRef]
- Malaguarnera, L. Influence of Resveratrol on the Immune Response. Nutrients 2019, 11, 946. [Google Scholar] [CrossRef]
- Russo, C.; Valle, M.S.; Malaguarnera, L.; Romano, I.R.; Malaguarnera, L. Comparison of Vitamin D and Resveratrol Performances in COVID-19. Nutrients 2023, 15, 2639. [Google Scholar] [CrossRef]
- Alway, S.E.; McCrory, J.L.; Kearcher, K.; Vickers, A.; Frear, B.; Gilleland, D.L.; Bonner, D.E.; Thomas, J.M.; Donley, D.A.; Lively, M.W.; et al. Resveratrol Enhances Exercise-Induced Cellular and Functional Adaptations of Skeletal Muscle in Older Men and Women. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2017, 72, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Montesano, A.; Luzi, L.; Senesi, P.; Mazzocchi, N.; Terruzzi, I. Resveratrol promotes myogenesis and hypertrophy in murine myoblasts. J. Transl. Med. 2013, 11, 310. [Google Scholar] [CrossRef] [PubMed]
- Júnior, I.I.; Zanetti, G.O.; Vieira, T.S.; Albuquerque, F.P.; Gomes, D.A.; Paula-Gomes, S.; Valentim, R.R.; Graca, F.A.; Kettlhut, I.C.; Navegantes, L.C.C.; et al. Resveratrol directly suppresses proteolysis possibly via PKA/CREB signaling in denervated rat skeletal muscle. An. Acad. Bras. Cienc. 2023, 95, e20220877. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, R.; Nakashima, R.; Yano, M.; Iwahara, N.; Asakura, S.; Nojima, I.; Saga, Y.; Kunimoto, R.; Horio, Y.; Kuno, A. Resveratrol, a SIRT1 activator, attenuates aging-associated alterations in skeletal muscle and heart in mice. J. Pharmacol. Sci. 2023, 152, 112–122. [Google Scholar] [CrossRef]
- Wang, D.T.; Yin, Y.; Yang, Y.J.; Lv, P.J.; Shi, Y.; Lu, L.; Wei, L.B. Resveratrol prevents TNF-alpha-induced muscle atrophy via regulation of Akt/mTOR/FoxO1 signaling in C2C12 myotubes. Int. Immunopharmacol. 2014, 19, 206–213. [Google Scholar] [CrossRef]
- Bennett, B.T.; Mohamed, J.S.; Alway, S.E. Effects of resveratrol on the recovery of muscle mass following disuse in the plantaris muscle of aged rats. PLoS ONE 2013, 8, e83518. [Google Scholar] [CrossRef]
- Berman, A.Y.; Motechin, R.A.; Wiesenfeld, M.Y.; Holz, M.K. The therapeutic potential of resveratrol: A review of clinical trials. NPJ Precis. Oncol. 2017, 1, 35. [Google Scholar] [CrossRef]
- Jackson, J.R.; Ryan, M.J.; Alway, S.E. Long-term supplementation with resveratrol alleviates oxidative stress but does not attenuate sarcopenia in aged mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2011, 66, 751–764. [Google Scholar] [CrossRef]
- Meng, X.; Zhou, J.; Zhao, C.N.; Gan, R.Y.; Li, H.B. Health Benefits and Molecular Mechanisms of Resveratrol: A Narrative Review. Foods 2020, 9, 340. [Google Scholar] [CrossRef]
- Kundu, J.K.; Shin, Y.K.; Kim, S.H.; Surh, Y.J. Resveratrol inhibits phorbol ester-induced expression of COX-2 and activation of NF-kappaB in mouse skin by blocking IkappaB kinase activity. Carcinogenesis 2006, 27, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Daiber, A.; Forstermann, U.; Li, H. Antioxidant effects of resveratrol in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1633–1646. [Google Scholar] [CrossRef] [PubMed]
- Qasem, R.J. The estrogenic activity of resveratrol: A comprehensive review of in vitro and in vivo evidence and the potential for endocrine disruption. Crit. Rev. Toxicol. 2020, 50, 439–462. [Google Scholar] [CrossRef] [PubMed]
- Sartori, R.; Romanello, V.; Sandri, M. Mechanisms of muscle atrophy and hypertrophy: Implications in health and disease. Nat. Commun. 2021, 12, 330. [Google Scholar] [CrossRef]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Malhotra, S.; Kumar, A. Nuclear factor-kappa B signaling in skeletal muscle atrophy. J. Mol. Med. 2008, 86, 1113–1126. [Google Scholar] [CrossRef]
- Toniolo, L.; Formoso, L.; Torelli, L.; Crea, E.; Bergamo, A.; Sava, G.; Giacomello, E. Long-term resveratrol treatment improves the capillarization in the skeletal muscles of ageing C57BL/6J mice. Int. J. Food Sci. Nutr. 2021, 72, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Dirks Naylor, A.J. Cellular effects of resveratrol in skeletal muscle. Life Sci. 2009, 84, 637–640. [Google Scholar] [CrossRef]
- Gliemann, L.; Olesen, J.; Bienso, R.S.; Schmidt, J.F.; Akerstrom, T.; Nyberg, M.; Lindqvist, A.; Bangsbo, J.; Hellsten, Y. Resveratrol modulates the angiogenic response to exercise training in skeletal muscles of aged men. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1111–H1119. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, Y.; Chen, J.; Xu, Y. Thrombospondin-1: A Key Protein That Induces Fibrosis in Diabetic Complications. J. Diabetes Res. 2020, 2020, 8043135. [Google Scholar] [CrossRef]
- Toniolo, L.; Concato, M.; Giacomello, E. Resveratrol, a Multitasking Molecule That Improves Skeletal Muscle Health. Nutrients 2023, 15, 3413. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, L. Vitamin D and microbiota: Two sides of the same coin in the immunomodulatory aspects. Int. Immunopharmacol. 2020, 79, 106112. [Google Scholar] [CrossRef] [PubMed]
- Heaney, R.P.; Armas, L.A.; Shary, J.R.; Bell, N.H.; Binkley, N.; Hollis, B.W. 25-Hydroxylation of vitamin D3: Relation to circulating vitamin D3 under various input conditions. Am. J. Clin. Nutr. 2008, 87, 1738–1742. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.J.; Pourshahidi, L.K.; McSorley, E.M.; Madigan, S.M.; Magee, P.J. Vitamin D: Recent advances and implications for athletes. Sports Med. 2015, 45, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Bing, R. Myopathia rachitica. Jb. Kinderheilk. 1908, 68, 649. [Google Scholar]
- Peacock, M. Phosphate Metabolism in Health and Disease. Calcif. Tissue Int. 2021, 108, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Patten, B.M.; Bilezikian, J.P.; Mallette, L.E.; Prince, A.; Engel, W.K.; Aurbach, G.D. Neuromuscular disease in primary hyperparathyroidism. Ann. Intern. Med. 1974, 80, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Pojednic, R.M.; Ceglia, L.; Olsson, K.; Gustafsson, T.; Lichtenstein, A.H.; Dawson-Hughes, B.; Fielding, R.A. Effects of 1,25-dihydroxyvitamin D3 and vitamin D3 on the expression of the vitamin d receptor in human skeletal muscle cells. Calcif. Tissue Int. 2015, 96, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Christakos, S.; Hewison, M.; Gardner, D.G.; Wagner, C.L.; Sergeev, I.N.; Rutten, E.; Pittas, A.G.; Boland, R.; Ferrucci, L.; Bikle, D.D. Vitamin D: Beyond bone. Ann. N. Y. Acad. Sci. 2013, 1287, 45–58. [Google Scholar] [CrossRef]
- Hii, C.S.; Ferrante, A. The Non-Genomic Actions of Vitamin D. Nutrients 2016, 8, 135. [Google Scholar] [CrossRef]
- Dzik, K.P.; Kaczor, J.J. Mechanisms of vitamin D on skeletal muscle function: Oxidative stress, energy metabolism and anabolic state. Eur. J. Appl. Physiol. 2019, 119, 825–839. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.J.; Daniel, Z.C.; Craggs, L.J.; Parr, T.; Brameld, J.M. Dose-dependent effects of vitamin D on transdifferentiation of skeletal muscle cells to adipose cells. J. Endocrinol. 2013, 217, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Garcia, L.A.; King, K.K.; Ferrini, M.G.; Norris, K.C.; Artaza, J.N. 1,25(OH)2vitamin D3 stimulates myogenic differentiation by inhibiting cell proliferation and modulating the expression of promyogenic growth factors and myostatin in C2C12 skeletal muscle cells. Endocrinology 2011, 152, 2976–2986. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J. Targeting the myostatin signaling pathway to treat muscle loss and metabolic dysfunction. J. Clin. Investig. 2021, 131, e148372. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Park, S.; Kim, Y.; Jung, J.; Lee, J.; Chang, Y.; Lee, S.P.; Park, B.C.; Wolfe, R.R.; Choi, C.S.; et al. Myostatin Inhibition-Induced Increase in Muscle Mass and Strength Was Amplified by Resistance Exercise Training, and Dietary Essential Amino Acids Improved Muscle Quality in Mice. Nutrients 2021, 13, 1508. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Ahmad, K.; Shaikh, S.; Lim, J.H.; Chun, H.J.; Ahmad, S.S.; Lee, E.J.; Choi, I. Identification and Evaluation of Traditional Chinese Medicine Natural Compounds as Potential Myostatin Inhibitors: An In Silico Approach. Molecules 2022, 27, 4303. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.S.; Lim, J.H.; Ahmad, K.; Chun, H.J.; Hur, S.J.; Lee, E.J.; Choi, I. Targeting myostatin using quercetin as a media supplement to improve myogenesis for cultured meat production: An in silico and in vitro study. Curr. Res. Food Sci. 2024, 8, 100678. [Google Scholar] [CrossRef] [PubMed]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.R.; Zheng, D.L.; Liu, P.M.; Yang, H.; Li, L.A.; Kuang, S.J.; Lai, Y.Y.; Rao, F.; Xue, Y.M.; Lin, J.J.; et al. High glucose induces Drp1-mediated mitochondrial fission via the Orai1 calcium channel to participate in diabetic cardiomyocyte hypertrophy. Cell Death Dis. 2021, 12, 216. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Dong, H.; Tsai, S.Y. Mitochondrial Properties in Skeletal Muscle Fiber. Cells 2023, 12, 2183. [Google Scholar] [CrossRef] [PubMed]
- Mukund, K.; Subramaniam, S. Skeletal muscle: A review of molecular structure and function, in health and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1462. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Romanello, V.; Sandri, M. The connection between the dynamic remodeling of the mitochondrial network and the regulation of muscle mass. Cell. Mol. Life Sci. 2021, 78, 1305–1328. [Google Scholar] [CrossRef]
- Ng, M.Y.W.; Wai, T.; Simonsen, A. Quality control of the mitochondrion. Dev. Cell 2021, 56, 881–905. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Varuzhanyan, G.; Pham, A.H.; Chan, D.C. Mitochondrial Dynamics is a Distinguishing Feature of Skeletal Muscle Fiber Types and Regulates Organellar Compartmentalization. Cell Metab. 2015, 22, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Favaro, G.; Romanello, V.; Varanita, T.; Andrea Desbats, M.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019, 10, 2576. [Google Scholar] [CrossRef] [PubMed]
- Montecinos-Franjola, F.; Ramachandran, R. Imaging Dynamin-Related Protein 1 (Drp1)-Mediated Mitochondrial Fission in Living Cells. Methods Mol. Biol. 2020, 2159, 205–217. [Google Scholar] [CrossRef]
- Cervantes-Silva, M.P.; Cox, S.L.; Curtis, A.M. Alterations in mitochondrial morphology as a key driver of immunity and host defence. EMBO Rep. 2021, 22, e53086. [Google Scholar] [CrossRef]
- Benefield, D.; Abdelmageed, Y.; Fowler, J.; Smith, S.; Arias-Parbul, K.; Dunning, C.; Rowe, G.C. Adult skeletal muscle peroxisome proliferator-activated receptor gamma -related coactivator 1 is involved in maintaining mitochondrial content. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2023, 324, R470–R479. [Google Scholar] [CrossRef] [PubMed]
- Cannavino, J.; Brocca, L.; Sandri, M.; Bottinelli, R.; Pellegrino, M.A. PGC1-alpha over-expression prevents metabolic alterations and soleus muscle atrophy in hindlimb unloaded mice. J. Physiol. 2014, 592, 4575–4589. [Google Scholar] [CrossRef] [PubMed]
- Talbot, J.; Maves, L. Skeletal muscle fiber type: Using insights from muscle developmental biology to dissect targets for susceptibility and resistance to muscle disease. Wiley Interdiscip. Rev. Dev. Biol. 2016, 5, 518–534. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Chu, C.T.; Kaufman, B.A. The mitochondrial transcription factor TFAM in neurodegeneration: Emerging evidence and mechanisms. FEBS Lett. 2018, 592, 793–811. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, L.; Gonzalez-Alvarado, M.N.; Motalleb, R.; Blomgren, K.; Borjesson, M.; Kuhn, H.G. Constitutive PGC-1alpha overexpression in skeletal muscle does not protect from age-dependent decline in neurogenesis. Sci. Rep. 2019, 9, 12320. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.D.; Fan, S.D.; Chen, X.Y.; Yan, X.L.; Zhang, X.Z.; Ma, B.W.; Yu, D.Y.; Xiao, W.Y.; Zhuang, C.L.; Yu, Z. Nrf2 deficiency exacerbates frailty and sarcopenia by impairing skeletal muscle mitochondrial biogenesis and dynamics in an age-dependent manner. Exp. Gerontol. 2019, 119, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Gouspillou, G.; Godin, R.; Piquereau, J.; Picard, M.; Mofarrahi, M.; Mathew, J.; Purves-Smith, F.M.; Sgarioto, N.; Hepple, R.T.; Burelle, Y.; et al. Protective role of Parkin in skeletal muscle contractile and mitochondrial function. J. Physiol. 2018, 596, 2565–2579. [Google Scholar] [CrossRef]
- Leduc-Gaudet, J.P.; Reynaud, O.; Hussain, S.N.; Gouspillou, G. Parkin overexpression protects from ageing-related loss of muscle mass and strength. J. Physiol. 2019, 597, 1975–1991. [Google Scholar] [CrossRef]
- Wu, D.; Zhang, H.; Li, F.; Liu, S.; Wang, Y.; Zhang, Z.; Wang, J.; Wu, Q. Resveratrol alleviates acute lung injury in mice by promoting Pink1/Parkin-related mitophagy and inhibiting NLRP3 inflammasome activation. Biochim. Biophys. Acta Gen. Subj. 2024, 1868, 130612. [Google Scholar] [CrossRef]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Alway, S.E.; Mohamed, J.S.; Myers, M.J. Mitochondria Initiate and Regulate Sarcopenia. Exerc. Sport Sci. Rev. 2017, 45, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Balaban, R.S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Aung, L.H.; Prabhakar, B.S.; Li, P. The mitochondrial ubiquitin ligase plays an anti-apoptotic role in cardiomyocytes by regulating mitochondrial fission. J. Cell. Mol. Med. 2016, 20, 2278–2288. [Google Scholar] [CrossRef] [PubMed]
- Lira, V.A.; Okutsu, M.; Zhang, M.; Greene, N.P.; Laker, R.C.; Breen, D.S.; Hoehn, K.L.; Yan, Z. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J. 2013, 27, 4184–4193. [Google Scholar] [CrossRef] [PubMed]
- Mofarrahi, M.; Guo, Y.; Haspel, J.A.; Choi, A.M.; Davis, E.C.; Gouspillou, G.; Hepple, R.T.; Godin, R.; Burelle, Y.; Hussain, S.N. Autophagic flux and oxidative capacity of skeletal muscles during acute starvation. Autophagy 2013, 9, 1604–1620. [Google Scholar] [CrossRef] [PubMed]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xiong, W.C.; Mei, L. Neuromuscular Junction Formation, Aging, and Disorders. Annu. Rev. Physiol. 2018, 80, 159–188. [Google Scholar] [CrossRef]
- Vos, M.; Lauwers, E.; Verstreken, P. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front. Synaptic Neurosci. 2010, 2, 139. [Google Scholar] [CrossRef]
- Martinez-Pena, Y.V.I.; Akaaboune, M. The Metabolic Stability of the Nicotinic Acetylcholine Receptor at the Neuromuscular Junction. Cells 2021, 10, 358. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, K.; Spendiff, S.; Lochmuller, H.; Horvath, R. Mitochondrial Mutations Can Alter Neuromuscular Transmission in Congenital Myasthenic Syndrome and Mitochondrial Disease. Int. J. Mol. Sci. 2023, 24, 8505. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.Q.; Mubaraki, A.; Yan, C.Z.; Provias, J.; Tarnopolsky, M.A. Neurogenic Muscle Biopsy Findings Are Common in Mitochondrial Myopathy. J. Neuropathol. Exp. Neurol. 2019, 78, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Sinam, I.S.; Siddique, Z.; Jeon, J.H.; Lee, I.K. The Link between Mitochondrial Dysfunction and Sarcopenia: An Update Focusing on the Role of Pyruvate Dehydrogenase Kinase 4. Diabetes Metab. J. 2023, 47, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Shur, N.F.; Creedon, L.; Skirrow, S.; Atherton, P.J.; MacDonald, I.A.; Lund, J.; Greenhaff, P.L. Age-related changes in muscle architecture and metabolism in humans: The likely contribution of physical inactivity to age-related functional decline. Ageing Res. Rev. 2021, 68, 101344. [Google Scholar] [CrossRef] [PubMed]
- Salucci, S.; Bartoletti Stella, A.; Battistelli, M.; Burattini, S.; Bavelloni, A.; Cocco, L.I.; Gobbi, P.; Faenza, I. How Inflammation Pathways Contribute to Cell Death in Neuro-Muscular Disorders. Biomolecules 2021, 11, 1109. [Google Scholar] [CrossRef] [PubMed]
- Gatti, M.; Dittlau, K.S.; Beretti, F.; Yedigaryan, L.; Zavatti, M.; Cortelli, P.; Palumbo, C.; Bertucci, E.; Van Den Bosch, L.; Sampaolesi, M.; et al. Human Neuromuscular Junction on a Chip: Impact of Amniotic Fluid Stem Cell Extracellular Vesicles on Muscle Atrophy and NMJ Integrity. Int. J. Mol. Sci. 2023, 24, 4944. [Google Scholar] [CrossRef] [PubMed]
- Pratt, J.; De Vito, G.; Narici, M.; Segurado, R.; Pessanha, L.; Dolan, J.; Conroy, J.; Boreham, C. Plasma C-Terminal Agrin Fragment as an Early Biomarker for Sarcopenia: Results From the GenoFit Study. J. Gerontol. Ser. A-Biol. 2021, 76, 2090–2096. [Google Scholar] [CrossRef] [PubMed]
- Gulino, R. Synaptic Dysfunction and Plasticity in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2023, 24, 4613. [Google Scholar] [CrossRef]
- Vecchio, D.; Ramdas, S.; Munot, P.; Pitt, M.; Beeson, D.; Knight, R.; Cruz, P.R.; Vincent, A.; Jayawant, S.; DeVile, C.; et al. Paediatric myasthenia gravis: Prognostic factors for drug free remission. Neuromuscul. Disord. 2020, 30, 120–127. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Martini, M.; Scicchitano, B.M.; Romanello, V.; Boncompagni, S.; Nicoletti, C.; Pietrangelo, L.; De Panfilis, S.; Catizone, A.; Bouche, M.; et al. Muscle Expression of SOD1(G93A) Triggers the Dismantlement of Neuromuscular Junction via PKC-Theta. Antioxid. Redox Signal 2018, 28, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Genin, E.C.; Hounoum, B.M.; Bannwarth, S.; Fragaki, K.; Lacas-Gervais, S.; Mauri-Crouzet, A.; Lespinasse, F.; Neveu, J.; Ropert, B.; Augé, G.; et al. Mitochondrial defect in muscle precedes neuromuscular junction degeneration and motor neuron death in CHCHD10 mouse. Acta Neuropathol. 2019, 138, 123–145. [Google Scholar] [CrossRef] [PubMed]
- So, E.; Mitchell, J.C.; Memmi, C.; Chennell, G.; Vizcay-Barrena, G.; Allison, L.; Shaw, C.E.; Vance, C. Mitochondrial abnormalities and disruption of the neuromuscular junction precede the clinical phenotype and motor neuron loss in hFUS transgenic mice. Hum. Mol. Genet. 2018, 27, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Pareyson, D.; Piscosquito, G.; Moroni, I.; Salsano, E.; Zeviani, M. Peripheral neuropathy in mitochondrial disorders. Lancet Neurol. 2013, 12, 1011–1024. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.Y.; Ranjit, R.; Richardson, A.; Van Remmen, H. Muscle mitochondrial catalase expression prevents neuromuscular junction disruption, atrophy, and weakness in a mouse model of accelerated sarcopenia. J. Cachexia Sarcopenia Muscle 2021, 12, 1582–1596. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Calvani, R.; Coelho, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Bonconpagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal Muscle Is a Primary Target of SOD1G93A-Mediated Toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yi, J.; Fu, R.; Liu, E.; Siddique, T.; Rios, E.; Deng, H.X. Hyperactive intracellular calcium signaling associated with localized mitochondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J. Biol. Chem. 2010, 285, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Serra, C.; Federici, M.; Buongiorno, A.; Senni, M.I.; Morelli, S.; Segratella, E.; Pascuccio, M.; Tiveron, C.; Mattei, E.; Tatangelo, L.; et al. Transgenic mice with dominant negative PKC-theta in skeletal muscle: A new model of insulin resistance and obesity. J. Cell. Physiol. 2003, 196, 89–97. [Google Scholar] [CrossRef]
- Arnold, A.S.; Gill, J.; Christe, M.; Ruiz, R.; McGuirk, S.; St-Pierre, J.; Tabares, L.; Handschin, C. Morphological and functional remodelling of the neuromuscular junction by skeletal muscle PGC-1alpha. Nat. Commun. 2014, 5, 3569. [Google Scholar] [CrossRef]
- Garcia, M.L.; Fernandez, A.; Solas, M.T. Mitochondria, motor neurons and aging. J. Neurol. Sci. 2013, 330, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.S.; Donlin-Asp, P.G.; Leitch, B.; Herzog, E.; Schuman, E.M. Local protein synthesis is a ubiquitous feature of neuronal pre- and postsynaptic compartments. Science 2019, 364, eaau3644. [Google Scholar] [CrossRef] [PubMed]
- Cioni, J.M.; Lin, J.Q.; Holtermann, A.V.; Koppers, M.; Jakobs, M.A.H.; Azizi, A.; Turner-Bridger, B.; Shigeoka, T.; Franze, K.; Harris, W.A.; et al. Late Endosomes Act as mRNA Translation Platforms and Sustain Mitochondria in Axons. Cell 2019, 176, 56–72. [Google Scholar] [CrossRef] [PubMed]
- Carnio, S.; LoVerso, F.; Baraibar, M.A.; Longa, E.; Khan, M.M.; Maffei, M.; Reischl, M.; Canepari, M.; Loefler, S.; Kern, H.; et al. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 2014, 8, 1509–1521. [Google Scholar] [CrossRef]
- Alamdari, N.; Aversa, Z.; Castillero, E.; Gurav, A.; Petkova, V.; Tizio, S.; Hasselgren, P.O. Resveratrol prevents dexamethasone-induced expression of the muscle atrophy-related ubiquitin ligases atrogin-1 and MuRF1 in cultured myotubes through a SIRT1-dependent mechanism. Biochem. Biophys. Res. Commun. 2012, 417, 528–533. [Google Scholar] [CrossRef]
- Mankowski, R.T.; Anton, S.D.; Buford, T.W.; Leeuwenburgh, C. Dietary Antioxidants as Modifiers of Physiologic Adaptations to Exercise. Med. Sci. Sports Exerc. 2015, 47, 1857–1868. [Google Scholar] [CrossRef] [PubMed]
- Shaito, A.; Al-Mansoob, M.; Ahmad, S.M.S.; Haider, M.Z.; Eid, A.H.; Posadino, A.M.; Pintus, G.; Giordo, R. Resveratrol-Mediated Regulation of Mitochondria Biogenesis-associated Pathways in Neurodegenerative Diseases: Molecular Insights and Potential Therapeutic Applications. Curr. Neuropharmacol. 2023, 21, 1184–1201. [Google Scholar] [CrossRef]
- Lombard, D.B.; Tishkoff, D.X.; Bao, J. Mitochondrial sirtuins in the regulation of mitochondrial activity and metabolic adaptation. Handb. Exp. Pharmacol. 2011, 206, 163–188. [Google Scholar] [CrossRef]
- Liao, Z.Y.; Chen, J.L.; Xiao, M.H.; Sun, Y.; Zhao, Y.X.; Pu, D.; Lv, A.K.; Wang, M.L.; Zhou, J.; Zhu, S.Y.; et al. The effect of exercise, resveratrol or their combination on Sarcopenia in aged rats via regulation of AMPK/Sirt1 pathway. Exp. Gerontol. 2017, 98, 177–183. [Google Scholar] [CrossRef]
- Ciccone, L.; Piragine, E.; Brogi, S.; Camodeca, C.; Fucci, R.; Calderone, V.; Nencetti, S.; Martelli, A.; Orlandini, E. Resveratrol-like Compounds as SIRT1 Activators. Int. J. Mol. Sci. 2022, 23, 15105. [Google Scholar] [CrossRef]
- Huang, Y.; Lu, J.; Zhan, L.; Wang, M.; Shi, R.; Yuan, X.; Gao, X.; Liu, X.; Zang, J.; Liu, W.; et al. Resveratrol-induced Sirt1 phosphorylation by LKB1 mediates mitochondrial metabolism. J. Biol. Chem. 2021, 297, 100929. [Google Scholar] [CrossRef] [PubMed]
- Abu Shelbayeh, O.; Arroum, T.; Morris, S.; Busch, K.B. PGC-1alpha Is a Master Regulator of Mitochondrial Lifecycle and ROS Stress Response. Antioxidants 2023, 12, 1075. [Google Scholar] [CrossRef]
- Iside, C.; Scafuro, M.; Nebbioso, A.; Altucci, L. SIRT1 Activation by Natural Phytochemicals: An Overview. Front. Pharmacol. 2020, 11, 1225. [Google Scholar] [CrossRef] [PubMed]
- Higashida, K.; Kim, S.H.; Jung, S.R.; Asaka, M.; Holloszy, J.O.; Han, D.H. Effects of resveratrol and SIRT1 on PGC-1alpha activity and mitochondrial biogenesis: A reevaluation. PLoS Biol. 2013, 11, e1001603. [Google Scholar] [CrossRef]
- Rajamohan, S.B.; Pillai, V.B.; Gupta, M.; Sundaresan, N.R.; Birukov, K.G.; Samant, S.; Hottiger, M.O.; Gupta, M.P. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol. Cell. Biol. 2009, 29, 4116–4129. [Google Scholar] [CrossRef]
- Canto, C.; Sauve, A.A.; Bai, P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol. Asp. Med. 2013, 34, 1168–1201. [Google Scholar] [CrossRef]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Lan, F.; Weikel, K.A.; Cacicedo, J.M.; Ido, Y. Resveratrol-Induced AMP-Activated Protein Kinase Activation Is Cell-Type Dependent: Lessons from Basic Research for Clinical Application. Nutrients 2017, 9, 751. [Google Scholar] [CrossRef]
- Wang, P.; Yang, Y.; Guo, J.; Ma, T.; Hu, Y.; Huang, L.; He, Y.; Xi, J. Resveratrol Inhibits Zinc Deficiency-Induced Mitophagy and Exerts Cardiac Cytoprotective Effects. Biol. Trace Elem. Res. 2024, 202, 1669–1682. [Google Scholar] [CrossRef]
- Li, Y.E.; Sowers, J.R.; Hetz, C.; Ren, J. Cell death regulation by MAMs: From molecular mechanisms to therapeutic implications in cardiovascular diseases. Cell Death Dis. 2022, 13, 504. [Google Scholar] [CrossRef] [PubMed]
- Perrone, M.; Caroccia, N.; Genovese, I.; Missiroli, S.; Modesti, L.; Pedriali, G.; Vezzani, B.; Vitto, V.A.M.; Antenori, M.; Lebiedzinska-Arciszewska, M.; et al. The role of mitochondria-associated membranes in cellular homeostasis and diseases. Int. Rev. Cell Mol. Biol. 2020, 350, 119–196. [Google Scholar] [CrossRef]
- Ren, X.; Chen, L.; Xie, J.; Zhang, Z.; Dong, G.; Liang, J.; Liu, L.; Zhou, H.; Luo, P. Resveratrol Ameliorates Mitochondrial Elongation via Drp1/Parkin/PINK1 Signaling in Senescent-Like Cardiomyocytes. Oxid. Med. Cell. Longev. 2017, 2017, 4175353. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Cui, Y.; Gao, J.L.; Li, R.; Jiang, X.H.; Tian, Y.X.; Wang, K.J.; Li, M.H.; Zhang, H.A.; Cui, J.Z. Neuroprotective effects of resveratrol against traumatic brain injury in rats: Involvement of synaptic proteins and neuronal autophagy. Mol. Med. Rep. 2016, 13, 5248–5254. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, J.; Maxwell, N.; Shapiro, D.; deCabo, R.; Valdez, G. Caloric Restriction Mimetics Slow Aging of Neuromuscular Synapses and Muscle Fibers. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2017, 73, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Bastianetto, S.; Menard, C.; Quirion, R. Neuroprotective action of resveratrol. Biochim. Biophys. Acta 2015, 1852, 1195–1201. [Google Scholar] [CrossRef]
- Hu, C.; Chen, C.; Chen, J.; Xiao, K.; Wang, J.; Shi, Q.; Ma, Y.; Gao, L.P.; Wu, Y.Z.; Liu, L.; et al. The low levels of nerve growth factor and its upstream regulatory kinases in prion infection is reversed by resveratrol. Neurosci. Res. 2021, 162, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Oda, H.; Ohta, S.; Ikeguchi, R.; Noguchi, T.; Kaizawa, Y.; Yurie, H.; Takeuchi, H.; Mitsuzawa, S.; Matsuda, S. Pretreatment of nerve grafts with resveratrol improves axonal regeneration following replantation surgery for nerve root avulsion injury in rats. Restor. Neurol. Neurosci. 2018, 36, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Giordo, R.; Nasrallah, G.K.; Posadino, A.M.; Galimi, F.; Capobianco, G.; Eid, A.H.; Pintus, G. Resveratrol-Elicited PKC Inhibition Counteracts NOX-Mediated Endothelial to Mesenchymal Transition in Human Retinal Endothelial Cells Exposed to High Glucose. Antioxidants 2021, 10, 224. [Google Scholar] [CrossRef]
- Toniolo, L.; Fusco, P.; Formoso, L.; Mazzi, A.; Canato, M.; Reggiani, C.; Giacomello, E. Resveratrol treatment reduces the appearance of tubular aggregates and improves the resistance to fatigue in aging mice skeletal muscles. Exp. Gerontol. 2018, 111, 170–179. [Google Scholar] [CrossRef]
- Widlund, A.L.; Baur, J.A.; Vang, O. mTOR: More targets of resveratrol? Expert. Rev. Mol. Med. 2013, 15, e10. [Google Scholar] [CrossRef] [PubMed]
- Seldeen, K.L.; Berman, R.N.; Pang, M.; Lasky, G.; Weiss, C.; MacDonald, B.A.; Thiyagarajan, R.; Redae, Y.; Troen, B.R. Vitamin D Insufficiency Reduces Grip Strength, Grip Endurance and Increases Frailty in Aged C57Bl/6J Mice. Nutrients 2020, 12, 3005. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Modesti, L.; Danese, A.; Angela Maria Vitto, V.; Ramaccini, D.; Aguiari, G.; Gafa, R.; Lanza, G.; Giorgi, C.; Pinton, P. Mitochondrial Ca(2+) Signaling in Health, Disease and Therapy. Cells 2021, 10, 1317. [Google Scholar] [CrossRef] [PubMed]
- Latham, C.M.; Brightwell, C.R.; Keeble, A.R.; Munson, B.D.; Thomas, N.T.; Zagzoog, A.M.; Fry, C.S.; Fry, J.L. Vitamin D Promotes Skeletal Muscle Regeneration and Mitochondrial Health. Front. Physiol. 2021, 12, 660498. [Google Scholar] [CrossRef] [PubMed]
- van der Meijden, K.; Bravenboer, N.; Dirks, N.F.; Heijboer, A.C.; den Heijer, M.; de Wit, G.M.; Offringa, C.; Lips, P.; Jaspers, R.T. Effects of 1,25(OH)2D3 and 25(OH)D3 on C2C12 Myoblast Proliferation, Differentiation, and Myotube Hypertrophy. J. Cell. Physiol. 2016, 231, 2517–2528. [Google Scholar] [CrossRef]
- Ryan, Z.C.; Craig, T.A.; Folmes, C.D.; Wang, X.; Lanza, I.R.; Schaible, N.S.; Salisbury, J.L.; Nair, K.S.; Terzic, A.; Sieck, G.C.; et al. 1alpha,25-Dihydroxyvitamin D3 Regulates Mitochondrial Oxygen Consumption and Dynamics in Human Skeletal Muscle Cells. J. Biol. Chem. 2016, 291, 1514–1528. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Ismail, A. Vitamin D treatment protects against and reverses oxidative stress induced muscle proteolysis. J. Steroid Biochem. Mol. Biol. 2015, 152, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, R.; Qiao, W.; Zhang, W.; Chen, J.; Mao, L.; Goltzman, D.; Miao, D. 1,25-Dihydroxyvitamin D exerts an antiaging role by activation of Nrf2-antioxidant signaling and inactivation of p16/p53-senescence signaling. Aging Cell 2019, 18, e12951. [Google Scholar] [CrossRef]
- Xiang, Y.; Fu, L.; Xiang, H.X.; Zheng, L.; Tan, Z.X.; Wang, L.X.; Cao, W.; Xu, D.X.; Zhao, H. Correlations among Pulmonary DJ-1, VDR and Nrf-2 in patients with Chronic Obstructive Pulmonary Disease: A Case-control Study. Int. J. Med. Sci. 2021, 18, 2449–2456. [Google Scholar] [CrossRef]
- Mathyssen, C.; Aelbrecht, C.; Serre, J.; Everaerts, S.; Maes, K.; Gayan-Ramirez, G.; Vanaudenaerde, B.; Janssens, W. Local expression profiles of vitamin D-related genes in airways of COPD patients. Respir. Res. 2020, 21, 137. [Google Scholar] [CrossRef] [PubMed]
- Safarpour, P.; Daneshi-Maskooni, M.; Vafa, M.; Nourbakhsh, M.; Janani, L.; Maddah, M.; Amiri, F.S.; Mohammadi, F.; Sadeghi, H. Vitamin D supplementation improves SIRT1, Irisin, and glucose indices in overweight or obese type 2 diabetic patients: A double-blind randomized placebo-controlled clinical trial. BMC Fam. Pract. 2020, 21, 26. [Google Scholar] [CrossRef]
- Nogueiras, R.; Habegger, K.M.; Chaudhary, N.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschop, M.H. Sirtuin 1 and sirtuin 3: Physiological modulators of metabolism. Physiol. Rev. 2012, 92, 1479–1514. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, Z.; Patonai, A.; Simon-Szabo, L.; Takacs, I. Interplay of Vitamin D and SIRT1 in Tissue-Specific Metabolism-Potential Roles in Prevention and Treatment of Non-Communicable Diseases Including Cancer. Int. J. Mol. Sci. 2023, 24, 6154. [Google Scholar] [CrossRef] [PubMed]
- Palacios, O.M.; Carmona, J.J.; Michan, S.; Chen, K.Y.; Manabe, Y.; Ward, J.L., III; Goodyear, L.J.; Tong, Q. Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1alpha in skeletal muscle. Aging 2009, 1, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Chen, K.; Abdel Khalek, W.; Ward, J.L., III; Yang, H.; Chabi, B.; Wrutniak-Cabello, C.; Tong, Q. Regulation of skeletal muscle oxidative capacity and muscle mass by SIRT3. PLoS ONE 2014, 9, e85636. [Google Scholar] [CrossRef]
- Kjobsted, R.; Hingst, J.R.; Fentz, J.; Foretz, M.; Sanz, M.N.; Pehmoller, C.; Shum, M.; Marette, A.; Mounier, R.; Treebak, J.T.; et al. AMPK in skeletal muscle function and metabolism. FASEB J. 2018, 32, 1741–1777. [Google Scholar] [CrossRef]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Brenmoehl, J.; Hoeflich, A. Dual control of mitochondrial biogenesis by sirtuin 1 and sirtuin 3. Mitochondrion 2013, 13, 755–761. [Google Scholar] [CrossRef]
- Chang, E.; Kim, Y. Vitamin D Ameliorates Fat Accumulation with AMPK/SIRT1 Activity in C2C12 Skeletal Muscle Cells. Nutrients 2019, 11, 2806. [Google Scholar] [CrossRef]
- Wang, X.; Hu, S.; Liu, L. Phosphorylation and acetylation modifications of FOXO3a: Independently or synergistically? Oncol. Lett. 2017, 13, 2867–2872. [Google Scholar] [CrossRef] [PubMed]
- Marcus, J.M.; Andrabi, S.A. SIRT3 Regulation Under Cellular Stress: Making Sense of the Ups and Downs. Front. Neurosci. 2018, 12, 799. [Google Scholar] [CrossRef] [PubMed]
- Salles, J.; Chanet, A.; Guillet, C.; Vaes, A.M.; Brouwer-Brolsma, E.M.; Rocher, C.; Giraudet, C.; Patrac, V.; Meugnier, E.; Montaurier, C.; et al. Vitamin D status modulates mitochondrial oxidative capacities in skeletal muscle: Role in sarcopenia. Commun. Biol. 2022, 5, 1288. [Google Scholar] [CrossRef] [PubMed]
- Skv, M.; Abraham, S.M.; Eshwari, O.; Golla, K.; Jhelum, P.; Maity, S.; Komal, P. Tremendous Fidelity of Vitamin D3 in Age-related Neurological Disorders. Mol. Neurobiol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Gezen-Ak, D.; Dursun, E.; Yilmazer, S. The Effect of Vitamin D Treatment On Nerve Growth Factor (NGF) Release From Hippocampal Neurons. Noro Psikiyatr. Ars. 2014, 51, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Neveu, I.; Naveilhan, P.; Baudet, C.; Brachet, P.; Metsis, M. 1,25-dihydroxyvitamin D3 regulates NT-3, NT-4 but not BDNF mRNA in astrocytes. Neuroreport 1994, 6, 124–126. [Google Scholar] [CrossRef] [PubMed]
- Pertile, R.A.N.; Cui, X.; Hammond, L.; Eyles, D.W. Vitamin D regulation of GDNF/Ret signaling in dopaminergic neurons. FASEB J. 2018, 32, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, A.H.; Shojaee, M.; Askari, R.; Abbasian, S.; Gentil, P. The effects of 12 weeks resistance training and vitamin D administration on neuromuscular joint, muscle strength and power in postmenopausal women. Physiol. Behav. 2024, 274, 114419. [Google Scholar] [CrossRef] [PubMed]
- Wrzosek, M.; Lukaszkiewicz, J.; Wrzosek, M.; Jakubczyk, A.; Matsumoto, H.; Piatkiewicz, P.; Radziwon-Zaleska, M.; Wojnar, M.; Nowicka, G. Vitamin D and the central nervous system. Pharmacol. Rep. 2013, 65, 271–278. [Google Scholar] [CrossRef]
- Butikofer, L.; Zurlinden, A.; Bolliger, M.F.; Kunz, B.; Sonderegger, P. Destabilization of the neuromuscular junction by proteolytic cleavage of agrin results in precocious sarcopenia. FASEB J. 2011, 25, 4378–4393. [Google Scholar] [CrossRef]
- Arakawa, M.; Wagatsuma, A. 1alpha, 25(OH)(2)D(3) regulates agrin-induced acetylcholine receptor clustering through upregulation of rapsyn expression in C2C12 myotubes. Biochem. Biophys. Res. Commun. 2020, 525, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Gifondorwa, D.J.; Thompson, T.D.; Wiley, J.; Culver, A.E.; Shetler, P.K.; Rocha, G.V.; Ma, Y.L.; Krishnan, V.; Bryant, H.U. Vitamin D and/or calcium deficient diets may differentially affect muscle fiber neuromuscular junction innervation. Muscle Nerve 2016, 54, 1120–1132. [Google Scholar] [CrossRef] [PubMed]
- Kougias, D.G.; Das, T.; Perez, A.B.; Pereira, S.L. A role for nutritional intervention in addressing the aging neuromuscular junction. Nutr. Res. 2018, 53, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Romeu Montenegro, K.; Amarante Pufal, M.; Newsholme, P. Vitamin D Supplementation and Impact on Skeletal Muscle Function in Cell and Animal Models and an Aging Population: What Do We Know So Far? Nutrients 2021, 13, 1110. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, C.; Valle, M.S.; D’Angeli, F.; Surdo, S.; Malaguarnera, L. Resveratrol and Vitamin D: Eclectic Molecules Promoting Mitochondrial Health in Sarcopenia. Int. J. Mol. Sci. 2024, 25, 7503. https://doi.org/10.3390/ijms25147503
Russo C, Valle MS, D’Angeli F, Surdo S, Malaguarnera L. Resveratrol and Vitamin D: Eclectic Molecules Promoting Mitochondrial Health in Sarcopenia. International Journal of Molecular Sciences. 2024; 25(14):7503. https://doi.org/10.3390/ijms25147503
Chicago/Turabian StyleRusso, Cristina, Maria Stella Valle, Floriana D’Angeli, Sofia Surdo, and Lucia Malaguarnera. 2024. "Resveratrol and Vitamin D: Eclectic Molecules Promoting Mitochondrial Health in Sarcopenia" International Journal of Molecular Sciences 25, no. 14: 7503. https://doi.org/10.3390/ijms25147503
APA StyleRusso, C., Valle, M. S., D’Angeli, F., Surdo, S., & Malaguarnera, L. (2024). Resveratrol and Vitamin D: Eclectic Molecules Promoting Mitochondrial Health in Sarcopenia. International Journal of Molecular Sciences, 25(14), 7503. https://doi.org/10.3390/ijms25147503