
Transcriptional Silencing of 35S rDNA in Tragopogon porrifolius Correlates with Cytosine Methylation in Sequence-Specific Manner



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
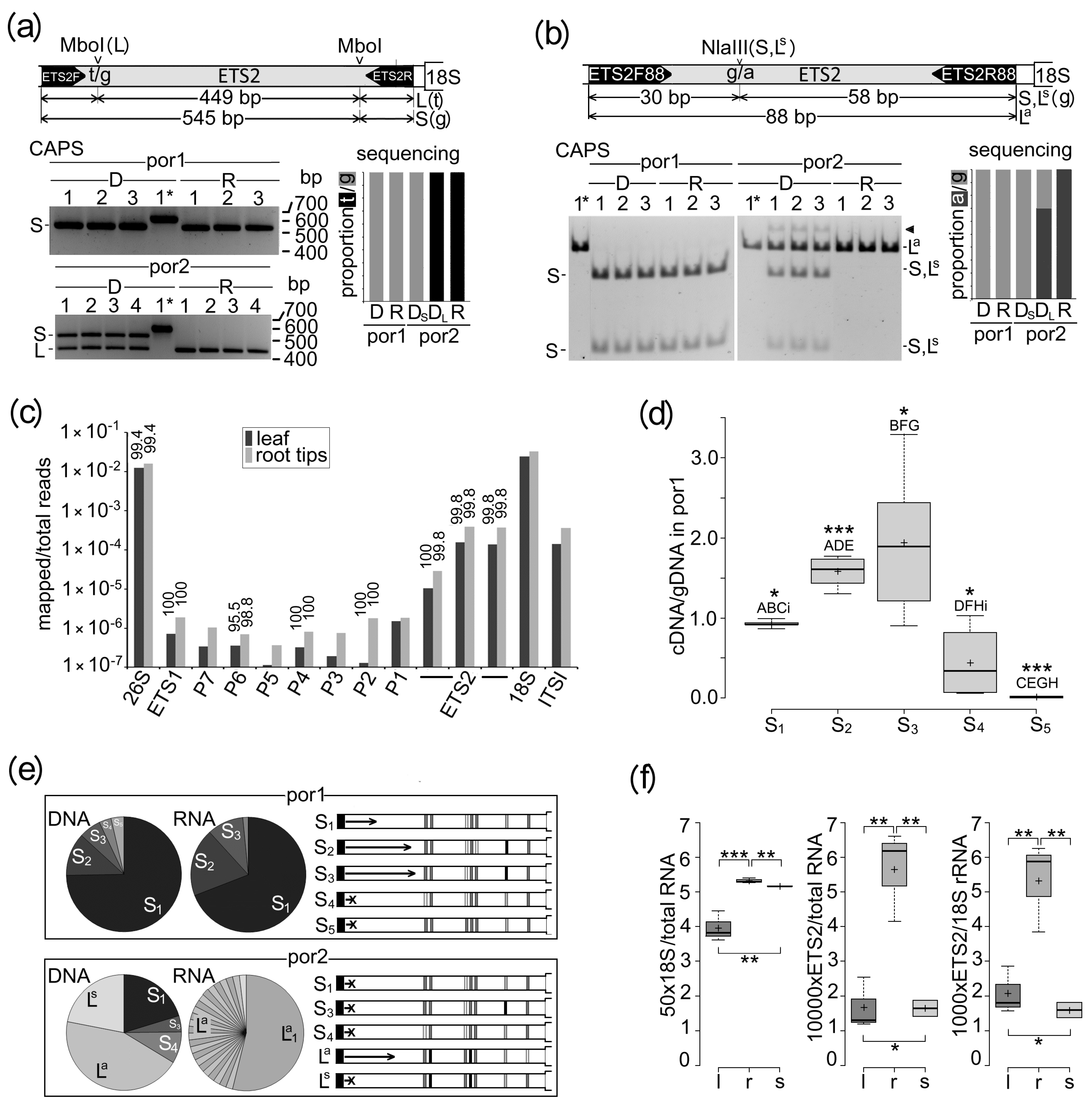
2.1. One Abundant rDNA Variant Was Detected in por1, Whereas Two rDNA Variants Occurred in por2
2.2. Multiple rDNA Variants within Each L- and S-rDNA Array Differed in Transcriptional Activity
2.3. Estimation of the Positions of Individual rDNA Variants within Chromosomal Complement
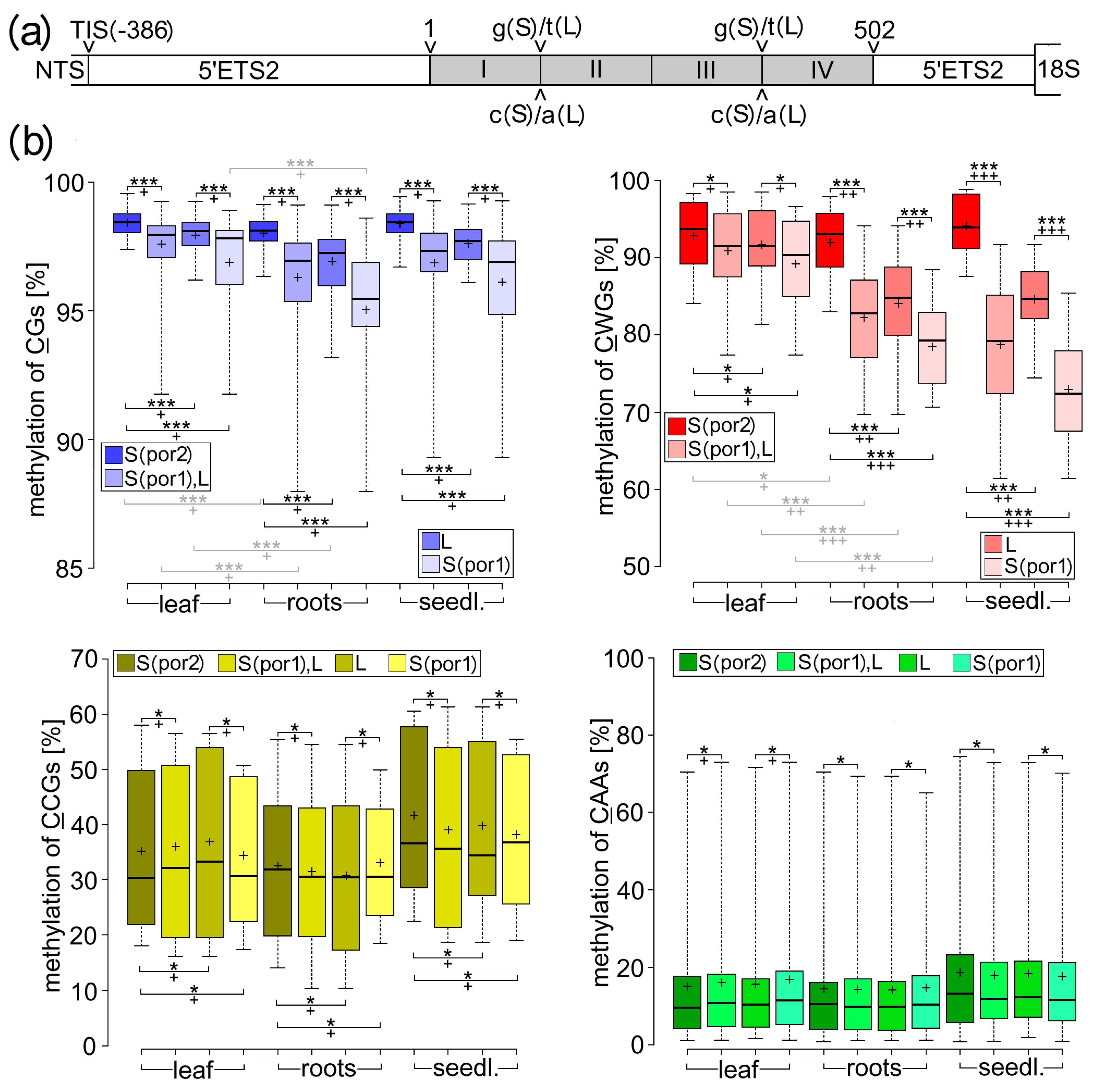
2.4. Transcriptional Variability between Individual rDNA Variants Correlated with Their Cytosine Methylation, Particularly in CWG Motifs
3. Discussion
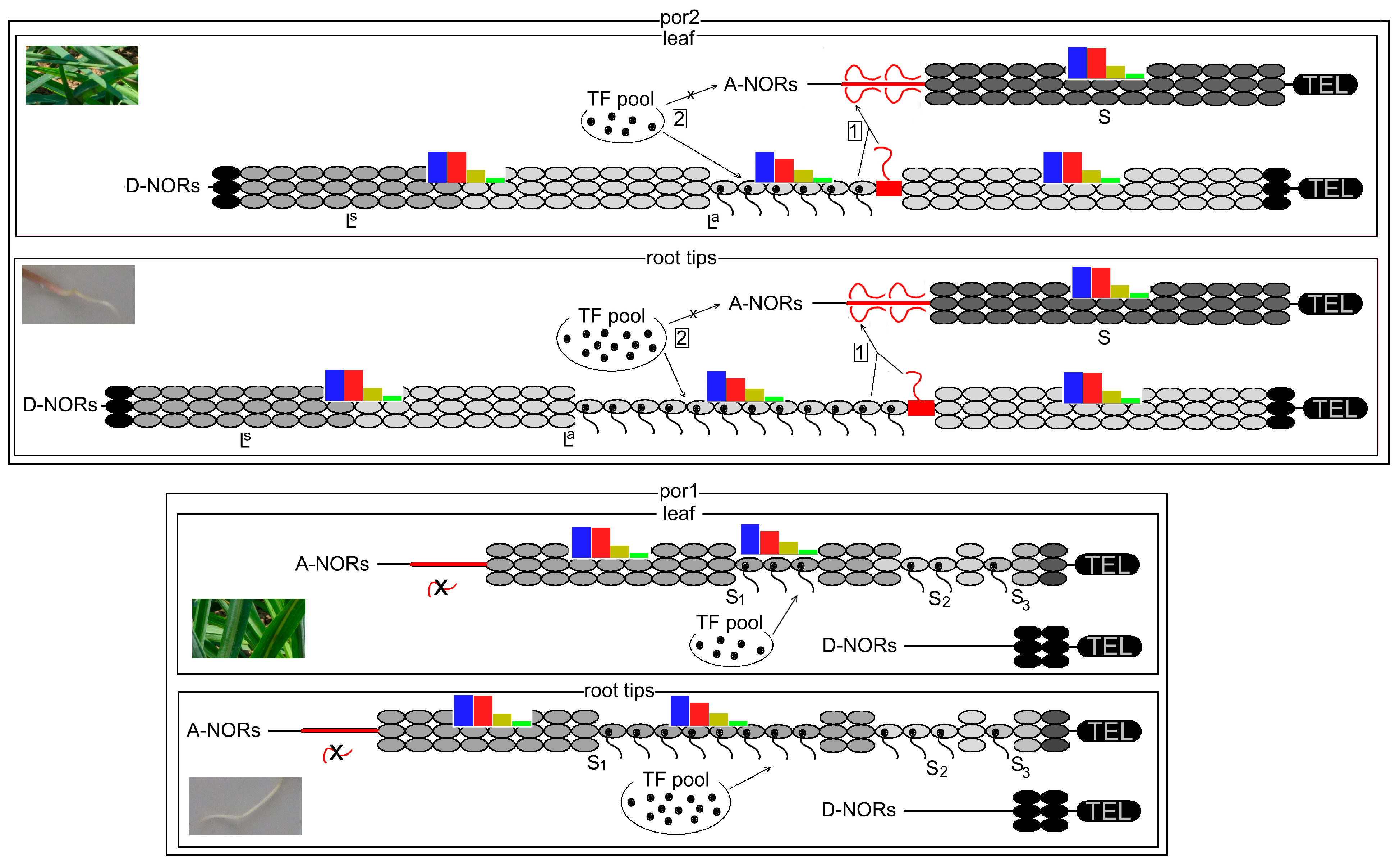
3.1. Bidirectional Nuclear Dominance in Non-Hybrid Diploid Tragopogon porrifolius ssp. Porrifolius
3.1.1. Competition between Promoters (Enhancers) for Limited Regulatory Elements
3.1.2. Intact D-NORs Mediate Silencing of A-NORs
3.1.3. Mutual Locations of Particular rDNA Variants within NORs
3.2. Molecular Mechanisms Leading to Sequence-Dependent Relationships between Cytosine Methylation and rDNA Transcription
4. Material and Methods
4.1. Plant Material and Drug Treatments
4.2. DNA and RNA Extractions and cDNA Preparations
4.3. PCR, CAPS, and Southern Hybridization
4.4. High-Throughput DNA Sequencing
4.5. Sequence Analyses
4.6. Fluorescence In Situ Hybridization
4.7. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Garcia, S.; Kovarik, A.; Leitch, A.R.; Garnatje, T. Cytogenetic features of rRNA genes across land plants: Analysis of the Plant rDNA database. Plant J. 2017, 89, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Rosato, M.; Kovarik, A.; Garilleti, R.; Rossello, J.A. Conserved Organisation of 45S rDNA Sites and rDNA Gene Copy Number among Major Clades of Early Land Plants. PLoS ONE 2016, 11, e0162544. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T. A new role of the rDNA and nucleolus in the nucleus-rDNA instability maintains genome integrity. Bioessays 2008, 30, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Ganley, A.R.D. Recombination regulation by transcription-induced cohesin dissociation in rDNA repeats. Science 2005, 309, 1581–1584. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.; Copenhaver, G.P.; Schlogelhofer, P. Meiotic DNA Repair in the Nucleolus Employs a Nonhomologous End-Joining Mechanism. Plant Cell 2019, 31, 2259–2275. [Google Scholar] [CrossRef] [PubMed]
- Tsang, E.; Carr, A.M. Replication fork arrest, recombination and the maintenance of ribosomal DNA stability. DNA Repair 2008, 7, 1613–1623. [Google Scholar] [CrossRef]
- Long, Q.; Rabanal, F.A.; Meng, D.Z.; Huber, C.D.; Farlow, A.; Platzer, A.; Zhang, Q.R.; Vilhjalmsson, B.J.; Korte, A.; Nizhynska, V.; et al. Massive genomic variation and strong selection in Arabidopsis thaliana lines from Sweden. Nat. Genet. 2013, 45, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Waters, E.R.; Schaal, B.A. Heat shock induces a loss of rRNA-encoding DNA repeats in Brassica nigra. Proc. Natl. Acad. Sci. USA 1996, 93, 1449–1452. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T. Regulation of ribosomal RNA gene copy number and its role in modulating genome integrity and evolutionary adaptability in yeast. Cell. Mol. Life Sci. 2011, 68, 1395–1403. [Google Scholar] [CrossRef]
- Paredes, S.; Branco, A.T.; Hartl, D.L.; Maggert, K.A.; Lemos, B. Ribosomal DNA Deletions Modulate Genome-Wide Gene Expression: “rDNA-Sensitive” Genes and Natural Variation. PLoS Genet. 2011, 7, e1001376. [Google Scholar] [CrossRef] [PubMed]
- Ganley, A.R.D.; Kobayashi, T. Highly efficient concerted evolution in the ribosomal DNA repeats: Total rDNA repeat variation revealed by whole-genome shotgun sequence data. Genome Res. 2007, 17, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Copenhaver, G.P.; Pikaard, C.S. Two-dimensional RFLP analyses reveal megabase-sized clusters of rRNA gene variants in Arabidopsis thaliana, suggesting local spreading of variants as the mode for gene homogenization during concerted evolution. Plant J. 1996, 9, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Havlova, K.; Dvorackova, M.; Peiro, R.; Abia, D.; Mozgova, I.; Vansacova, L.; Gutierrez, C.; Fajkus, J. Variation of 45S rDNA intergenic spacers in Arabidopsis thaliana. Plant Mol. Biol. 2016, 92, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Matyasek, R.; Kuderova, A.; Kutilkova, E.; Kucera, M.; Kovarik, A. Intragenomic heterogeneity of intergenic ribosomal DNA spacers in Cucurbita moschata is determined by DNA minisatellites with variable potential to form non-canonical DNA conformations. DNA Res. 2019, 26, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Galian, J.A.; Rosato, M.; Rossello, J.A. Incomplete sequence homogenization in 45S rDNA multigene families: Intermixed IGS heterogeneity within the single NOR locus of the polyploid species Medicago arborea (Fabaceae). Ann. Bot. 2014, 114, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.; Sestini, G.; Elgert, C.; von Haeseler, A.; Schlogelhofer, P. Sequencing of the Arabidopsis NOR2 reveals its distinct organization and tissue-specific rRNA ribosomal variants. Nat. Commun. 2021, 12, 387. [Google Scholar] [CrossRef] [PubMed]
- Saez-Vasquez, J.; Delseny, M. Ribosome Biogenesis in Plants: From Functional 45S Ribosomal DNA Organization to Ribosome Assembly Factors. Plant Cell 2019, 31, 1945–1967. [Google Scholar] [CrossRef] [PubMed]
- Hemleben, V.; Zentgraf, U. Structural organization and regulation of transcription by RNA polymerase I of plant nuclear ribosomal RNA genes. Plant Promot. Transcr. Factors 1994, 20, 3–24. [Google Scholar]
- Grummt, I. The nucleolus-guardian of cellular homeostasis and genome integrity. Chromosoma 2013, 122, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Tucker, S.; Vitins, A.; Pikaard, C.S. Nucleolar dominance and ribosomal RNA gene silencing. Curr. Opin. Cell Biol. 2010, 22, 351–356. [Google Scholar] [CrossRef]
- Xie, W.B.; Ling, T.; Zhou, Y.G.; Feng, W.J.; Zhu, Q.Y.; Stunnenberg, H.G.; Grummt, I.; Tao, W. The chromatin remodeling complex NuRD establishes the poised state of rRNA genes characterized by bivalent histone modifications and altered nucleosome positions. Proc. Natl. Acad. Sci. USA 2012, 109, 8161–8166. [Google Scholar] [CrossRef]
- Durut, N.; Abou-Ellail, M.; Pontvianne, F.; Das, S.; Kojima, H.; Ukai, S.; de Bures, A.; Comella, P.; Nidelet, S.; Rialle, S.; et al. A Duplicated Nucleolin Gene with Antagonistic Activity Is Required for Chromatin Organization of Silent 45S rDNA in Arabidopsis. Plant Cell 2014, 26, 1330–1344. [Google Scholar] [CrossRef] [PubMed]
- Earley, K.; Lawrence, R.J.; Pontes, O.; Reuther, R.; Enciso, A.J.; Silva, M.; Neves, N.; Gross, M.; Viegas, W.; Pikaard, C.S. Erasure of histone acetylation by Arabidopsis HDA6 mediates large-scale gene silencing in nucleolar dominance. Genes Dev. 2006, 20, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Pontvianne, F.; Blevins, T.; Chandrasekhara, C.; Feng, W.; Stroud, H.; Jacobsen, S.E.; Michaels, S.D.; Pikaard, C.S. Histone methyltransferases regulating rRNA gene dose and dosage control in Arabidopsis. Genes Dev. 2012, 26, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Preuss, S.B.; Costa-Nunes, P.; Tucker, S.; Pontes, O.; Lawrence, R.J.; Mosher, R.; Kasschau, K.D.; Carrington, J.C.; Baulcombe, D.C.; Viegas, W.; et al. Multimegabase Silencing in Nucleolar Dominance Involves siRNA-Directed DNA Methylation and Specific Methylcytosine-Binding Proteins. Mol. Cell 2008, 32, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Schmitz, K.M.; Sandoval, J.; Grummt, I. Intergenic transcripts originating from a subclass of ribosomal DNA repeats silence ribosomal RNA genes in trans. EMBO Rep. 2010, 11, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Pikaard, C.S.; Chandrasekhara, C.; McKinlay, A.; Enganti, R.; Fultz, D. Reaching for the off switch in nucleolar dominance. Plant J. 2023, 115, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H. Cell-type-specific regulation of RNA polymerase I transcription: A new frontier. Bioessays 2006, 28, 719–725. [Google Scholar] [CrossRef]
- Shiba, H.; Takayania, S. RNA silencing systems and their relevance to allele-specific DNA methylation in plants. Biosci. Biotechnol. Biochem. 2007, 71, 2632–2646. [Google Scholar] [CrossRef]
- Schmitz, R.J.; Lewis, Z.A.; Galli, M.G. DNA Methylation: Shared and Divergent Features across Eukaryotes. Trends Genet. 2019, 35, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Disteche, C.M.; Berletch, J.B. X-chromosome inactivation and escape. J. Genet. 2015, 94, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Satyaki, P.R.V.; Gehring, M. DNA methylation and imprinting in plants: Machinery and mechanisms. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Dhayalan, A.; Rajavelu, A.; Rathert, P.; Tamas, R.; Jurkowska, R.Z.; Ragozin, S.; Jeltsch, A. The Dnmt3a PWWP Domain Reads Histone 3 Lysine 36 Trimethylation and Guides DNA Methylation. J. Biol. Chem. 2010, 285, 26114–26120. [Google Scholar] [CrossRef]
- Du, J.M.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, E.; Kleckner, N. DNA sequence homology induces cytosine-to-thymine mutation by a heterochromatin-related pathway in Neurospora. Nat. Genet. 2017, 49, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Matzke, M.A.; Mosher, R.A. RNA-directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, G.H.; Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef]
- Li, Y.; Kumar, S.; Qian, W.Q. Active DNA demethylation: Mechanism and role in plant development. Plant Cell Rep. 2018, 37, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Mavrodiev, E.V.; Soltis, P.S.; Gitzendanner, M.A.; Baldini, R.M.; Soltis, D.E. Polyphyly of Tragopogon porrifolius L. (asteraceae), a European native with intercontinental disjuncts. Int. J. Plant Sci. 2007, 168, 889–904. [Google Scholar] [CrossRef]
- Ownbey, M.; McCollum, G.D. The chromosomes of Tragopogon. Rhodora 1954, 56, 7–21. [Google Scholar]
- Wilson, F.D. Karyotypes of Tragopogon (Compositae, Lactuceae). Brittonia 1983, 35, 341–350. [Google Scholar] [CrossRef]
- Pires, J.C.; Lim, K.Y.; Kovarik, A.; Matyasek, R.; Boyd, A.; Leitch, A.R.; Leitch, I.J.; Bennett, M.D.; Soltis, P.S.; Soltis, D.E. Molecular cytogenetic analysis of recently evolved Tragopogon (Asteraceae) allopolyploids reveal a karyotype that is additive of the diploid progenitors. Am. J. Bot. 2004, 91, 1022–1035. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Pikaard, C.S. Epigenetic silencing of RNA polymerase I transcription: A role for DNA methylation and histone modification in nucleolar dominance. Genes Dev. 1997, 11, 2124–2136. [Google Scholar] [CrossRef] [PubMed]
- Gultepe, M.; Coskuncelebi, K.; Makbul, S.; Vladimirov, V. Chromosome counts of Tragopogon L. (Asteraceae) from Turkey. Caryologia 2015, 68, 193–199. [Google Scholar] [CrossRef]
- Mavrodiev, E.V.; Tancig, M.; Sherwood, A.M.; Gitzendanner, M.A.; Rocca, J.; Soltis, P.S.; Soltis, D.E. Phylogeny of Tragopogon L. (Asteraceae) based on internal and external transcribed spacer sequence data. Int. J. Plant Sci. 2005, 166, 117–133. [Google Scholar] [CrossRef]
- Matyasek, R.; Tate, J.A.; Lim, Y.K.; Srubarova, H.; Koh, J.; Leitch, A.R.; Soltis, D.E.; Soltis, P.S.; Kovarik, A. Concerted evolution of rDNA in recently formed tragopogon allotetraploids is typically associated with an inverse correlation between gene copy number and expression. Genetics 2007, 176, 2509–2519. [Google Scholar] [CrossRef] [PubMed]
- Dobesova, E.; Malinska, H.; Matyasek, R.; Leitch, A.R.; Soltis, D.E.; Soltis, P.S.; Kovarik, A. Silenced rRNA genes are activated and substitute for partially eliminated active homeologs in the recently formed allotetraploid, Tragopogon mirus (Asteraceae). Heredity 2015, 114, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Subrahmanyam, N.C.; Azad, A.A. Nucleoli and Ribosomal-Rna Cistron Numbers in Hordeum Species and Interspecific Hybrids Exhibiting Suppression of Secondary Constriction. Chromosoma 1978, 69, 265–273. [Google Scholar] [CrossRef]
- Maroof, M.A.S.; Allard, R.W.; Zhang, Q.F. Genetic Diversity and Ecogeographical Differentiation among Ribosomal DNA Alleles in Wild and Cultivated Barley. Proc. Natl. Acad. Sci. USA 1990, 87, 8486–8490. [Google Scholar] [CrossRef]
- Felle, M.; Exler, J.H.; Merkl, R.; Dachauer, K.; Brehm, A.; Grummt, I.; Langst, G. DNA sequence encoded repression of rRNA gene transcription in chromatin. Nucleic Acids Res. 2010, 38, 5304–5314. [Google Scholar] [CrossRef] [PubMed]
- Manelyte, L.; Strohner, R.; Gross, T.; Langst, G. Chromatin Targeting Signals, Nucleosome Positioning Mechanism and Non-Coding RNA-Mediated Regulation of the Chromatin Remodeling Complex NoRC. PLoS Genet. 2014, 10, e1004157. [Google Scholar] [CrossRef] [PubMed]
- Putnam, C.D.; Pikaard, C.S. Cooperative Binding of the Xenopus Rna Polymerase-I Transcription Factor Xubf to Repetitive Ribosomal Gene Enhancers. Mol. Cell Biol. 1992, 12, 4970–4980. [Google Scholar] [CrossRef] [PubMed]
- Pikaard, C.S. Ribosomal Gene Promoter Domains Can Function as Artificial Enhancers of Rna-Polymerase-I Transcription, Supporting a Promoter Origin for Natural Enhancers in Xenopus. Proc. Natl. Acad. Sci. USA 1994, 91, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Caudy, A.A.; Pikaard, C.S. Xenopus ribosomal RNA gene Intergenic Spacer elements conferring transcriptional enhancement and nucleolar dominance-like competition in oocytes. J. Biol. Chem. 2002, 277, 31577–31584. [Google Scholar] [CrossRef] [PubMed]
- Reeder, R.H.; Roan, J.G. The Mechanism of Nucleolar Dominance in Xenopus Hybrids. Cell 1984, 38, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Zentgraf, U.; Hemleben, V. Complex-Formation of Nuclear Proteins with the Rna Polymerase-I Promoter and Repeated Elements in the External Transcribed Spacer of Cucumis-Sativus Ribosomal DNA. Nucleic Acids Res. 1992, 20, 3685–3691. [Google Scholar] [CrossRef] [PubMed]
- Komarova, N.Y.; Grimm, G.W.; Hemleben, V.; Volkov, R.A. Molecular evolution of 35S rDNA and taxonomic status of Lycopersicon within Solanum sect. Petota. Plant Syst. Evol. 2008, 276, 59–71. [Google Scholar] [CrossRef]
- Martini, G.; Odell, M.; Flavell, R.B. Partial Inactivation of Wheat Nucleolus Organizers by the Nucleolus Organizer Chromosomes from Aegilops-Umbellulata. Chromosoma 1982, 84, 687–700. [Google Scholar] [CrossRef]
- Silva, M.; Pereira, H.S.; Bento, M.; Santos, A.P.; Shaw, P.; Delgado, M.; Neves, N.; Viegas, W. Interplay of Ribosomal DNA Loci in Nucleolar Dominance: Dominant NORs are Up-Regulated by Chromatin Dynamics in the Wheat-Rye System. PLoS ONE 2008, 3, e3824. [Google Scholar] [CrossRef] [PubMed]
- Matyasek, R.; Dobesova, E.; Huska, D.; Jezkova, I.; Soltis, P.S.; Soltis, D.E.; Kovarik, A. Interpopulation hybridization generates meiotically stable rDNA epigenetic variants in allotetraploid Tragopogon mirus. Plant J. 2016, 85, 362–377. [Google Scholar] [CrossRef] [PubMed]
- Komarova, N.Y.; Grabe, T.; Huigen, D.J.; Hemleben, V.; Volkov, R.A. Organization, differential expression and methylation of rDNA in artificial Solanum allopolyploids. Plant Mol. Biol. 2004, 56, 439–463. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Pikaard, C.S. Transcriptional analysis of nucleolar dominance in polyploid plants: Biased expression/silencing of progenitor rRNA genes is developmentally regulated in Brassica. Proc. Natl. Acad. Sci. USA 1997, 94, 3442–3447. [Google Scholar] [CrossRef] [PubMed]
- Wanzenbock, E.M.; Schofer, C.; Schweizer, D.; Bachmair, A. Ribosomal transcription units integrated via T-DNA transformation associate with the nucleolus and do not require upstream repeat sequences for activity in Arabidopsis thaliana. Plant J. 1997, 11, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Lopez, F.B.; Fort, A.; Tadini, L.; Probst, A.V.; McHale, M.; Friel, J.; Ryder, P.; Pontvianne, F.; Pesaresi, P.; Sulpice, R.; et al. Gene dosage compensation of rRNA transcript levels in Arabidopsis thaliana lines with reduced ribosomal gene copy number. Plant Cell 2021, 33, 1135–1150. [Google Scholar] [CrossRef] [PubMed]
- Matzke, M.; Kanno, T.; Claxinger, L.; Huettel, B.; Matzke, A.J.M. RNA-mediated chromatin-based silencing in plants. Curr. Opin. Cell Biol. 2009, 21, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Abou-Ellail, M.; Cooke, R.; Saez-Vasquez, J. Variations on a team Major and minor variants of Arabidopsis thaliana rDNA genes. Nucleus 2011, 2, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Earley, K.W.; Pontvianne, F.; Wierzbicki, A.T.; Blevins, T.; Tucker, S.; Costa-Nunes, P.; Pontes, O.; Pikaard, C.S. Mechanisms of HDA6-mediated rRNA gene silencing: Suppression of intergenic Pol II transcription and differential effects on maintenance versus siRNA-directed cytosine methylation. Genes Dev. 2010, 24, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Vydzhak, O.; Luke, B.; Schindler, N. Non-coding RNAs at the Eukaryotic rDNA Locus: RNA-DNA Hybrids and Beyond. J. Mol. Biol. 2020, 432, 4287–4304. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, M.; Yamagata, H.; Kamiya, K.; Nakamura, M.; Saji, S.; Kanamori, H.; Wu, J.Z.; Matsumoto, T.; Sasaki, T. Sequence comparison of distal and proximal ribosomal DNA arrays in rice (Oryza sativa L.) chromosome 9S and analysis of their flanking regions. Theor. Appl. Genet. 2006, 113, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Chester, M.; Sykorova, E.; Fajkus, J.; Leitch, A.R. Single Integration and Spread of a Copia-Like Sequence Nested in rDNA Intergenic Spacers of Allium cernuum (Alliaceae). Cytogenet. Genome Res. 2010, 129, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.C.; Zhang, X.Z.; Garcia, S.; Leitch, A.R.; Kovarik, A. Intragenomic rDNA variation-the product of concerted evolution, mutation, or something in between? Heredity 2023, 131, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Kovarik, A.; Dadejova, M.; Lim, Y.K.; Chase, M.W.; Clarkson, J.J.; Knapp, S.; Leitch, A.R. Evolution of rDNA in allopolyploids: A potential link between rDNA homogenization and epigenetics. Ann. Bot. 2008, 101, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.W.; Liu, J.; Liu, F.Q.; Zhong, X.H. Depositing centromere repeats induces heritable intragenic heterochromatin establishment and spreading in Arabidopsis. Nucleic Acids Res. 2023, 51, 6039–6054. [Google Scholar] [CrossRef]
- Fultz, D.; Mckinlay, A.; Enganti, R.; Pikaard, C.S. Sequence and epigenetic landscapes of active and silent nucleolus organizer regions in. Sci. Adv. 2023, 9, eadj4509. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Han, F.P. Asymmetric Epigenetic Modification and Elimination of rDNA Sequences by Polyploidization in Wheat. Plant Cell 2014, 26, 4311–4327. [Google Scholar] [CrossRef] [PubMed]
- Domb, K.; Katz, A.; Harris, K.D.; Yaari, R.; Kaisler, E.; Nguyen, V.H.; Hong, U.V.T.; Griess, O.; Heskiau, K.G.; Ohad, N.; et al. DNA methylation mutants in Physcomitrella patens elucidate individual roles of CG and non-CG methylation in genome regulation. Proc. Natl. Acad. Sci. USA 2020, 117, 33700–33710. [Google Scholar] [CrossRef] [PubMed]
- Zabet, N.R.; Catoni, M.; Prischi, F.; Paszkowski, J. Cytosine methylation at CpCpG sites triggers accumulation of non-CpG methylation in gene bodies. Nucleic Acids Res. 2017, 45, 3777–3784. [Google Scholar] [CrossRef] [PubMed]
- Bewick, A.J.; Ji, L.X.; Niederhuth, C.E.; Willing, E.M.; Hofmeister, B.T.; Shi, X.L.; Wang, L.; Lu, Z.F.; Rohr, N.A.; Hartwig, B.; et al. On the origin and evolutionary consequences of gene body DNA methylation. Proc. Natl. Acad. Sci. USA 2016, 113, 9111–9116. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.G.; Zhang, H.M.; Julian, R.; Tang, K.; Xie, S.J.; Zhu, J.K. Regulatory link between DNA methylation and active demethylation in. Proc. Natl. Acad. Sci. USA 2015, 112, 3553–3557. [Google Scholar] [CrossRef]
- Cao, X.F.; Jacobsen, S.E. Role of the arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. 2002, 12, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.H.; Du, J.M.; Hale, C.J.; Gallego-Bartolome, J.; Feng, S.H.; Vashisht, A.A.; Chory, J.; Wohlschlegel, J.A.; Patel, D.J.; Jacobsen, S.E. Molecular Mechanism of Action of Plant DRM De Novo DNA Methyltransferases. Cell 2014, 157, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Saze, H.; Scheid, O.M.; Paszkowski, J. Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat. Genet. 2003, 34, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Borges, F.; Donoghue, M.T.A.; LeBlanc, C.; Wear, E.E.; Tanurdzic, M.; Berube, B.; Brooks, A.; Thompson, W.F.; Hanley-Bowdoin, L.; Martienssen, R.A. Loss of Small-RNA-Directed DNA Methylation in the Plant Cell Cycle Promotes Germline Reprogramming and Somaclonal Variation. Curr. Biol. 2021, 31, 591–600.e4. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.C.; Yu, C.W.; Duan, J.; Luo, M.; Wang, K.C.; Tian, G.; Cui, Y.H.; Wu, K.Q. HDA6 Directly Interacts with DNA Methyltransferase MET1 and Maintains Transposable Element Silencing in Arabidopsis. Plant Physiol. 2012, 158, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Du, J.M.; Zhong, X.H.; Bernatavichute, Y.V.; Stroud, H.; Feng, S.H.; Caro, E.; Vashisht, A.A.; Terragni, J.; Chin, H.G.; Tu, A.; et al. Dual Binding of Chromomethylase Domains to H3K9me2-Containing Nucleosomes Directs DNA Methylation in Plants. Cell 2012, 151, 167–180. [Google Scholar] [CrossRef]
- Inagaki, S.; Miura-Kamio, A.; Nakamura, Y.; Lu, F.L.; Cui, X.; Cao, X.F.; Kimura, H.; Saze, H.; Kakutani, T. Autocatalytic differentiation of epigenetic modifications within the Arabidopsis genome. EMBO J. 2010, 29, 3496–3506. [Google Scholar] [CrossRef] [PubMed]
- Widman, N.; Feng, S.; Jacobsen, S.E.; Pellegrini, M. Epigenetic differences between shoots and roots in Arabidopsis reveals tissue-specific regulation. Epigenetics 2014, 9, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Gouil, Q.; Baulcombe, D.C. DNA Methylation Signatures of the Plant Chromomethyltransferases. PLoS Genet. 2016, 12, e1006526. [Google Scholar] [CrossRef] [PubMed]
- Yaari, R.; Noy-Malka, C.; Wiedemann, G.; Gershovitz, N.A.; Reski, R.; Katz, A.; Ohad, N. DNA Methyltransferase 1 is involved in (m)CG and (m)CCG DNA methylation and is essential for sporophyte development in Physcomitrella patens. Plant. Mol. Biol. 2015, 88, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Stroud, H.; Do, T.; Du, J.M.; Zhong, X.H.; Feng, S.H.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-CG methylation patterns shape the epigenetic landscape in. Nat. Struct. Mol. Biol. 2014, 21, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Eichten, S.R.; Hermanson, P.J.; Zaunbrecher, V.M.; Song, J.W.; Wendt, J.; Rosenbaum, H.; Madzima, T.F.; Sloan, A.E.; Huang, J.; et al. Genetic Perturbation of the Maize Methylome. Plant Cell 2014, 26, 4602–4616. [Google Scholar] [CrossRef] [PubMed]
- Kovarik, A.; Van Houdt, H.; Holy, A.; Depicker, A. Drug-induced hypomethylation of a posttranscriptionally silenced transgene locus of tobacco leads to partial release of silencing. FEBS Lett. 2000, 467, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Miao, Y.X.; Cai, B.T.; Yi, Q.P.; Tian, X.J.; Wang, Q.H.; Ma, D.; Luo, Q.; Tan, F.; Hu, Y.F. A histone deacetylase inhibitor enhances rice immunity by derepressing the expression of defense-related genes. Front. Plant Sci. 2022, 13, 1041095. [Google Scholar] [CrossRef]
- Matyasek, R.; Fulnecek, J.; Leitch, A.R.; Kovarik, A. Analysis of two abundant, highly related satellites in the allotetraploid Nicotiana arentsii using double-strand conformation polymorphism analysis and sequencing. New Phytol. 2011, 192, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, G.; Hasterok, R. BAC ‘landing’ on chromosomes of Brachypodium distachyon for comparative genome alignment. Nat. Protoc. 2007, 2, 88–98. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matyášek, R.; Kalfusová, R.; Kuderová, A.; Řehůřková, K.; Sochorová, J.; Kovařík, A. Transcriptional Silencing of 35S rDNA in Tragopogon porrifolius Correlates with Cytosine Methylation in Sequence-Specific Manner. Int. J. Mol. Sci. 2024, 25, 7540. https://doi.org/10.3390/ijms25147540
Matyášek R, Kalfusová R, Kuderová A, Řehůřková K, Sochorová J, Kovařík A. Transcriptional Silencing of 35S rDNA in Tragopogon porrifolius Correlates with Cytosine Methylation in Sequence-Specific Manner. International Journal of Molecular Sciences. 2024; 25(14):7540. https://doi.org/10.3390/ijms25147540
Chicago/Turabian StyleMatyášek, Roman, Radka Kalfusová, Alena Kuderová, Kateřina Řehůřková, Jana Sochorová, and Aleš Kovařík. 2024. "Transcriptional Silencing of 35S rDNA in Tragopogon porrifolius Correlates with Cytosine Methylation in Sequence-Specific Manner" International Journal of Molecular Sciences 25, no. 14: 7540. https://doi.org/10.3390/ijms25147540
APA StyleMatyášek, R., Kalfusová, R., Kuderová, A., Řehůřková, K., Sochorová, J., & Kovařík, A. (2024). Transcriptional Silencing of 35S rDNA in Tragopogon porrifolius Correlates with Cytosine Methylation in Sequence-Specific Manner. International Journal of Molecular Sciences, 25(14), 7540. https://doi.org/10.3390/ijms25147540