
Identifying Diagnostic Markers and Constructing Predictive Models for Oxidative Stress in Multiple Sclerosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Result
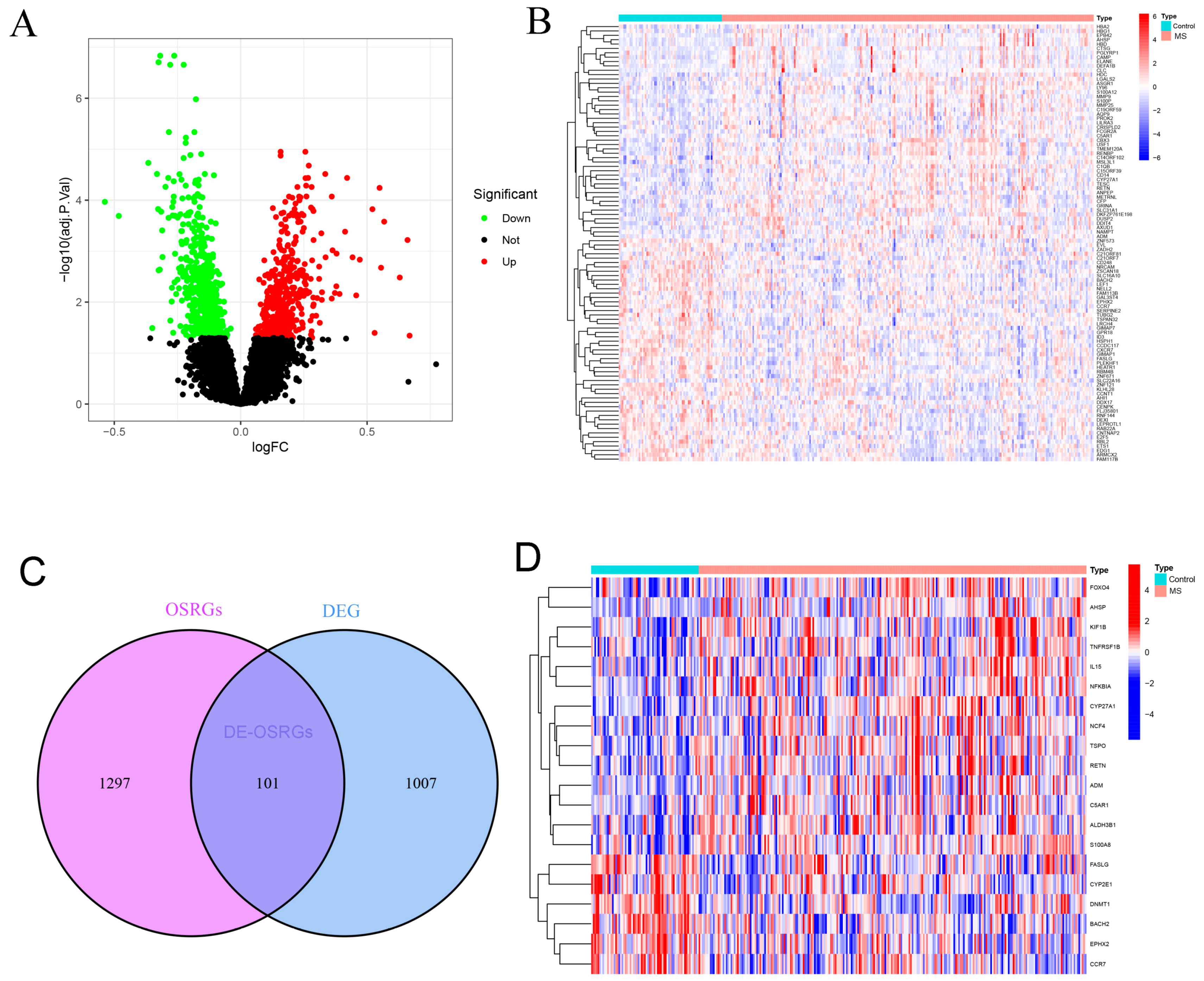
2.1. Identification of DE-OSRGs in Multiple Sclerosis
2.2. Functional Enrichment Analysis of DE-OSRGs in Multiple Sclerosis
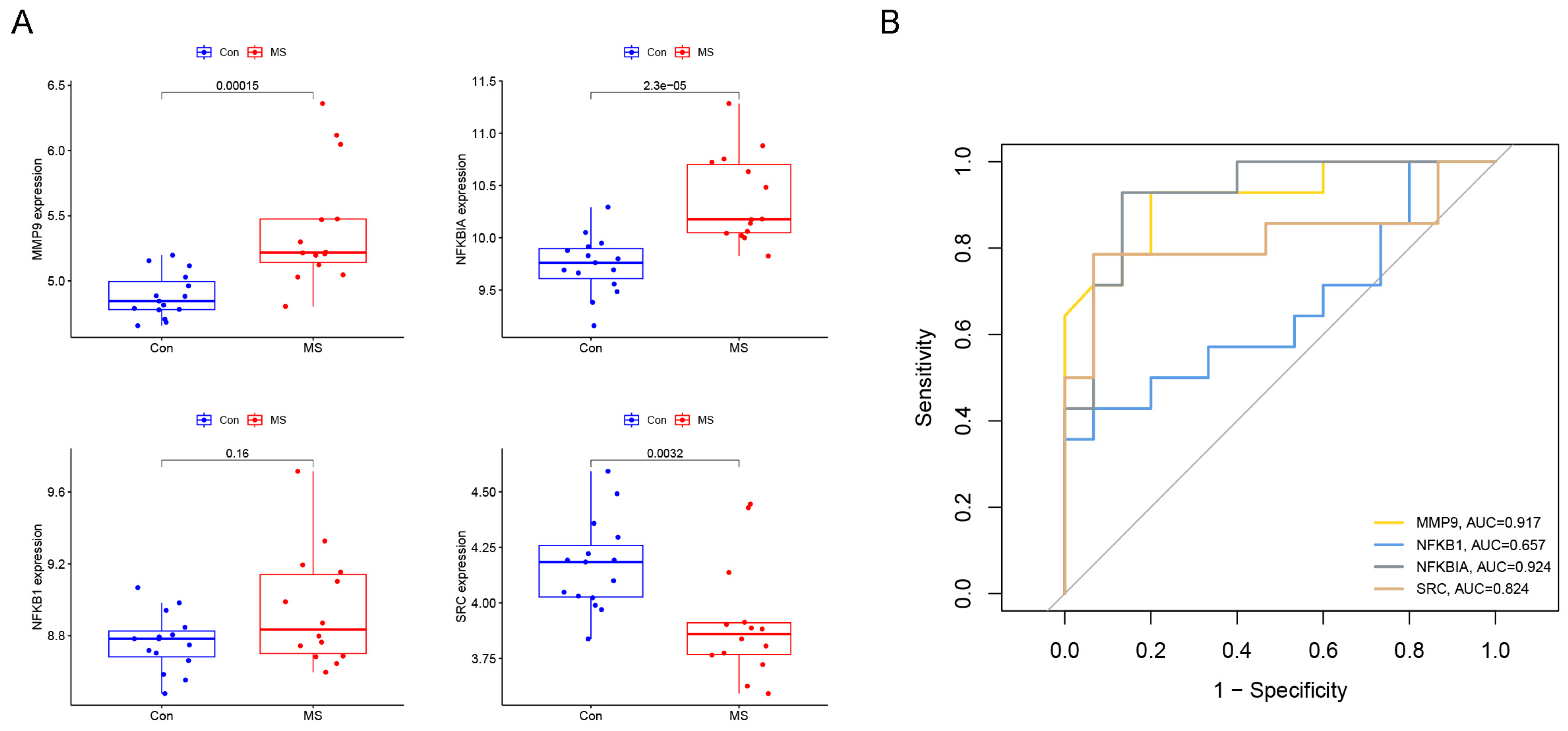
2.3. Identification of Diagnostic Genes for Multiple Sclerosis
2.4. Establishment and Evaluation of Multiple Sclerosis Diagnostic Prediction Model
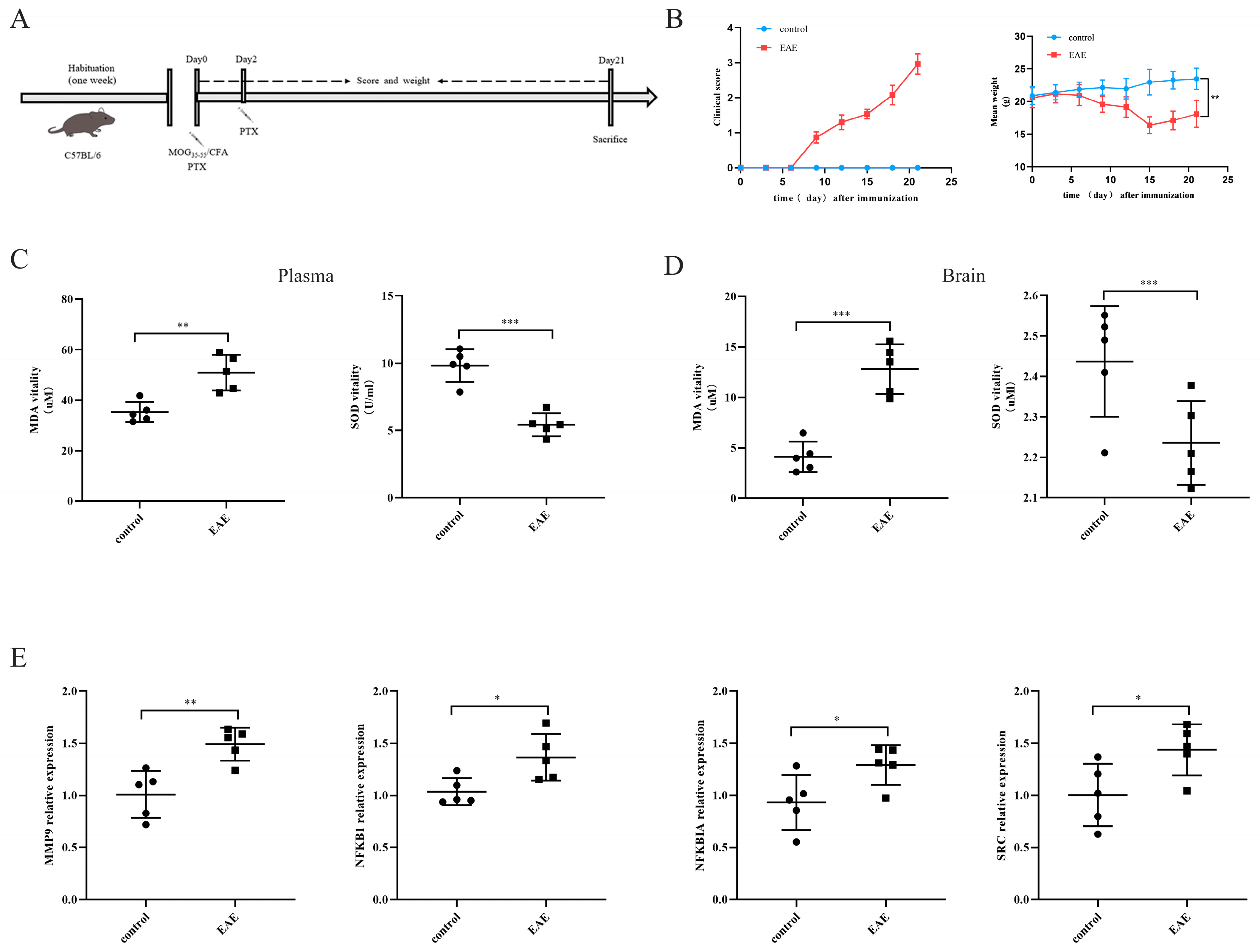
2.5. Expressions of the Four Characteristic Genes in the EAE Model
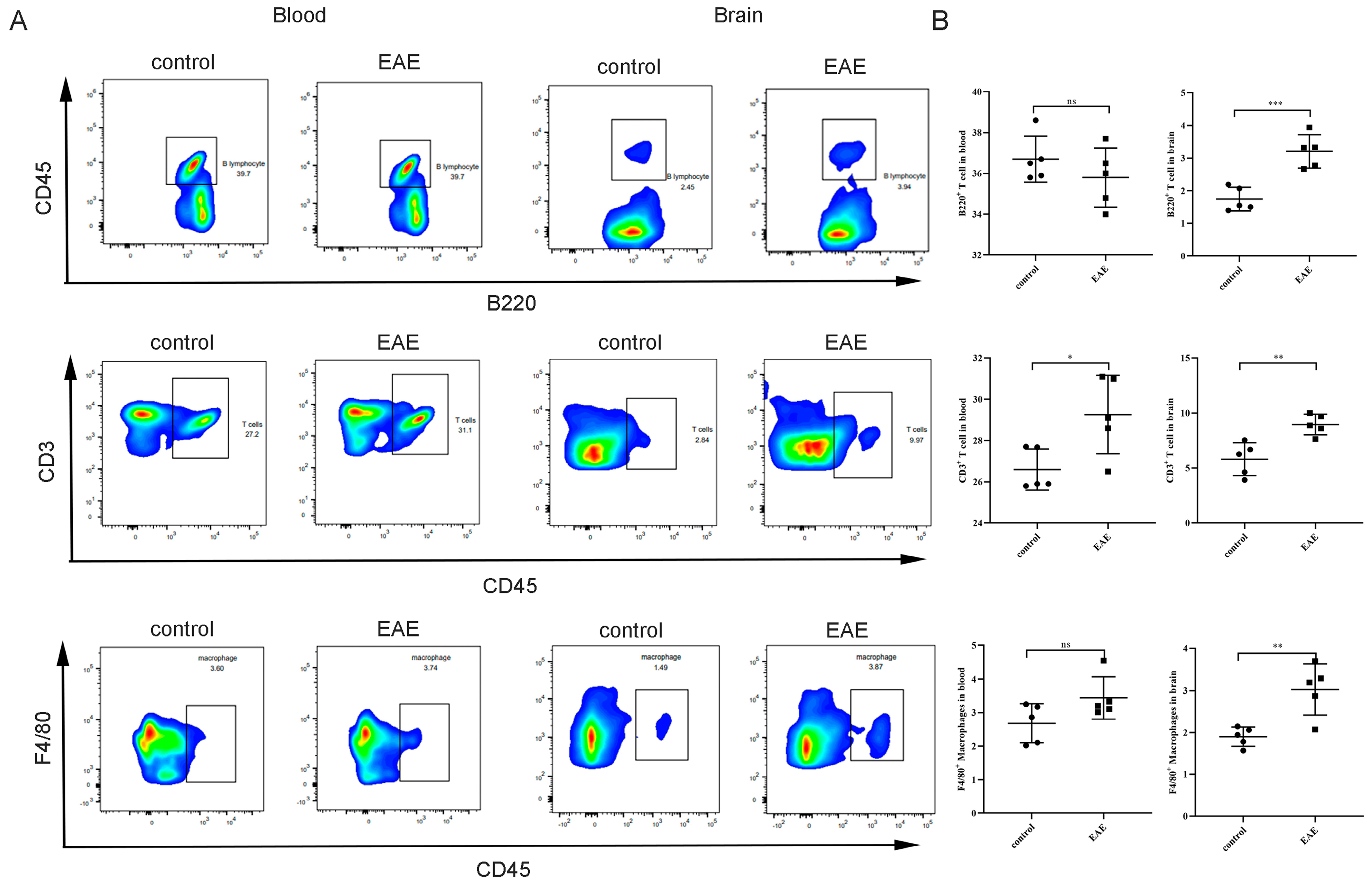
2.6. Association between Characteristic Genes and the Proportion of Infiltrating Immune Cells
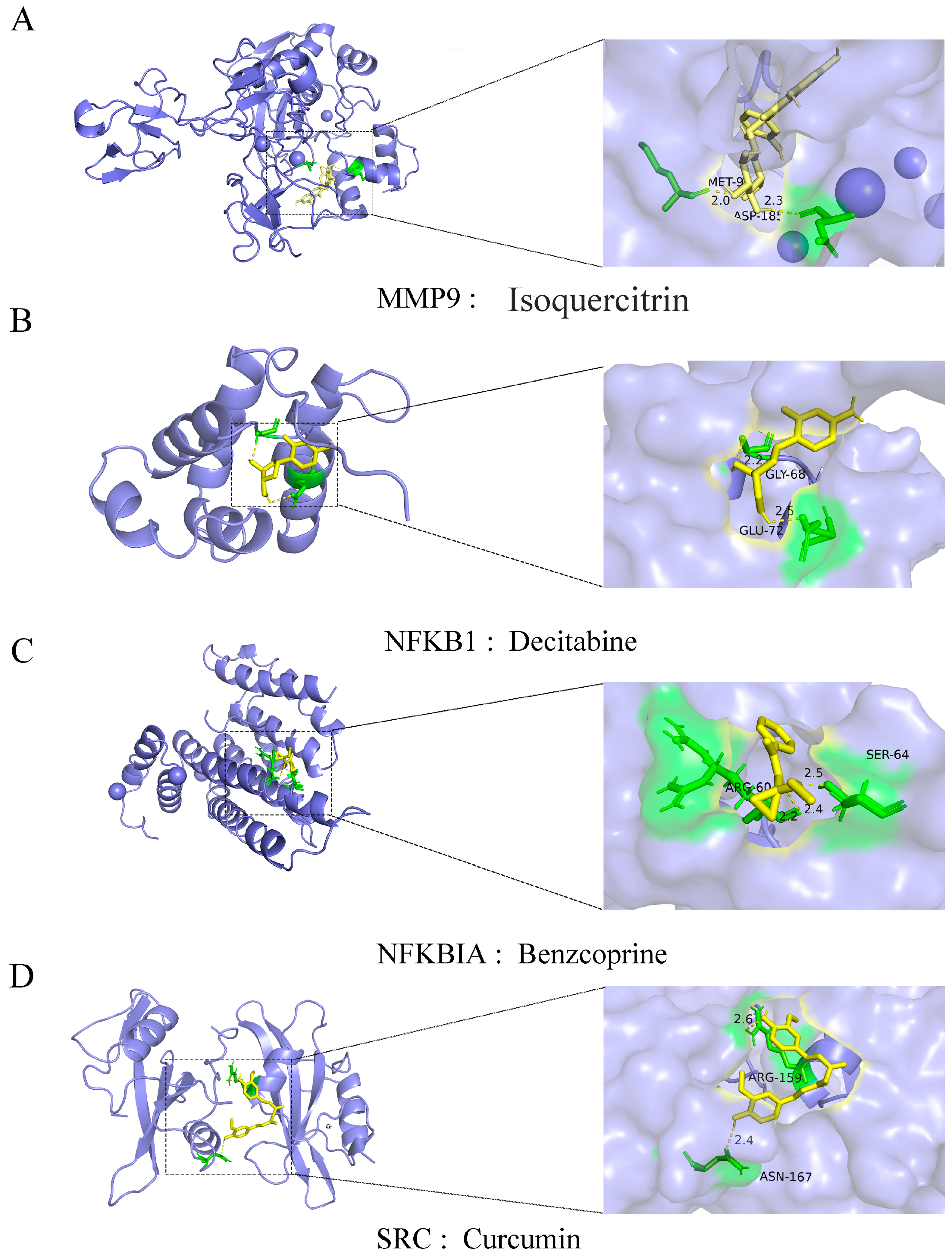
2.7. Feature Genes and Small Molecular Compound Docking
3. Discussion
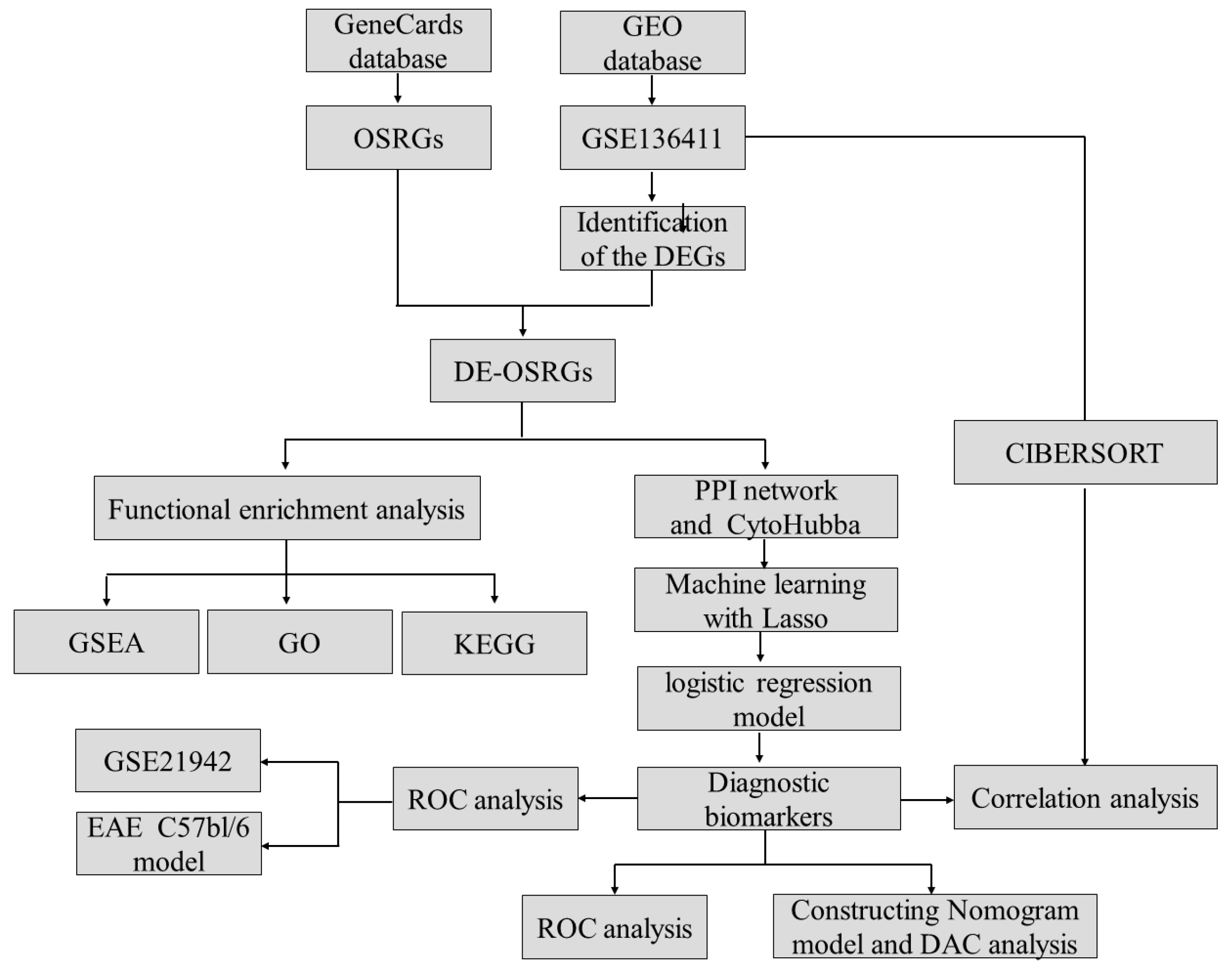
4. Materials and Methods
4.1. Data Sources
4.2. Identification of Differentially Expressed Genes (DEGs)
4.3. Pathway Enrichment Analysis
4.4. Optimal Diagnostic Biomarker Selection and Model Building
4.5. Immune Infiltration Analysis
4.6. EAE Model and Scoring
4.7. Chemical Index Detection
4.8. RNA Extraction and qRT-PCR
4.9. H&E Staining
4.10. Flow Cytometry Analysis
4.11. Drugs Screened and Docking
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jellinger, K.A. Cognitive impairment in multiple sclerosis: From phenomenology to neurobiological mechanisms. J. Neural Transm. 2024, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.F. Overview and diagnosis of multiple sclerosis. Am. J. Manag. Care 2016, 22 (Suppl. S6), s141–s150. [Google Scholar] [PubMed]
- Silva, B.A.; Leal, M.C.; Farías, M.I.; Erhardt, B.; Galeano, P.; Pitossi, F.J.; Ferrari, C.C. Environmental enrichment improves cognitive symptoms and pathological features in a focal model of cortical damage of multiple sclerosis. Brain Res. 2020, 1727, 146520. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, A.A.; El-Sharkawy, K.A.; Mahmoudy, A.M.; Hashim, N.A. Burden of multiple sclerosis: Impact on the patient, family and society. Mult. Scler. Relat. Disord. 2022, 63, 103864. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, X.; Li, H.; Sun, Z.; Zhong, Y. Gender differences in the burden of multiple sclerosis in China from 1990 to 2019 and its 25-year projection: An analysis of the Global Burden of Diseases Study. Health Sci. Rep. 2023, 6, e1738. [Google Scholar] [CrossRef] [PubMed]
- Konen, F.F.; Möhn, N.; Witte, T.; Schefzyk, M.; Wiestler, M.; Lovric, S.; Hufendiek, K.; Schwenkenbecher, P.; Sühs, K.W.; Friese, M.A.; et al. Treatment of autoimmunity: The impact of disease-modifying therapies in multiple sclerosis and comorbid autoimmune disorders. Autoimmun. Rev. 2023, 22, 103312. [Google Scholar] [CrossRef] [PubMed]
- Lorefice, L.; Mellino, P.; Fenu, G.; Cocco, E. How to measure the treatment response in progressive multiple sclerosis: Current perspectives and limitations in clinical settings’. Mult. Scler. Relat. Disord. 2023, 76, 104826. [Google Scholar] [CrossRef] [PubMed]
- Vasić, M.; Topić, A.; Marković, B.; Milinković, N.; Dinčić, E. Oxidative stress-related risk of the multiple sclerosis development. J. Med. Biochem. 2023, 42, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Huang, Q.; Zhao, D.; Lian, F.; Li, X.; Qi, W. The impact of oxidative stress-induced mitochondrial dysfunction on diabetic microvascular complications. Front. Endocrinol. 2023, 14, 1112363. [Google Scholar] [CrossRef]
- Sanabria-Castro, A.; Alape-Girón, A.; Flores-Díaz, M.; Echeverri-McCandless, A.; Parajeles-Vindas, A. Oxidative stress involvement in the molecular pathogenesis and progression of multiple sclerosis: A literature review. Rev. Neurosci. 2024, 35, 355–371. [Google Scholar] [CrossRef]
- Wadhwa, R.; Gupta, R.; Maurya, P.K. Oxidative Stress and Accelerated Aging in Neurodegenerative and Neuropsychiatric Disorder. Curr. Pharm. Des. 2018, 24, 4711–4725. [Google Scholar] [CrossRef] [PubMed]
- Minutoli, L.; Puzzolo, D.; Rinaldi, M.; Irrera, N.; Marini, H.; Arcoraci, V.; Bitto, A.; Crea, G.; Pisani, A.; Squadrito, F.; et al. ROS-Mediated NLRP3 Inflammasome Activation in Brain, Heart, Kidney, and Testis Ischemia/Reperfusion Injury. Oxid. Med. Cell Longev. 2016, 2016, 2183026. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.V.; Javadov, S.; Sommer, N. Cellular ROS and Antioxidants: Physiological and Pathological Role. Antioxidants 2024, 13, 602. [Google Scholar] [CrossRef] [PubMed]
- Arif, A.; Quds, R.; Mahmood, R. Bioallethrin enhances generation of ROS, damages DNA, impairs the redox system and causes mitochondrial dysfunction in human lymphocytes. Sci. Rep. 2021, 11, 8300. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- van den Bosch, A.; Fransen, N.; Mason, M.; Rozemuller, A.J.; Teunissen, C.; Smolders, J.; Huitinga, I. Neurofilament Light Chain Levels in Multiple Sclerosis Correlate With Lesions Containing Foamy Macrophages and with Acute Axonal Damage. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Werner, P.; Pitt, D.; Raine, C.S. Multiple sclerosis: Altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann. Neurol. 2001, 50, 169–180. [Google Scholar] [CrossRef]
- Groppo, E.; Baglio, F.; Cattaneo, D.; Tavazzi, E.; Bergsland, N.; Di Tella, S.; Parelli, R.; Carpinella, I.; Grosso, C.; Capra, R.; et al. Multidisciplinary Rehabilitation is Efficacious and Induces Neural Plasticity in Multiple Sclerosis even when Complicated by Progressive Multifocal Leukoencephalopathy. Front. Neurol. 2017, 8, 491. [Google Scholar] [CrossRef]
- Theodoropoulou, E.; Alfredsson, L.; Piehl, F.; Marabita, F.; Jagodic, M. Different epigenetic clocks reflect distinct pathophysiological features of multiple sclerosis. Epigenomics 2019, 11, 1429–1439. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, M.; Xu, L.; Zhong, M.; Wang, Y.; Luan, M.; Li, X.; Zheng, X. Cerebellar and/or Brainstem Lesions Indicate Poor Prognosis in Multiple Sclerosis: A Systematic Review. Front. Neurol. 2022, 13, 874388. [Google Scholar] [CrossRef]
- Brück, W. The pathology of multiple sclerosis is the result of focal inflammatory demyelination with axonal damage. J. Neurol. 2005, 252 (Suppl. S5), v3–v9. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Metz, I.; Amor, S.; van der Valk, P.; Stadelmann, C.; Brück, W. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol. 2013, 125, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Ramos-González, E.J.; Bitzer-Quintero, O.K.; Ortiz, G.; Hernández-Cruz, J.J.; Ramírez-Jirano, L.J. Relationship between inflammation and oxidative stress and its effect on multiple sclerosis. Neurologia 2024, 39, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Mostert, J.; Arutjunyan, A.; Stepanov, M.; Teelken, A.; Heersema, D.; De Keyser, J. Peripheral blood leukocyte NO production and oxidative stress in multiple sclerosis. Mult. Scler. 2008, 14, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.; Wilcockson, D.C.; Campion, S.; Lunnon, K.; Perry, V.H. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J. Neurosci. 2005, 25, 9275–9284. [Google Scholar] [CrossRef] [PubMed]
- Padureanu, R.; Albu, C.V.; Mititelu, R.R.; Bacanoiu, M.V.; Docea, A.O.; Calina, D.; Padureanu, V.; Olaru, G.; Sandu, R.E.; Malin, R.D.; et al. Oxidative Stress and Inflammation Interdependence in Multiple Sclerosis. J. Clin. Med. 2019, 8, 1815. [Google Scholar] [CrossRef]
- Wigerblad, G.; Kaplan, M.J. Neutrophil extracellular traps in systemic autoimmune and autoinflammatory diseases. Nat. Rev. Immunol. 2023, 23, 274–288. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; Wang, N.; Tall, A.R.; Tabas, I. Mitochondrial Oxidative Stress Promotes Atherosclerosis and Neutrophil Extracellular Traps in Aged Mice. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e99–e107. [Google Scholar] [CrossRef] [PubMed]
- Signorile, A.; Ferretta, A.; Ruggieri, M.; Paolicelli, D.; Lattanzio, P.; Trojano, M.; De Rasmo, D. Mitochondria, Oxidative Stress, cAMP Signalling and Apoptosis: A Crossroads in Lymphocytes of Multiple Sclerosis, a Possible Role of Nutraceutics. Antioxidants 2020, 10, 21. [Google Scholar] [CrossRef]
- Ontaneda, D.; Tallantyre, E.; Kalincik, T.; Planchon, S.M.; Evangelou, N. Early highly effective versus escalation treatment approaches in relapsing multiple sclerosis. Lancet Neurol. 2019, 18, 973–980. [Google Scholar] [CrossRef]
- Prokaeva, A.; Arkhipov, I.; Dorchinets, E.; Korobko, D.; Malkova, N. Multiple sclerosis: Modern diagnostic markers and prognostic factors of disease progression. Сибирский Научный Медицинский Журнал 2024, 44, 39–51. [Google Scholar] [CrossRef]
- Wang, X.; Yu, Y.Y.; Lieu, S.; Yang, F.; Lang, J.; Lu, C.; Werb, Z.; Hu, D.; Miclau, T.; Marcucio, R.; et al. MMP9 regulates the cellular response to inflammation after skeletal injury. Bone 2013, 52, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Cai, X.; Chen, S.; Fu, W.; Chai, D.; Zhang, H.; Zhang, Y. 7-Methoxy-1-Tetralone Induces Apoptosis, Suppresses Cell Proliferation and Migration in Hepatocellular Carcinoma via Regulating c-Met, p-AKT, NF-κB, MMP2, and MMP9 Expression. Front. Oncol. 2020, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chuang, C.Y.; Hawkins, C.L.; Davies, M.J. Activation and Inhibition of Human Matrix Metalloproteinase-9 (MMP9) by HOCl, Myeloperoxidase and Chloramines. Antioxidants 2022, 11, 1616. [Google Scholar] [CrossRef] [PubMed]
- Nazdik, M.K.; Taheri, M.; Sajjadi, E.; Arsang-Jang, S.; Koohpar, Z.K.; Inoko, H.; Sayad, A. Increased expression ratio of matrix metalloproteinase-9 (MMP9) and tissue inhibitor of matrix metalloproteinase (TIMP-1) RNA levels in Iranian multiple sclerosis patients. Hum. Antibodies 2016, 24, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Hamedani, S.Y.; Taheri, M.; Sajjadi, E.; Omrani, M.D.; Mazdeh, M.; Arsang-Jang, S.; Panah, A.S.; Sayad, A. Up regulation of MMP9 gene expression in female patients with multiple sclerosis. Hum. Antibodies 2016, 24, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Zhang, W.; Jin, X.; Li, X.; Sun, S.; Zhao, Y.; Jia, Q.; Li, L.; Liu, Y.; Liu, D.; et al. Genetic Screening of Haploid Neural Stem Cells Reveals that Nfkbia and Atp2b4 are Key Regulators of Oxidative Stress in Neural Precursors. Adv. Sci. 2024, e2309292. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; Feng, Z.; Li, Q.; Qi, Z.; Zhang, Y. Inhibition of ROS-mediated activation Src-MAPK/AKT signaling by orientin alleviates H2O2-induced apoptosis in PC12 cells. Drug Des. Dev. Ther. 2018, 12, 3973–3984. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Huang, M.W.; Xiao, C.X.; Long, F.; Wang, Y.; Liu, S.Y.; Jia, W.W.; Wu, W.J.; Yang, D.; Hu, J.F.; et al. Matairesinol Suppresses Neuroinflammation and Migration Associated with Src and ERK1/2-NF-κB Pathway in Activating BV2 Microglia. Neurochem. Res. 2017, 42, 2850–2860. [Google Scholar] [CrossRef]
- Cartwright, T.; Perkins, N.D.; Wilson, C.L. NFKB1: A suppressor of inflammation, ageing and cancer. FEBS J. 2016, 283, 1812–1822. [Google Scholar] [CrossRef]
- Guo, Y.; Liu, Z.; Wang, M. NFKB1-mediated downregulation of microRNA-106a promotes oxidative stress injury and insulin resistance in mice with gestational hypertension. Cytotechnology 2021, 73, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Jia, K.; Zhou, Y.; Chen, L.; Wang, F.; Yi, X.; Huang, Y.; Ge, Y.; Chen, X.; Liao, D.; et al. Rutin hydrate relieves neuroinflammation in zebrafish models: Involvement of NF-κB pathway as a central network. Fish Shellfish Immunol. 2023, 141, 109062. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Choi, I.Y.; Lee, P.; Adany, P.; Hughes, A.J.; Belliston, S.; Denney, D.R.; Lynch, S.G. In vivo evidence of oxidative stress in brains of patients with progressive multiple sclerosis. Mult. Scler. 2018, 24, 1029–1038. [Google Scholar] [CrossRef]
- Traugott, U.; Reinherz, E.L.; Raine, C.S. Multiple sclerosis. Distribution of T cells, T cell subsets and Ia-positive macrophages in lesions of different ages. J. Neuroimmunol. 1983, 4, 201–221. [Google Scholar] [CrossRef]
- Rodríguez Murúa, S.; Farez, M.F.; Quintana, F.J. The Immune Response in Multiple Sclerosis. Annu. Rev. Pathol. 2022, 17, 121–139. [Google Scholar] [CrossRef]
- Slomski, A. Drug Slows Disability Progression in Multiple Sclerosis. JAMA 2018, 319, 2069. [Google Scholar] [CrossRef]
- Hryniewicz-Jankowska, A.; Wierzbicki, J.; Tabola, R.; Stach, K.; Sossey-Alaoui, K.; Augoff, K. The Effect of Neddylation Inhibition on Inflammation-Induced MMP9 Gene Expression in Esophageal Squamous Cell Carcinoma. Int. J. Mol. Sci. 2021, 22, 1716. [Google Scholar] [CrossRef]
- Jiang, K.; Chen, H.; Fang, Y.; Chen, L.; Zhong, C.; Bu, T.; Dai, S.; Pan, X.; Fu, D.; Qian, Y.; et al. Exosomal ANGPTL1 attenuates colorectal cancer liver metastasis by regulating Kupffer cell secretion pattern and impeding MMP9 induced vascular leakiness. J. Exp. Clin. Cancer Res. 2021, 40, 21. [Google Scholar] [CrossRef] [PubMed]
- Appleton, K.; Mackay, H.J.; Judson, I.; Plumb, J.A.; McCormick, C.; Strathdee, G.; Lee, C.; Barrett, S.; Reade, S.; Jadayel, D.; et al. Phase I and pharmacodynamic trial of the DNA methyltransferase inhibitor decitabine and carboplatin in solid tumors. J. Clin. Oncol. 2007, 25, 4603–4609. [Google Scholar] [CrossRef]
- Boi, M.; Gaudio, E.; Bonetti, P.; Kwee, I.; Bernasconi, E.; Tarantelli, C.; Rinaldi, A.; Testoni, M.; Cascione, L.; Ponzoni, M.; et al. The BET Bromodomain Inhibitor OTX015 Affects Pathogenetic Pathways in Preclinical B-cell Tumor Models and Synergizes with Targeted Drugs. Clin. Cancer Res. 2015, 21, 1628–1638. [Google Scholar] [CrossRef]
- Fan, X.; Huang, J.; Zhang, W.; Su, Z.; Li, J.; Wu, Z.; Zhang, P. A Multifunctional, Tough, Stretchable, and Transparent Curcumin Hydrogel with Potent Antimicrobial, Antioxidative, Anti-inflammatory, and Angiogenesis Capabilities for Diabetic Wound Healing. ACS Appl. Mater. Interfaces 2024, 16, 9749–9767. [Google Scholar] [CrossRef]
- Marton, L.T.; Barbalho, S.M.; Sloan, K.P.; Sloan, L.A.; Goulart, R.A.; Araújo, A.C.; Bechara, M.D. Curcumin, autoimmune and inflammatory diseases: Going beyond conventional therapy—A systematic review. Crit. Rev. Food Sci. Nutr. 2022, 62, 2140–2157. [Google Scholar] [CrossRef] [PubMed]
- Cores, Á.; Carmona-Zafra, N.; Martín-Cámara, O.; Sánchez, J.D.; Duarte, P.; Villacampa, M.; Bermejo-Bescós, P.; Martín-Aragón, S.; León, R.; Menéndez, J.C. Curcumin-Piperlongumine Hybrids with a Multitarget Profile Elicit Neuroprotection in In Vitro Models of Oxidative Stress and Hyperphosphorylation. Antioxidants 2021, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Irving, A.A.; Marling, S.J.; Seeman, J.; Plum, L.A.; DeLuca, H.F. UV light suppression of EAE (a mouse model of multiple sclerosis) is independent of vitamin D and its receptor. Proc. Natl. Acad. Sci. USA 2019, 116, 22552–22555. [Google Scholar] [CrossRef] [PubMed]
- Nitsch, L.; Petzinna, S.; Zimmermann, J.; Schneider, L.; Krauthausen, M.; Heneka, M.T.; Getts, D.R.; Becker, A.; Müller, M. Astrocyte-specific expression of interleukin 23 leads to an aggravated phenotype and enhanced inflammatory response with B cell accumulation in the EAE model. J. Neuroinflammation 2021, 18, 101. [Google Scholar] [CrossRef]
- Manglani, M.; Gossa, S.; McGavern, D.B. Leukocyte Isolation from Brain, Spinal Cord, and Meninges for Flow Cytometric Analysis. Curr. Protoc. Immunol. 2018, 121, e44. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Wang, F.; Zhao, Q.; Zhang, L.; Chen, S.; Wang, S. Identifying Diagnostic Markers and Constructing Predictive Models for Oxidative Stress in Multiple Sclerosis. Int. J. Mol. Sci. 2024, 25, 7551. https://doi.org/10.3390/ijms25147551
Ma Y, Wang F, Zhao Q, Zhang L, Chen S, Wang S. Identifying Diagnostic Markers and Constructing Predictive Models for Oxidative Stress in Multiple Sclerosis. International Journal of Molecular Sciences. 2024; 25(14):7551. https://doi.org/10.3390/ijms25147551
Chicago/Turabian StyleMa, Yantuanjin, Fang Wang, Qiting Zhao, Lili Zhang, Shunmei Chen, and Shufen Wang. 2024. "Identifying Diagnostic Markers and Constructing Predictive Models for Oxidative Stress in Multiple Sclerosis" International Journal of Molecular Sciences 25, no. 14: 7551. https://doi.org/10.3390/ijms25147551
APA StyleMa, Y., Wang, F., Zhao, Q., Zhang, L., Chen, S., & Wang, S. (2024). Identifying Diagnostic Markers and Constructing Predictive Models for Oxidative Stress in Multiple Sclerosis. International Journal of Molecular Sciences, 25(14), 7551. https://doi.org/10.3390/ijms25147551