
Computational NMR Study of Benzothienoquinoline Heterohelicenes †


Abstract
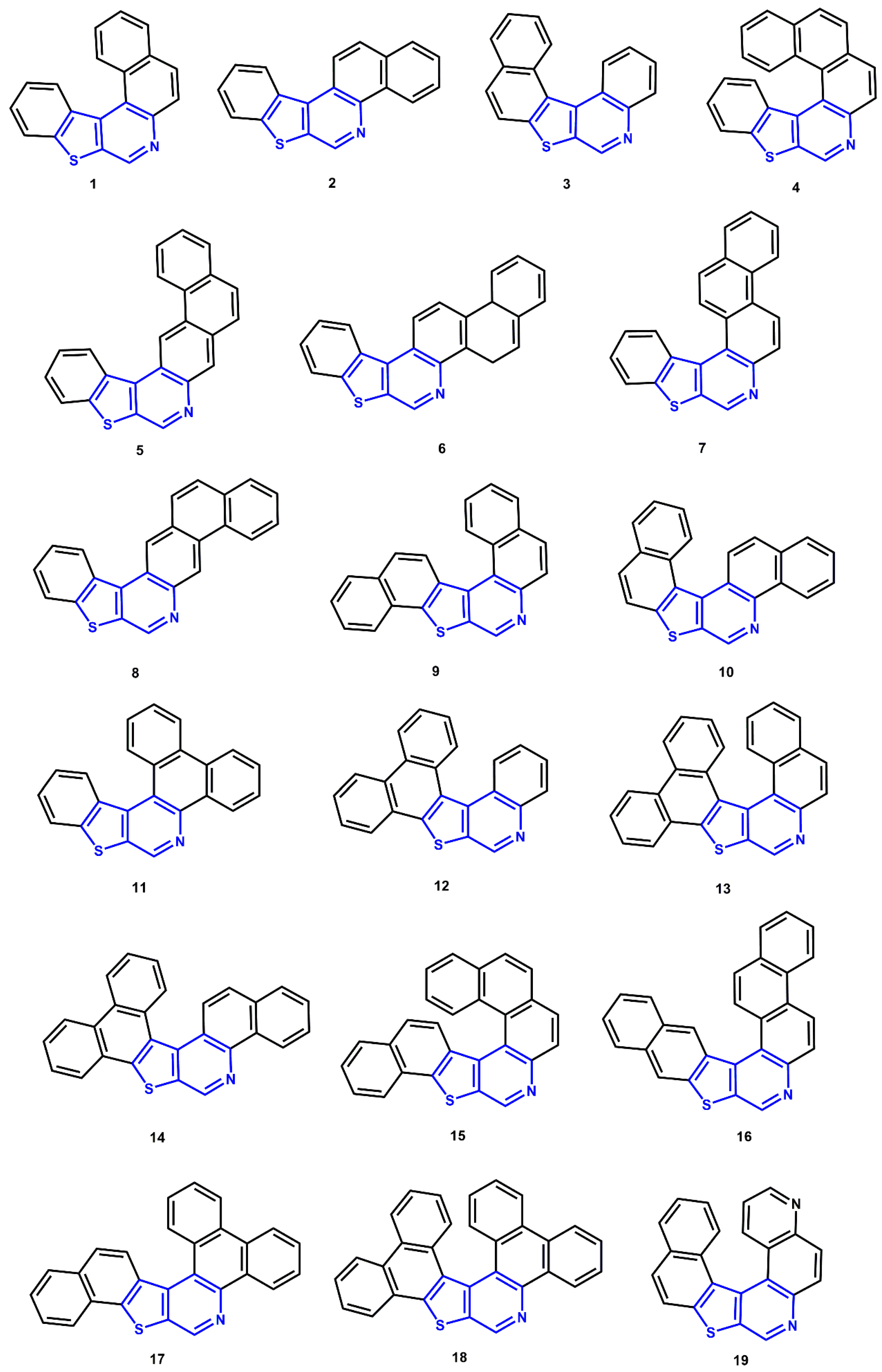
:1. Introduction
2. Computational Details
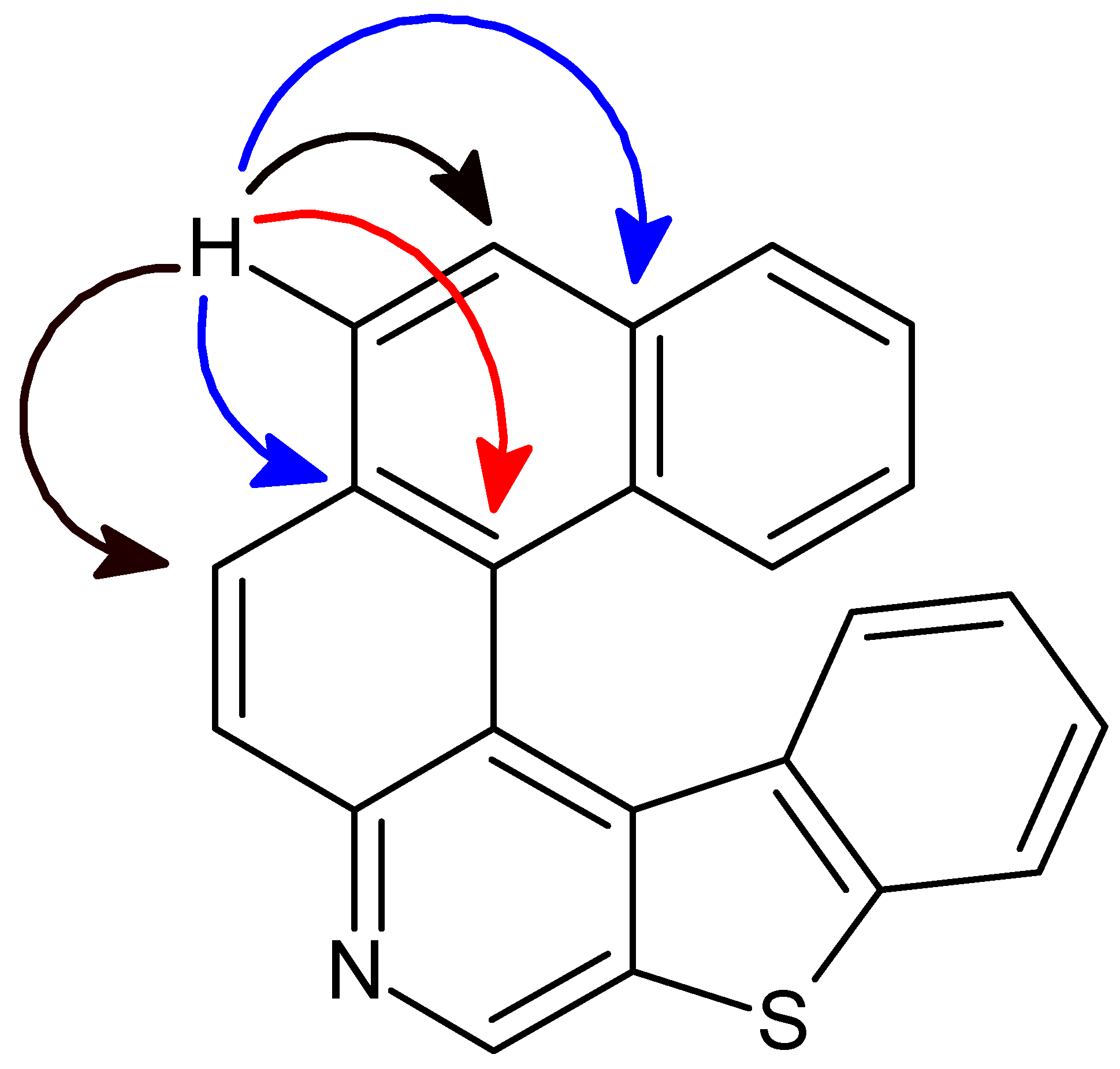
3. Results and Discussion
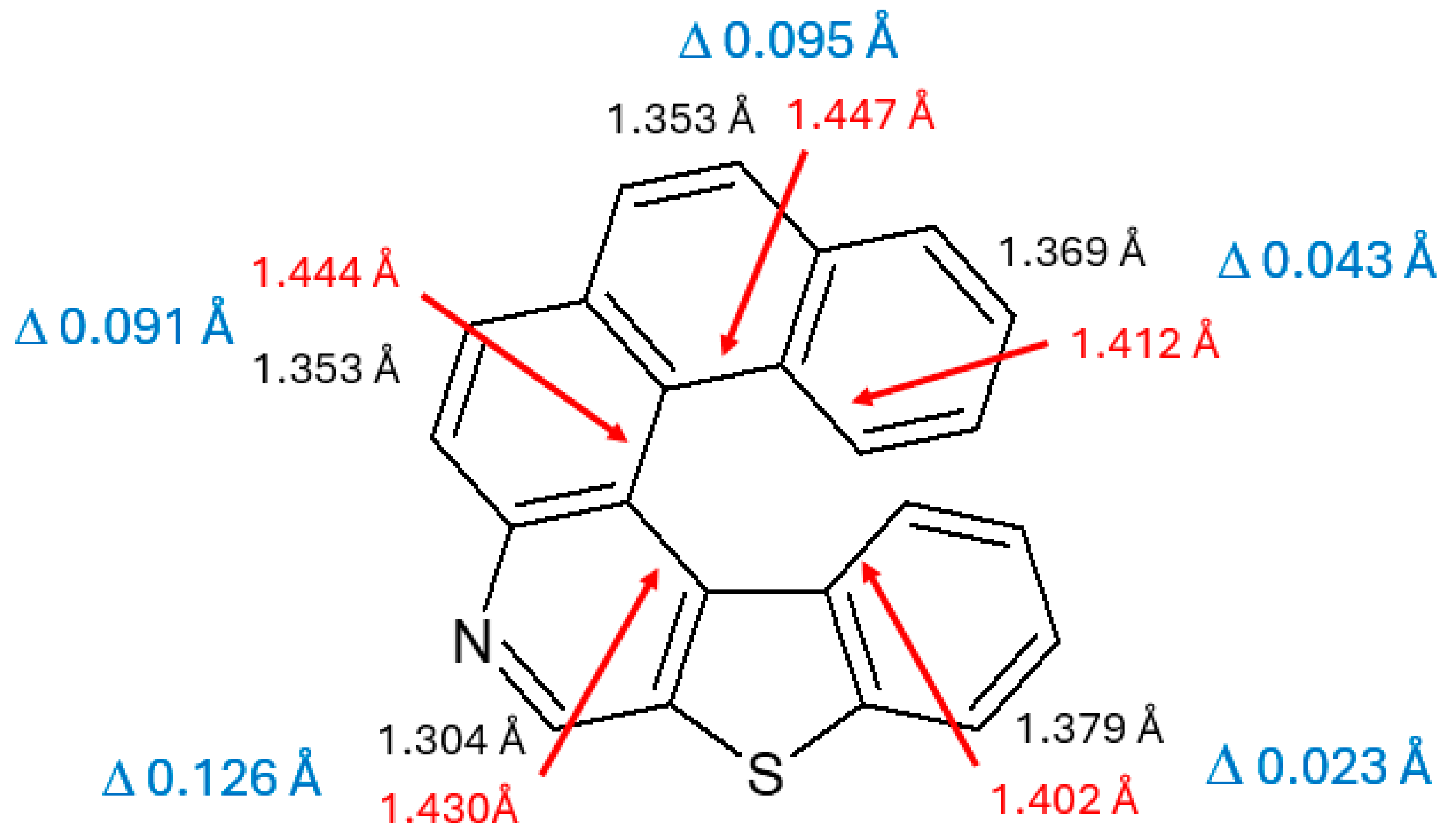
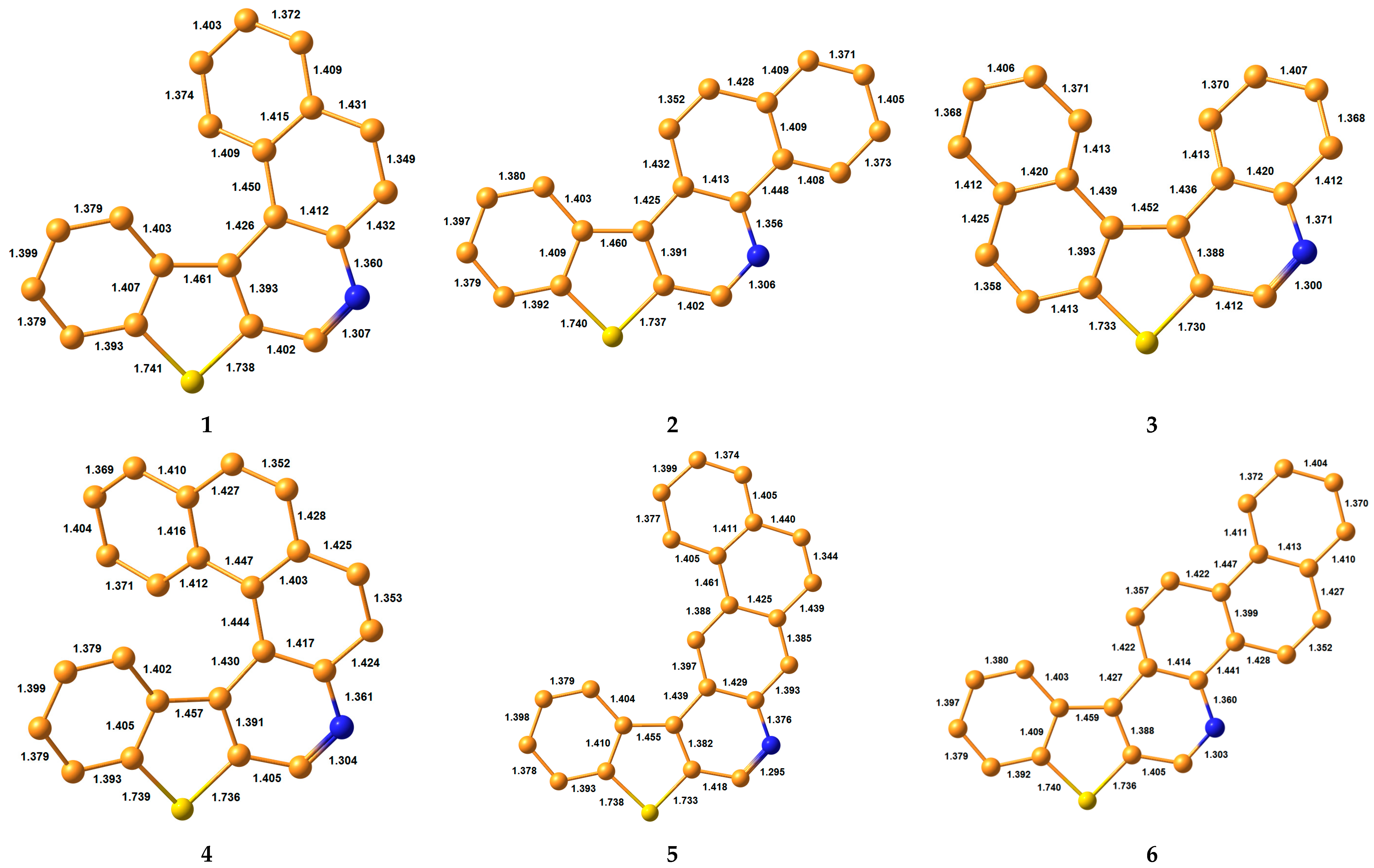
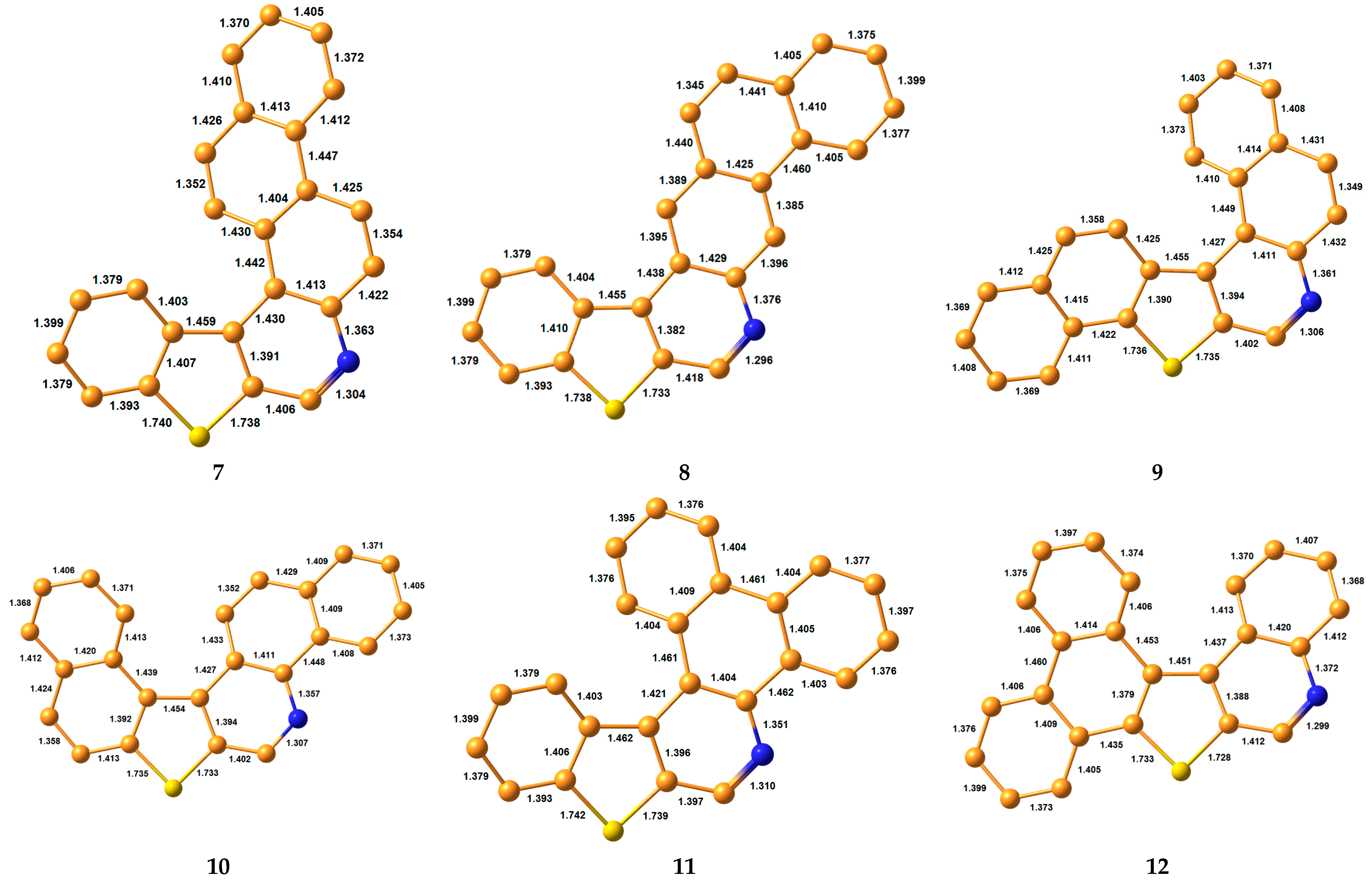
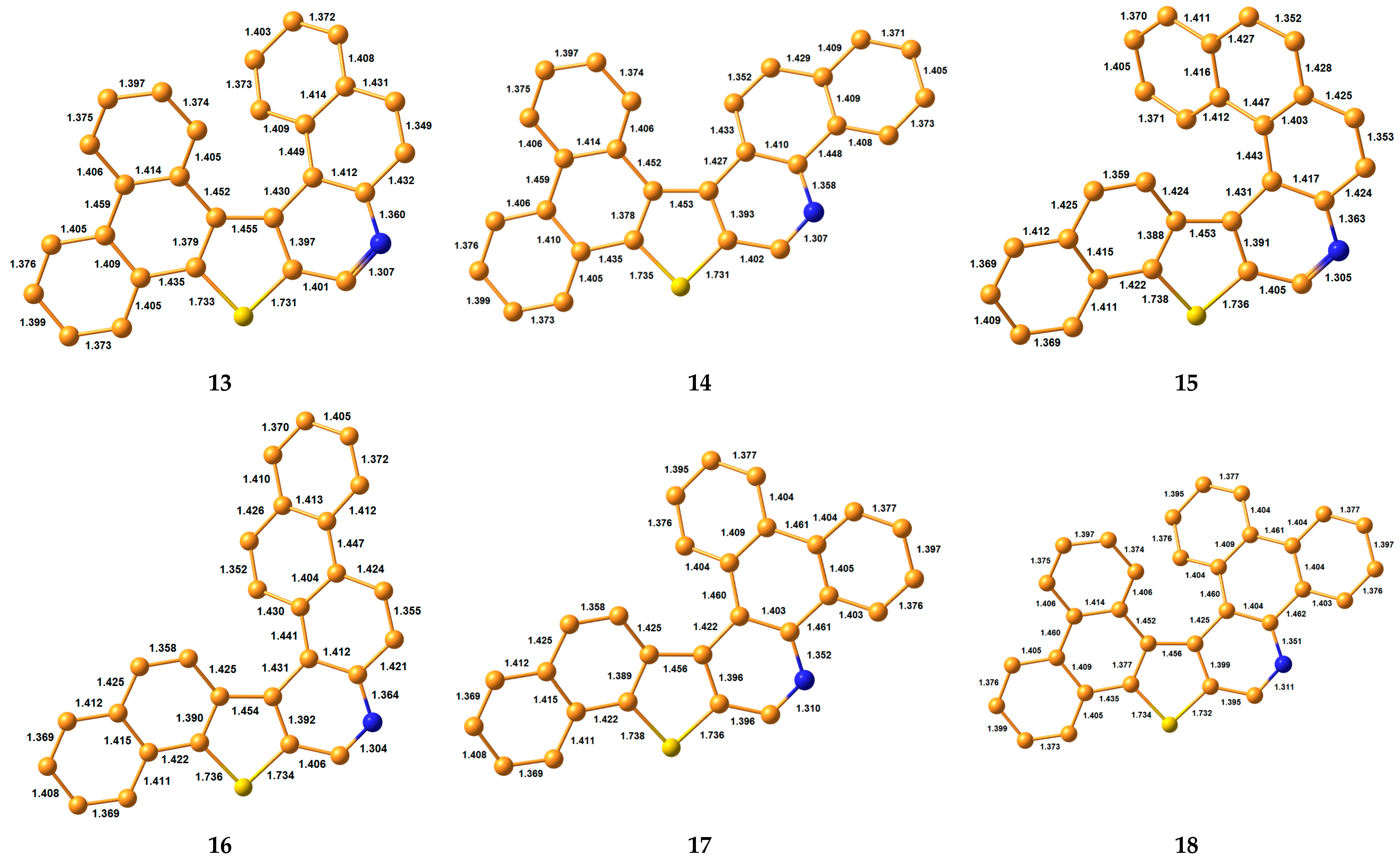
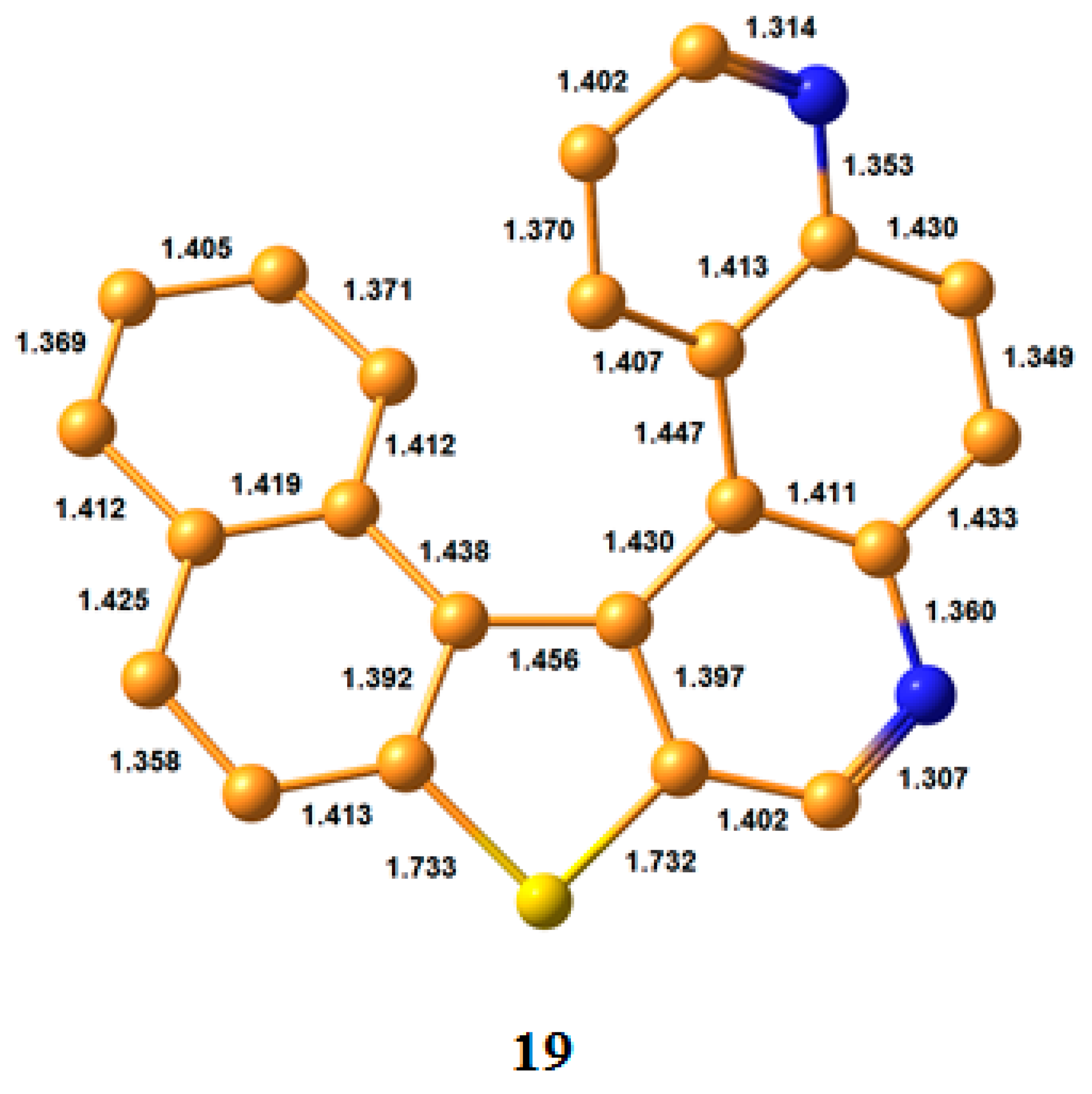
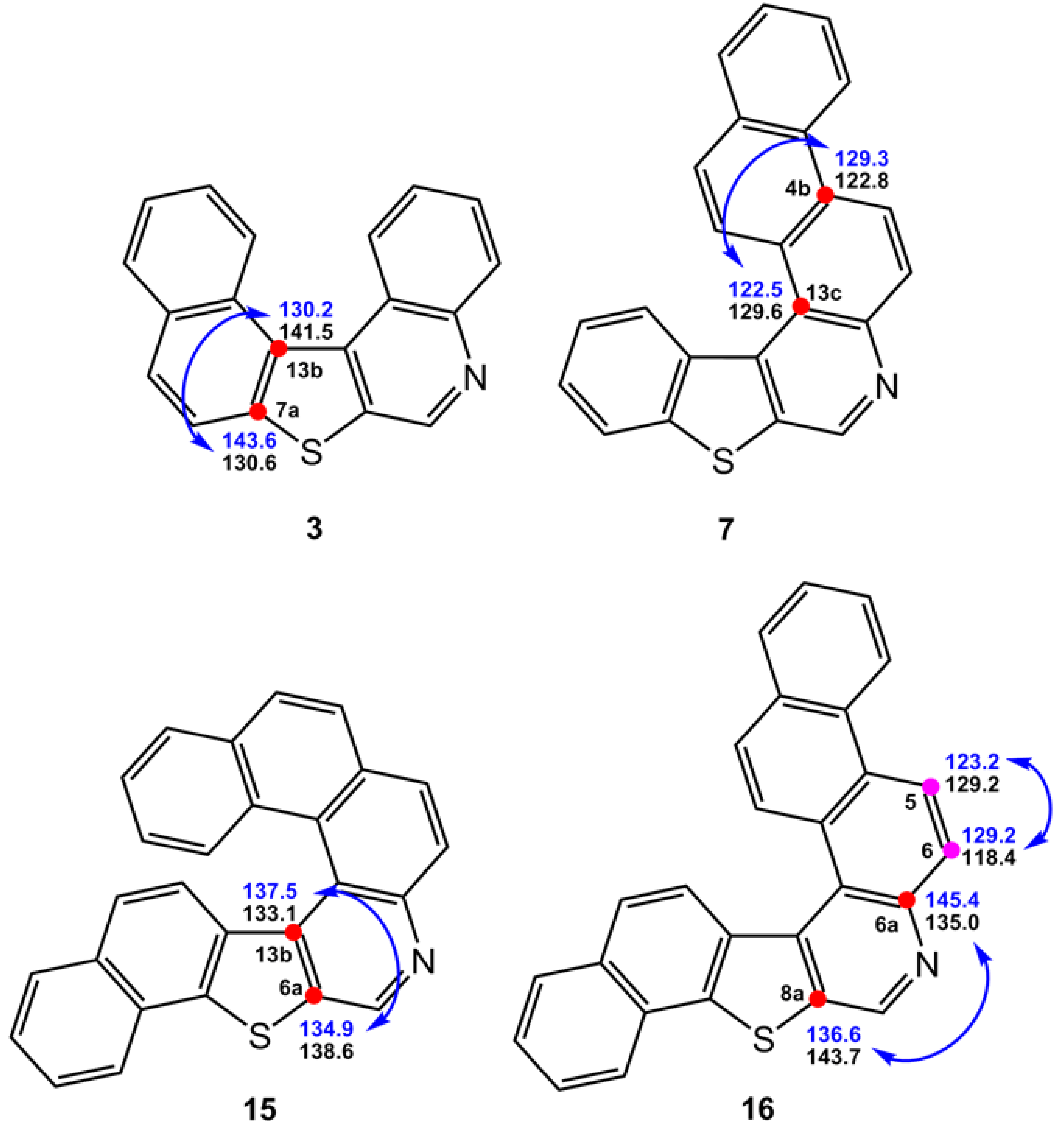
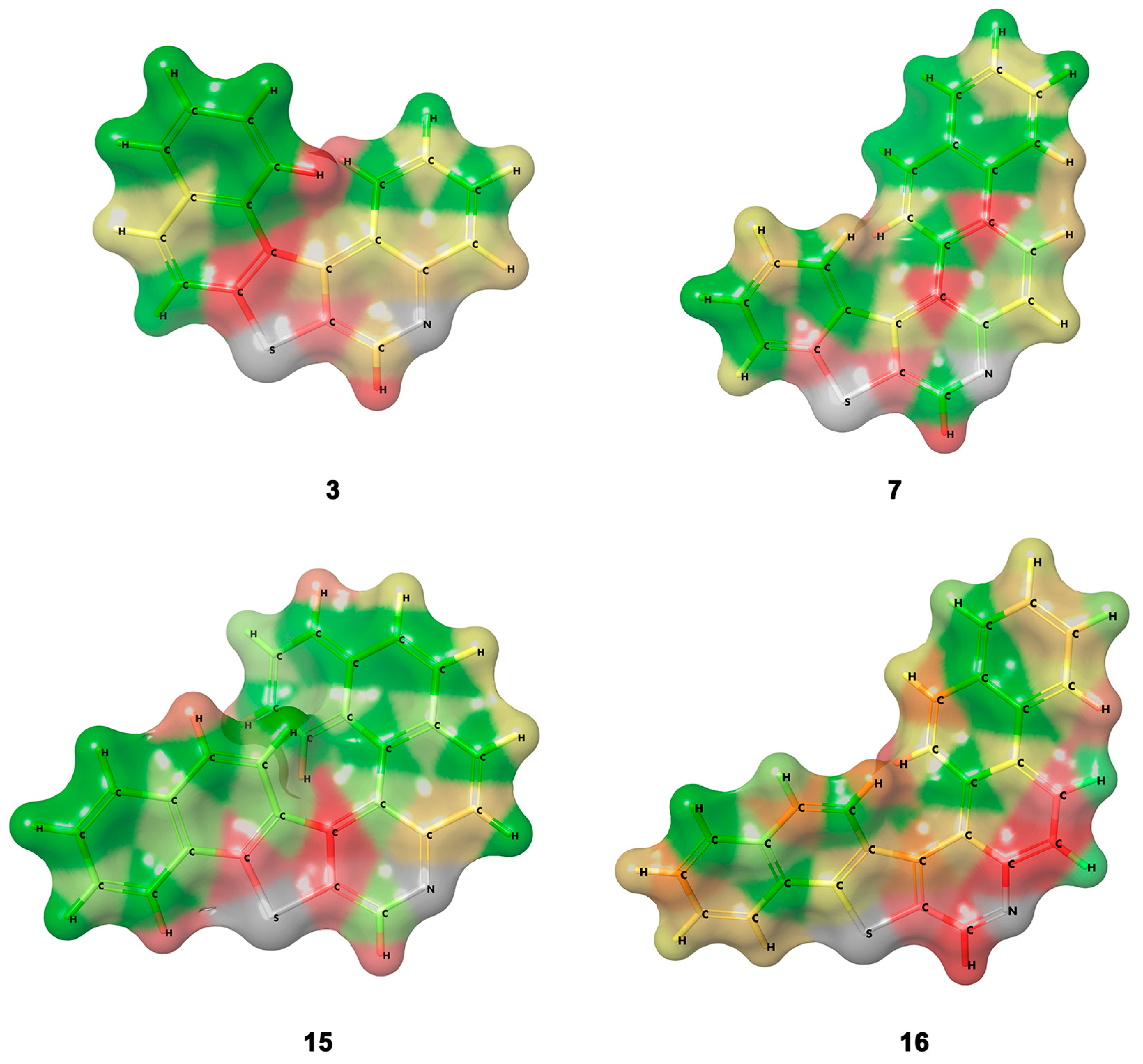
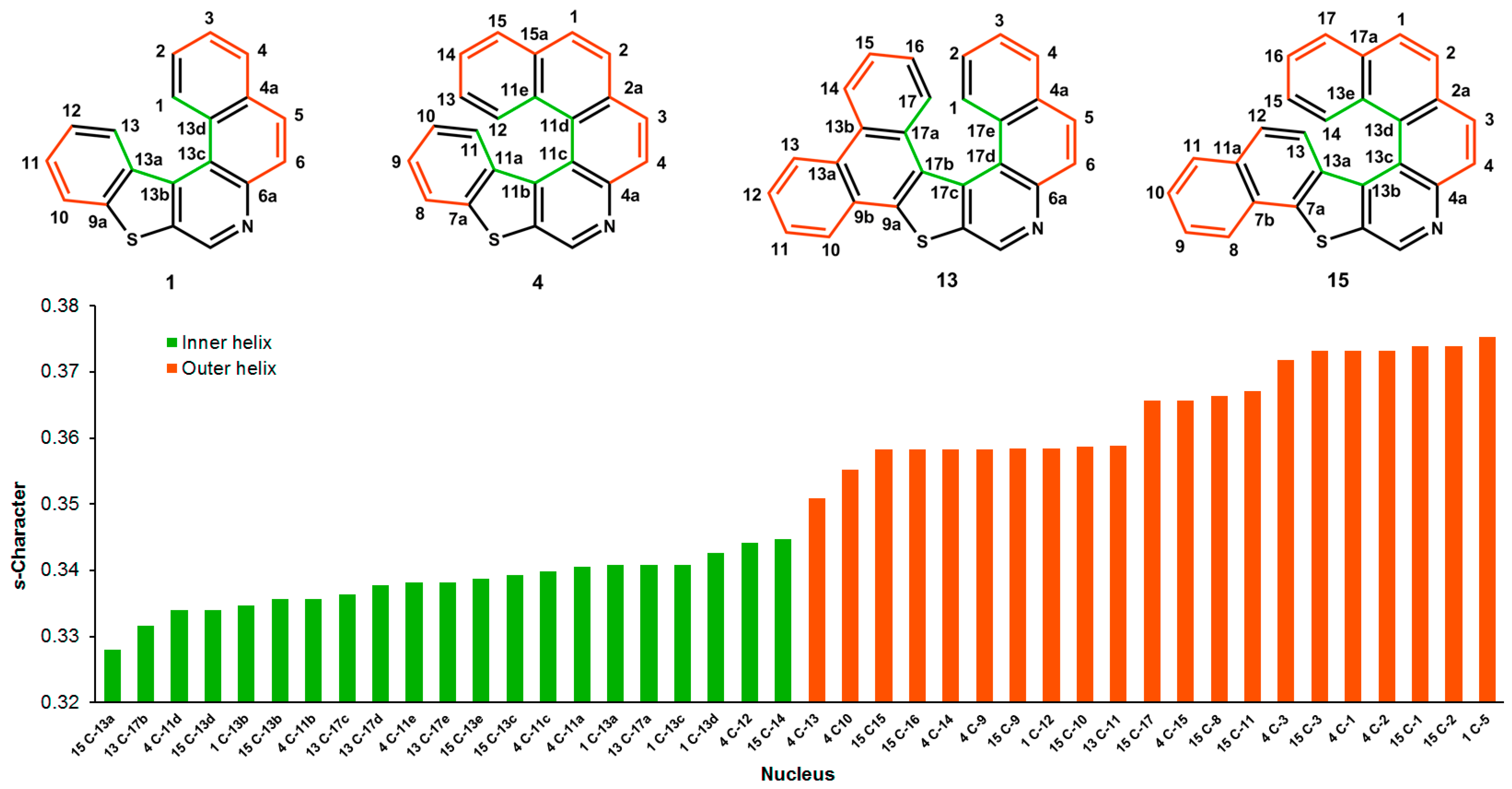
3.1. Bond Lengths
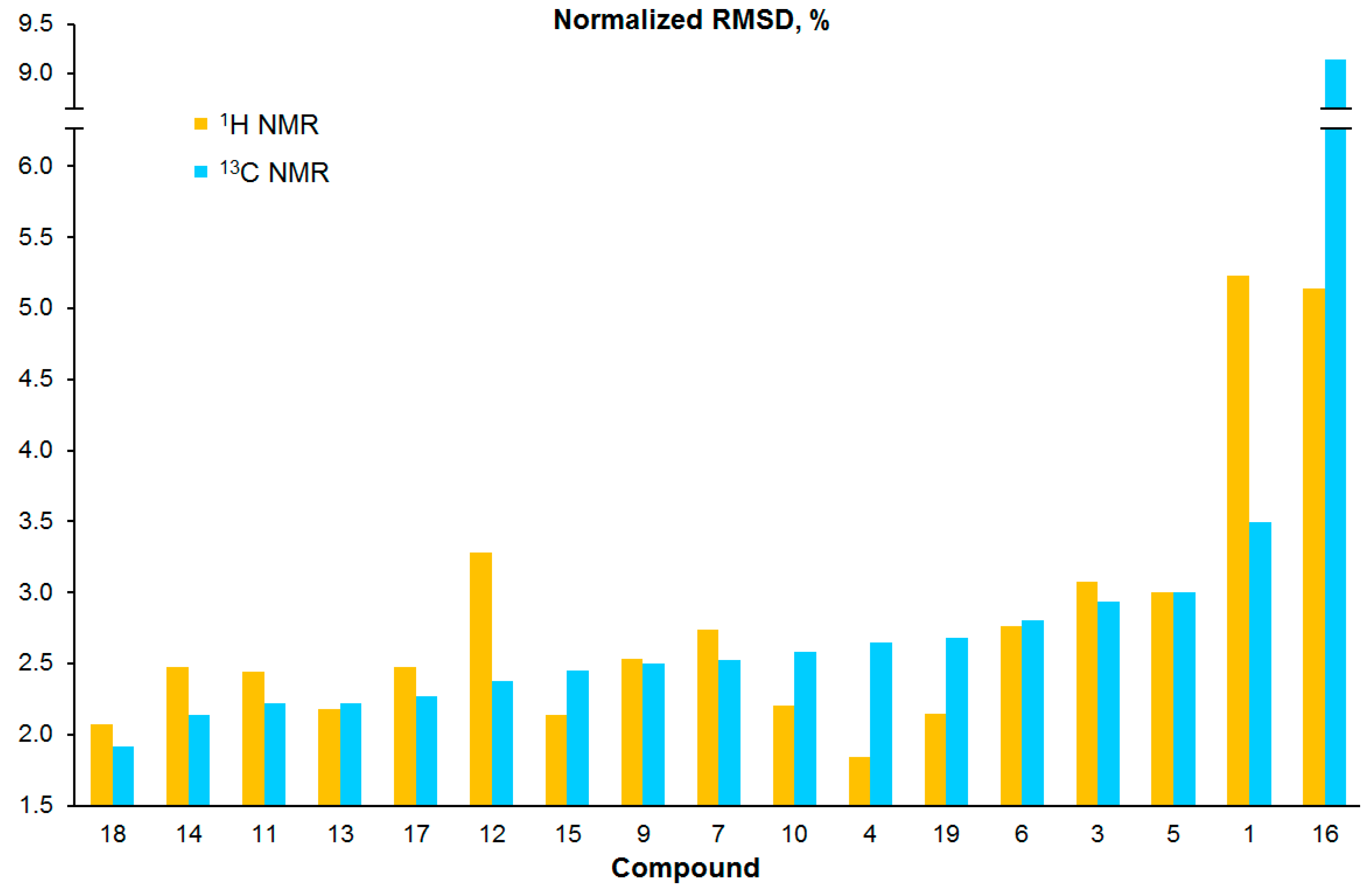
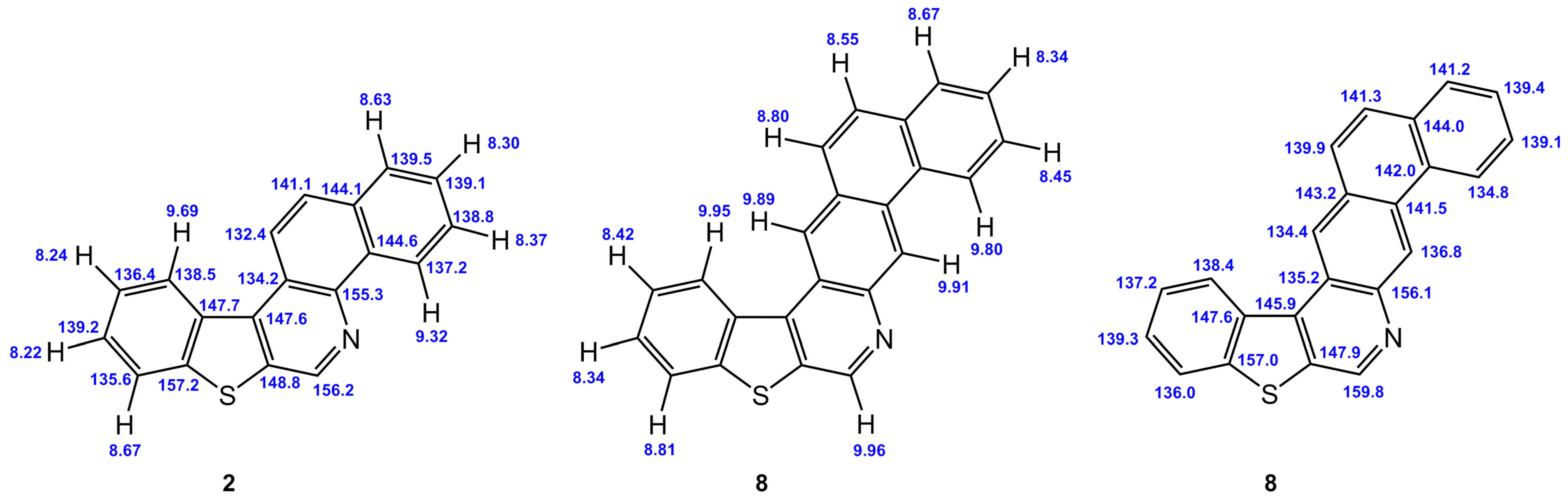
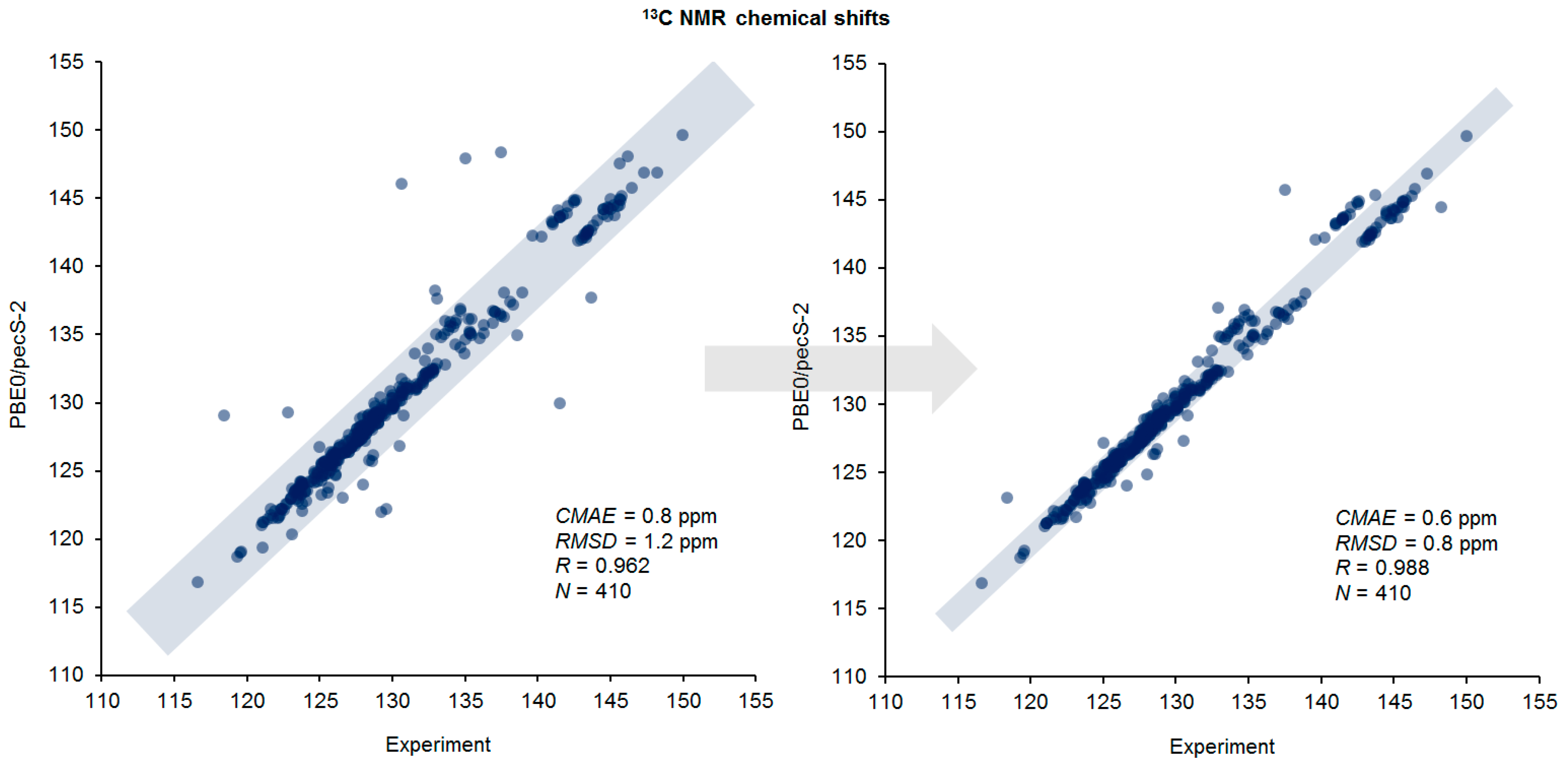
3.2. Chemical Shifts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Musmar, M.J.; Willcott, M.R.; Martin, G.E.; Isao, M.; Lee, M.L.; Hurd, R.E.; Johnson, L.F.; Castle, R.N. Autocorrelated 13C-13C double quantum coherence two-dimensional NMR spectroscopy: Utilization of a modified version of the technique as an adjunct in the total assignment of the 1H- and 13C-NMR spectra of the mutagen phenanthro[3,4-b]thiophene. J. Heterocycl. Chem. 1983, 20, 1661–1669. [Google Scholar] [CrossRef]
- Musmar, M.J.; Zektzer, A.S.; Martin, G.E.; Gampe, R.T., Jr.; Lee, M.L.; Tedjamulia, M.L.; Caslte, R.N.; Hurd, R.E. Benzo[b]phenanthro[4,3-d]thiophene: Spectral assignment by two-dimensional NMR methods and tertiary structure determination. Magn. Reson. Chem. 1987, 24, 1039–1043. [Google Scholar] [CrossRef]
- Shockcor, J.P.; Crouch, R.C.; Martin, G.E.; Cheir, A.; Luo, J.-K.; Caslte, R.N. Total assignment of the 1H and 13C NMR spectra of benzo[f][1]benzothieno[2,3-c]quinoline by inverse-detected two-dimensional NMR techniques. J. Heterocycl. Chem. 1991, 28, 2035–2039. [Google Scholar] [CrossRef]
- Luo, J.-K.; Zektzer, A.S.; Caslte, R.N.; Crouch, R.C.; Shockcor, J.P.; Martin, G.E. The synthesis of novel polycyclic heterocyclic ring systems via photocyclization. 11. Synthesis and total assignment of the 1H and 13C NMR spectra of isomeric benzothienonaphthoquinolines using multiple inverse detected two-dimensional NMR methods. J. Heterocycl. Chem. 1993, 30, 453–460. [Google Scholar] [CrossRef]
- Newman, M.S.; Lednicer, D. The Synthesis and Resolution of Hexahelicene. J. Am. Chem. Soc. 1956, 78, 4765–4770. [Google Scholar] [CrossRef]
- Mackay, I.R.; Robertson, J.M.; Sime, J.G. The crystal structure of hexahelicene. J. Chem. Soc. D 1969, 1470–1471. [Google Scholar] [CrossRef]
- Semenov, V.A.; Martin, G.E.; Krivdin, L.B. Computational studies of [1]benzothieno[2,3-c]naphtho[1,2-f]quinolone. Magn. Reson. Chem. 2023, 61, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.-K.; Castle, R.N. The synthesis of novel polycyclic heterocyclic ring systems via photocyclization. 2. Benzo[h][1]benzothieno[2,3-c]quinoline and benzo[h][1]benzothieno[2,3-c]quinoline. J. Heterocycl. Chem. 1990, 27, 1031–1033. [Google Scholar] [CrossRef]
- Luo, J.-K.; Castle, S.L.; Castle, R.N. The Synthesis of Novel Polycyclic Heterocyclic Ring Systems via Photocyclization. 12. Benzo[h]naphtho-[2′,1′:4,5]thieno[2,3-c]quinoline and benzo[f]-naphtho[2′,1′:4,5]thieno[2,3-c]quinoline. J. Heterocycl. Chem. 1993, 30, 653–658. [Google Scholar] [CrossRef]
- Luo, J.-K.; Castle, R.N. The synthesis of novel polycyclic heterocyclic ring systems via photocyclization. 5. [1]Benzothieno[2,3-c]naphtho[2,1-f]-quinoline and [1]benzothieno[2,3-c]naphtho[1,2-g]quinoline. J. Heterocycl. Chem. 1991, 28, 1825–1830. [Google Scholar] [CrossRef]
- Sasaki, K.; Castle, L.W.; Castle, R.N. Complete assignment of the 1H- and 13C-NMR spectra of [1]benzothieno[2,3-c]naphtho[1,2-h]quinoline and [1]benzothieno[2,3-c]naphtho[1,2-h][1,2,4]triazolo[4,3-a]quinoline. Concerted use of two-dimensional NMR techniques. J. Heterocycl. Chem. 1994, 31, 65–71. [Google Scholar] [CrossRef]
- Martin, G.E.; Spitzer, T.D.; Crouch, R.C.; Luo, J.-K.; Castle, R.N. Inverted and suppressed direct response HMQC-TOCSY spectra—A convenient method of spectral editing. J. Heterocycl. Chem. 1992, 29, 577–582. [Google Scholar] [CrossRef]
- Luo, J.-K.; Federspiel, R.F.; Castle, R.N. The synthesis of novel polycyclic heterocyclic ring system via photocyclization. 18. Benzo[h]naphtho-[1′,2′:4,5]thieno[2,3-c]quinoline and benzo[h]naphtho-[1′,2′:4,5]thieno[2,3-c] [1,2,4]triazolo[4,3-a]quinoline. J. Heterocycl. Chem. 1996, 33, 923–926. [Google Scholar] [CrossRef]
- Luo, J.-K.; Cabal, M.P.; Federspiel, R.F.; Castle, R.N. The synthesis of novel polycyclic heterocyclic ring systems via photocyclization. 22. Dibenzo[f,h]benzothieno[2,3-c]quinoline, dibenzo[f,h]benzothieno[2,3-c][1,2,4]triazolo[4,3-a]quinoline and dibenzo[f,h]naphtho[2′,1′:4,5]thieno[2,3-c]quinoline. J. Heterocycl. Chem. 2000, 37, 997–1001. [Google Scholar] [CrossRef]
- Castle, L.W.; Johnston, M.D., Jr.; Camoutsis, C.L.; Castle, R.N. Assignments of the 1H and 13C NMR spectra of pseudo-symmetric heterocycles using the HMQC-TOCSY experiment to differentiate overlapping spin systems. J. Heterocycl. Chem. 1992, 29, 1805–1815. [Google Scholar] [CrossRef]
- Luo, J.-K.; Federspiel, R.F.; Castle, R.N. The synthesis of novel polycyclic heterocyclic ring systems via photocyclization. 21. Naptho[2′,1′:4,5]thieno[2,3-c]naphtho[1,2-f]-quinoline,naphtho[2′,1′:4,5]thieno[2,3-c]naphtho[1,2-f][1,2,4]-triazolo[4,3-a]quinolineandnaphtho[2′,1′:4,5]thieno[2,3-c]naphtho[1,2-f]-tetrazolo[1,5-a]quinoline. J. Heterocycl. Chem. 2000, 37, 171–174. [Google Scholar]
- Luo, J.-K.; Cabal, M.-P.; Federspiel, R.F.; Castle, R.N. The synthesis of novel polycyclic heterocyclic ring systems via photocyclization. 23. Naphtho[2′,1′:4,5]thieno-[2,3-c]naphtho[2,1-f]quinoline. J. Heterocycl. Chem. 2001, 38, 137–140. [Google Scholar] [CrossRef]
- Luo, J.-K.; Castle, R.N. The synthesis of novel polycyclic heterocyclic ring systems via photocyclization. 13. Dibenzo-[f,h]phenanthro[9′,10′:4,5]thieno[2,3-c]quinoline. J. Heterocycl. Chem. 1993, 30, 1167–1172. [Google Scholar] [CrossRef]
- Musmar, M.J.; Zektzer, A.S.; Castle, R.N.; Kent Dalley, N. The synthesis of novel polycyclic heterocyclic ring systems via photocyclization. 10. Synthesis and structure determination of naphtho[1′,2′:4,5]thieno[3,2-a]-4,7-phenanthroline. J. Heterocycl. Chem. 1993, 30, 487–492. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, A.; Cohen, R.D.; Del Poggetto, G.; Huang, Z.; Tang, H.; Martin, G.E.; Sherer, E.C.; Reibarkh, M.; Wang, X. Unequivocal identification of two-bond heteronuclear correlations in natural products at nanomole scale by i-HMBC. Nat. Commun. 2023, 14, 1842. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B. GAUSSIAN 09, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009; Available online: http://www.gaussian.com (accessed on 7 July 2024).
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. THEOCHEM 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Cossi, M.; Barone, V. An accurate density functional method for the study of magnetic properties: The PBE0 model. J. Mol. Struct. THEOCHEM 1999, 1, 145–157. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward chemical accuracy in the computation of NMR shieldings: The PBE0 model. Chem. Phys. Lett. 1998, 298, 113–119. [Google Scholar] [CrossRef]
- Rusakov, Y.; Rusakova, I.L. New pecS-n (n = 1, 2) basis sets for quantum chemical calculations of the NMR chemical shifts of H, C, N, and O nuclei. J. Chem. Phys. 2022, 156, 244112. [Google Scholar] [CrossRef] [PubMed]
- Rusakov, Y.Y.; Semenov, V.A.; Rusakova, I.L. On the efficiency of the Density Functional Theory (DFT)-based computational protocol for 1H and 13C Nuclear Magnetic Resonance (NMR) chemical shifts of natural products: Studying the accuracy of the pecS-n (n = 1, 2) Basis Sets. Int. J. Mol. Sci. 2023, 24, 14623. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.K.; Becker, E.D.; Cabral de Menezes, S.M.; Granger, P.; Hoffman, R.E.; Zilm, K.W. Further conventions for NMR shielding and chemical shifts (IUPAC Recommendations 2008). Pure Appl. Chem. 2008, 80, 59–84. [Google Scholar] [CrossRef]
- Weinhold, F. Encyclopedia of Computational Chemistry; Schleyer, P.v.R., Allinger, N.L., Clark, T., Gasteiger, J., Kollman, P.A., Schaefer, H.F., III, Schreiner, P.R., Eds.; John Wiley & Sons: Chichester, UK, 1998; Volume 3, p. 1792. [Google Scholar]
- Weinhold, F.; Carpenter, J.E. The Structure of Small Molecules and Ions; Naaman, R., Vager, Z., Eds.; Plenum: New York, NY, USA, 1988; p. 227. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Carpenter, J.E.; Weinhold, F. Analysis of the geometry of the hydroxymethyl radical by the “different hybrids for different spins” natural bond orbital procedure. J. Mol. Struct. THEOCHEM 1988, 169, 41–62. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Outer Helix | Inner Helix | ||||
|---|---|---|---|---|---|
| Bond | Fraction of p a | s-Character | Bond | Fraction of p a | s-Character |
| Compound: 1 | |||||
| C1-C2 | 1.73 | 0.37 | C13-C13a | 1.83 | 0.35 |
| C2-C3 | 1.85 | 0.35 | C13a-C13b | 2.06 | 0.33 |
| C3-C4 | 1.74 | 0.36 | C13b-C13c | 1.93 | 0.34 |
| C4-C4a | 1.90 | 0.34 | C13c-C13d | 1.95 | 0.34 |
| C4a-C5 | 2.00 | 0.33 | C13d-C1 | 1.90 | 0.35 |
| C5-C6 | 1.67 | 0.38 | |||
| C10-C11 | 1.77 | 0.36 | |||
| C11-C12 | 1.82 | 0.35 | |||
| C12-C13 | 1.76 | 0.36 | |||
| Compound: 4 | |||||
| C1-C2 | 1.68 | 0.37 | C11-C11a | 1.83 | 0.35 |
| C2-C2a | 1.97 | 0.34 | C11a-C11b | 2.06 | 0.33 |
| C2a-C3 | 1.97 | 0.34 | C11b-C11c | 1.91 | 0.34 |
| C3-C4 | 1.69 | 0.37 | C11c-C11d | 1.98 | 0.34 |
| C8-C9 | 1.77 | 0.36 | C11d-C11e | 2.01 | 0.33 |
| C9-C10 | 1.82 | 0.36 | C11e-C12 | 1.91 | 0.34 |
| C13-C14 | 1.85 | 0.35 | |||
| C14-C15 | 1.74 | 0.37 | |||
| C15-C15a | 1.92 | 0.34 | |||
| C15a-C1 | 1.99 | 0.33 | |||
| Compound: 13 | |||||
| C1-C2 | 3.47 | 0.37 | C17-C17a | 3.74 | 0.35 |
| C2-C3 | 3.69 | 0.35 | C17a-C17b | 4.02 | 0.33 |
| C3-C4 | 3.48 | 0.36 | C17b-C17c | 4.06 | 0.33 |
| C4-C4a | 3.80 | 0.34 | C17c-C17d | 3.85 | 0.34 |
| C4a-C5 | 3.99 | 0.33 | C17d-C17e | 4.01 | 0.33 |
| C5-C6 | 3.33 | 0.38 | C17e-C1 | 3.82 | 0.34 |
| C9b-C10 | 3.78 | 0.35 | |||
| C10-C11 | 3.50 | 0.36 | |||
| C11-C12 | 3.65 | 0.35 | |||
| C12-C13 | 3.55 | 0.36 | |||
| C13-C13a | 3.79 | 0.35 | |||
| C13b-C14 | 3.77 | 0.35 | |||
| C14-C15 | 3.50 | 0.36 | |||
| C15-C16 | 3.67 | 0.35 | |||
| C16-C17 | 3.50 | 0.36 | |||
| Compound: 15 | |||||
| C1-C2 | 1.68 | 0.37 | C13a-C13b | 2.05 | 0.33 |
| C2-C2a | 1.97 | 0.34 | C13b-C13c | 1.92 | 0.34 |
| C2a-C3 | 1.97 | 0.34 | C13c-C13d | 1.99 | 0.34 |
| C3-C4 | 1.68 | 0.37 | C13d-C13e | 2.01 | 0.33 |
| C7b-C8 | 1.93 | 0.34 | C13e-C14 | 1.90 | 0.34 |
| C8-C9 | 1.73 | 0.37 | |||
| C9-C10 | 1.86 | 0.35 | |||
| C10-C11 | 1.73 | 0.37 | |||
| C11-C11a | 1.93 | 0.34 | |||
| C11a-C12 | 1.95 | 0.34 | |||
| C12-C13 | 1.68 | 0.37 | |||
| C15-C16 | 1.85 | 0.35 | |||
| C16-C17 | 1.74 | 0.37 | |||
| C17-C17a | 1.91 | 0.34 | |||
| C17a-C1 | 1.98 | 0.34 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semenov, V.A.; Martin, G.E.; Krivdin, L.B. Computational NMR Study of Benzothienoquinoline Heterohelicenes. Int. J. Mol. Sci. 2024, 25, 7733. https://doi.org/10.3390/ijms25147733
Semenov VA, Martin GE, Krivdin LB. Computational NMR Study of Benzothienoquinoline Heterohelicenes. International Journal of Molecular Sciences. 2024; 25(14):7733. https://doi.org/10.3390/ijms25147733
Chicago/Turabian StyleSemenov, Valentin A., Gary E. Martin, and Leonid B. Krivdin. 2024. "Computational NMR Study of Benzothienoquinoline Heterohelicenes" International Journal of Molecular Sciences 25, no. 14: 7733. https://doi.org/10.3390/ijms25147733