

Novel AT2 Cell Subpopulations and Diagnostic Biomarkers in IPF: Integrating Machine Learning with Single-Cell Analysis


Abstract
1. Introduction
2. Results
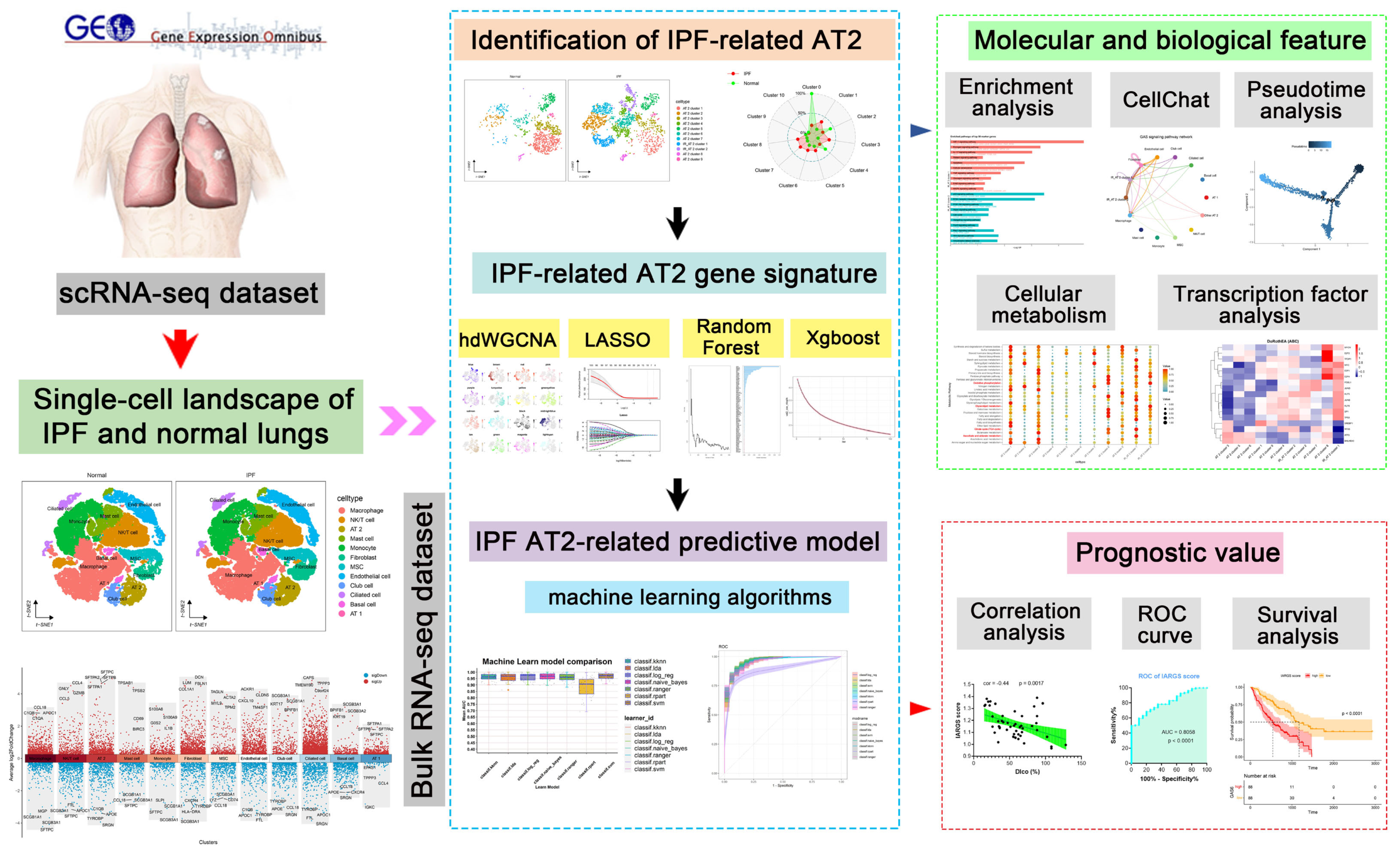
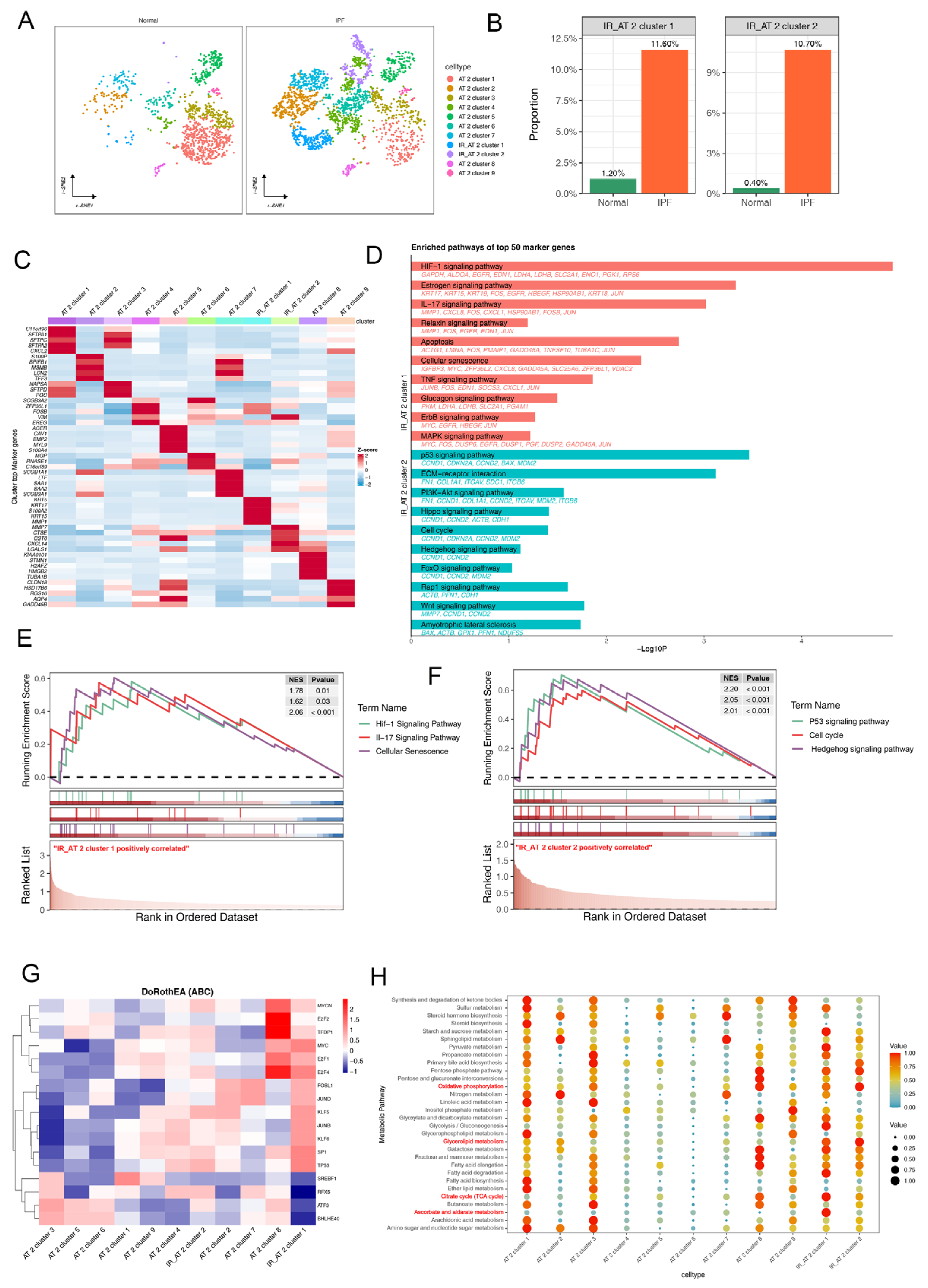
2.1. Single-Cell Transcriptional Landscape of AT2 Cells in Normal and Fibrotic Lung Tissues
2.2. Pseudo-Time Trajectory Analysis and Cell Communication Analysis
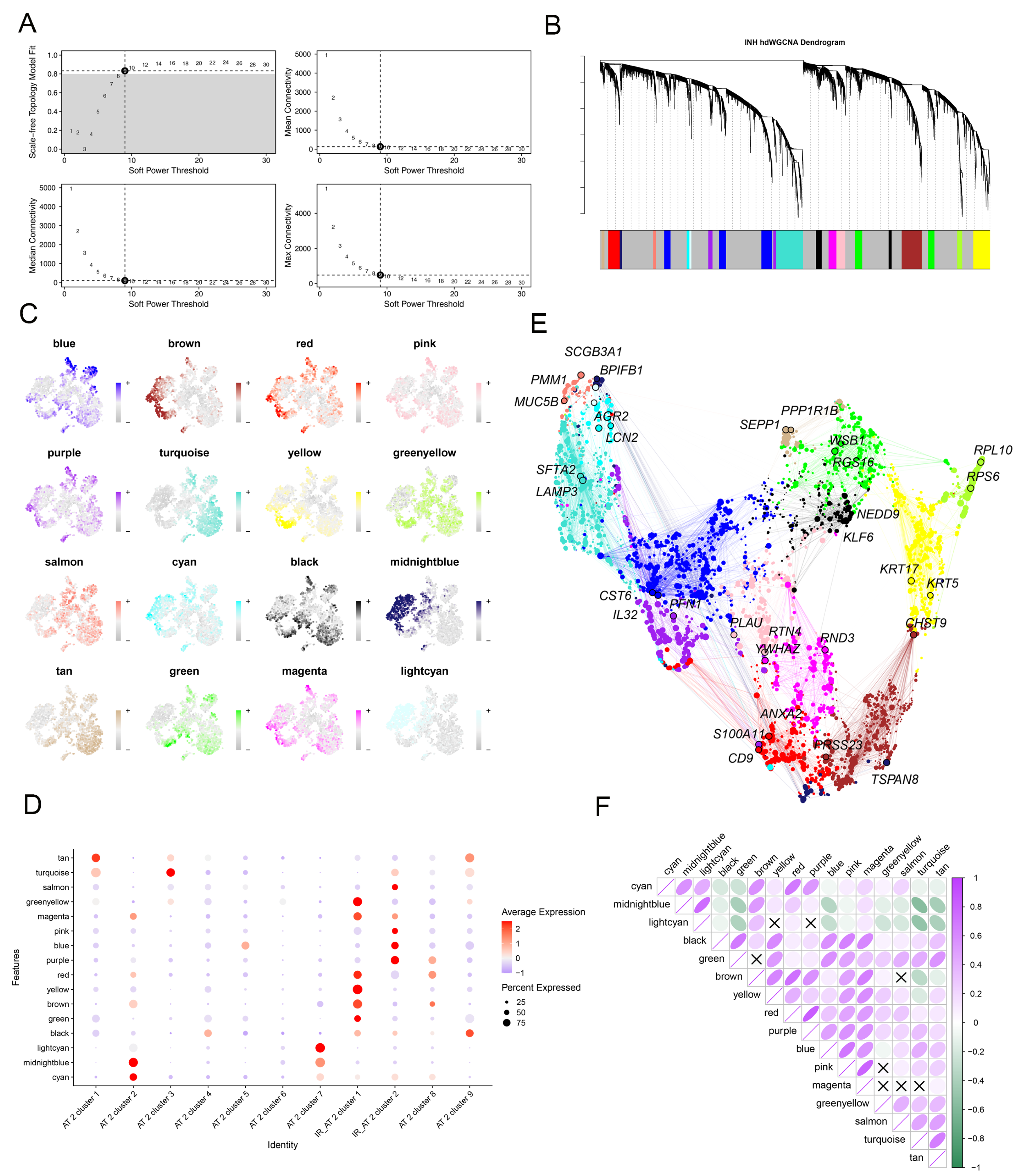
2.3. Identification of the Crucial Modules Related to IPF-Related AT2 by hdWGCNA
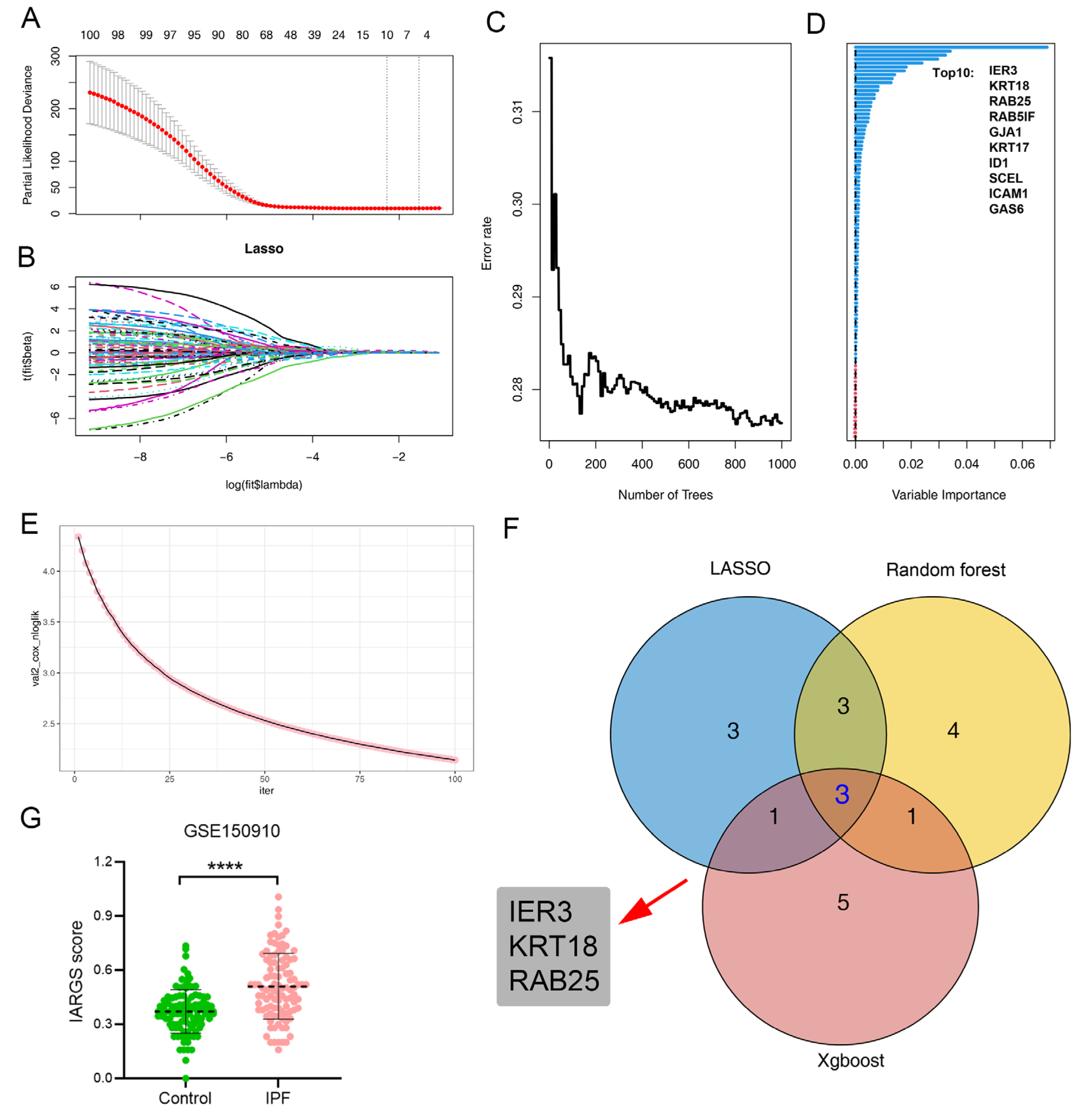
2.4. Various Machine Learning Algorithms Identifies Signature Genes for IPF-Related AT2 Cell
2.5. Machine Learning-Based Construction of the Predictive Model for IPF
3. Discussion
4. Materials and Methods
4.1. Data Acquisition and Processing
4.2. Trajectory and Cell–Cell Communication Analysis
4.3. Enrichment Analysis
4.4. High Dimensional Weighted Gene Co-Expression Network Analysis (hdWGCNA)
4.5. Construction of Machine Learning Model
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Spagnolo, P.; Kropski, J.A.; Jones, M.G.; Lee, J.S.; Rossi, G.; Karampitsakos, T.; Maher, T.M.; Tzouvelekis, A.; Ryerson, C.J. Idiopathic pulmonary fibrosis: Disease mechanisms and drug development. Pharmacol. Ther. 2021, 222, 107798. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M. Interstitial Lung Disease: A Review. JAMA 2024, 331, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.L.; Rojas, M.; Pardo, A.; Selman, M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov. 2017, 16, 755–772. [Google Scholar] [CrossRef] [PubMed]
- Katzen, J.; Beers, M.F. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J. Clin. Investig. 2020, 130, 5088–5099. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Guan, X.; Carraro, G.; Parimon, T.; Liu, X.; Huang, G.; Mulay, A.; Soukiasian, H.J.; David, G.; Weigt, S.S.; et al. Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Salton, F.; Ruaro, B.; Confalonieri, P.; Confalonieri, M. Epithelial-Mesenchymal Transition: A Major Pathogenic Driver in Idiopathic Pulmonary Fibrosis? Medicina 2020, 56, 608. [Google Scholar] [CrossRef] [PubMed]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, K.; Shah, P.P.; Morley, M.P.; Loebel, C.; Santini, G.T.; Katzen, J.; Basil, M.C.; Lin, S.M.; Planer, J.D.; Cantu, E.; et al. Biophysical forces mediated by respiration maintain lung alveolar epithelial cell fate. Cell 2023, 186, 1478–1492.e1415. [Google Scholar] [CrossRef]
- Parimon, T.; Chen, P.; Stripp, B.R.; Liang, J.; Jiang, D.; Noble, P.W.; Parks, W.C.; Yao, C. Senescence of alveolar epithelial progenitor cells: A critical driver of lung fibrosis. Am. J. Physiol. Cell Physiol. 2023, 325, C483–C495. [Google Scholar] [CrossRef]
- Enomoto, Y.; Katsura, H.; Fujimura, T.; Ogata, A.; Baba, S.; Yamaoka, A.; Kihara, M.; Abe, T.; Nishimura, O.; Kadota, M.; et al. Autocrine TGF-β-positive feedback in profibrotic AT2-lineage cells plays a crucial role in non-inflammatory lung fibrogenesis. Nat. Commun. 2023, 14, 4956. [Google Scholar] [CrossRef] [PubMed]
- Li, L.F.; Yu, C.C.; Huang, C.Y.; Wu, H.P.; Chu, C.M.; Liu, P.C.; Liu, Y.Y. Attenuation of Ventilation-Enhanced Epithelial-Mesenchymal Transition through the Phosphoinositide 3-Kinase-γ in a Murine Bleomycin-Induced Acute Lung Injury Model. Int. J. Mol. Sci. 2023, 24, 5538. [Google Scholar] [CrossRef] [PubMed]
- Olajuyin, A.M.; Zhang, X.; Ji, H.L. Alveolar type 2 progenitor cells for lung injury repair. Cell Death Discov. 2019, 5, 63. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, Y. Heterogeneous groups of alveolar type II cells in lung homeostasis and repair. Am. J. Physiol. Cell Physiol. 2020, 319, C991–C996. [Google Scholar] [CrossRef] [PubMed]
- Unterman, A.; Zhao, A.Y.; Neumark, N.; Schupp, J.C.; Ahangari, F.; Cosme, C., Jr.; Sharma, P.; Flint, J.; Stein, Y.; Ryu, C.; et al. Single-Cell Profiling Reveals Immune Aberrations in Progressive Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2024. [Google Scholar] [CrossRef]
- Konkimalla, A.; Konishi, S.; Macadlo, L.; Kobayashi, Y.; Farino, Z.J.; Miyashita, N.; El Haddad, L.; Morowitz, J.; Barkauskas, C.E.; Agarwal, P.; et al. Transitional cell states sculpt tissue topology during lung regeneration. Cell Stem Cell 2023, 30, 1486–1502.e1489. [Google Scholar] [CrossRef]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.T.; Funari, V.A.; Gokey, J.J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016, 1, e90558. [Google Scholar] [CrossRef] [PubMed]
- Deo, R.C. Machine Learning in Medicine. Circulation 2015, 132, 1920–1930. [Google Scholar] [CrossRef] [PubMed]
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 2022, 23, 40–55. [Google Scholar] [CrossRef]
- Reel, P.S.; Reel, S.; Pearson, E.; Trucco, E.; Jefferson, E. Using machine learning approaches for multi-omics data analysis: A review. Biotechnol. Adv. 2021, 49, 107739. [Google Scholar] [CrossRef]
- Mei, Q.; Liu, Z.; Zuo, H.; Yang, Z.; Qu, J. Idiopathic Pulmonary Fibrosis: An Update on Pathogenesis. Front. Pharmacol. 2021, 12, 797292. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, X.; Ma, Q.; Wang, Q.; Wu, J.; Yu, H.; Li, K.; Li, Y.; Wang, J.; Zhang, Q.; et al. Glutamine Metabolism Is Required for Alveolar Regeneration during Lung Injury. Biomolecules 2022, 12, 728. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Zhang, J.; Qing, G.; Liu, D.; Wang, X.; Chen, Y.; Li, Y.; Guo, S. S100A2 Silencing Relieves Epithelial-Mesenchymal Transition in Pulmonary Fibrosis by Inhibiting the Wnt/β-Catenin Signaling Pathway. DNA Cell Biol. 2021, 40, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Craig, V.J.; Zhang, L.; Hagood, J.S.; Owen, C.A. Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 53, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Ahmadvand, N.; Khosravi, F.; Lingampally, A.; Wasnick, R.; Vazquez-Armendariz, A.I.; Carraro, G.; Heiner, M.; Rivetti, S.; Lv, Y.; Wilhelm, J.; et al. Identification of a novel subset of alveolar type 2 cells enriched in PD-L1 and expanded following pneumonectomy. Eur. Respir. J. 2021, 58, 2004168. [Google Scholar] [CrossRef]
- Kindler, O.; Pulkkinen, O.; Cherstvy, A.G.; Metzler, R. Burst statistics in an early biofilm quorum sensing model: The role of spatial colony-growth heterogeneity. Sci. Rep. 2019, 9, 12077. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, X.; Yang, C.; Wang, S.; Shen, H. Essential role of IL-17 in acute exacerbation of pulmonary fibrosis induced by non-typeable Haemophilus influenzae. Theranostics 2022, 12, 5125–5137. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, C.; Cao, H.; Chen, L.; Hou, J.; Xiang, Z.; Hu, K.; Han, X. The hedgehog and Wnt/β-catenin system machinery mediate myofibroblast differentiation of LR-MSCs in pulmonary fibrogenesis. Cell Death Dis. 2018, 9, 639. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Y. Epithelial stem cells and niches in lung alveolar regeneration and diseases. Chin. Med. J. Pulm. Crit. Care Med. 2024, 2, 17–26. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, L.; Ma, S.; Cheng, L.; Yu, G. Repair and regeneration of the alveolar epithelium in lung injury. FASEB J. 2024, 38, e23612. [Google Scholar] [CrossRef]
- Wilson, C.; Mertens, T.C.; Shivshankar, P.; Bi, W.; Collum, S.D.; Wareing, N.; Ko, J.; Weng, T.; Naikawadi, R.P.; Wolters, P.J.; et al. Sine oculis homeobox homolog 1 plays a critical role in pulmonary fibrosis. JCI Insight 2022, 7, e142984. [Google Scholar] [CrossRef] [PubMed]
- Bellan, M.; Cittone, M.G.; Tonello, S.; Rigamonti, C.; Castello, L.M.; Gavelli, F.; Pirisi, M.; Sainaghi, P.P. Gas6/TAM System: A Key Modulator of the Interplay between Inflammation and Fibrosis. Int. J. Mol. Sci. 2019, 20, 5070. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Yang, Y.; Han, X. Machine Learning and Single-Cell Analysis Identify Molecular Features of IPF-Associated Fibroblast Subtypes and Their Implications on IPF Prognosis. Int. J. Mol. Sci. 2023, 25, 94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhao, S.; Yao, Z.; Wang, L.; Shao, J.; Chen, A.; Zhang, F.; Zheng, S. Autophagy regulates turnover of lipid droplets via ROS-dependent Rab25 activation in hepatic stellate cell. Redox Biol. 2017, 11, 322–334. [Google Scholar] [CrossRef] [PubMed]
- Morse, C.; Tabib, T.; Sembrat, J.; Buschur, K.L.; Bittar, H.T.; Valenzi, E.; Jiang, Y.; Kass, D.J.; Gibson, K.; Chen, W.; et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur. Respir. J. 2019, 54, 1802441. [Google Scholar] [CrossRef] [PubMed]
- Hanley, C.J.; Waise, S.; Ellis, M.J.; Lopez, M.A.; Pun, W.Y.; Taylor, J.; Parker, R.; Kimbley, L.M.; Chee, S.J.; Shaw, E.C.; et al. Single-cell analysis reveals prognostic fibroblast subpopulations linked to molecular and immunological subtypes of lung cancer. Nat. Commun. 2023, 14, 387. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]
- Morabito, S.; Reese, F.; Rahimzadeh, N.; Miyoshi, E.; Swarup, V. hdWGCNA identifies co-expression networks in high-dimensional transcriptomics data. Cell Rep. Methods 2023, 3, 100498. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Year | Area | Species | Platform | Data Type | Number of Samples | |
|---|---|---|---|---|---|---|---|
| Normal | IPF | ||||||
| GSE128033 | 2019 | USA | Homo | GPL18573 | scRNA-seq | 10 | 8 |
| GSE150910 | 2020 | USA | Homo | GPL24676 | Bulk RNA-seq | 103 | 103 |
| GSE32537 | 2011 | USA | Homo | GPL6244 | Bulk RNA-seq | 39 | 131 |
| GSE110147 | 2018 | Canada | Homo | GPL6244 | Bulk RNA-seq | 11 | 22 |
| GSE70866 | 2015 | Germany | Homo | GPL14550 | Bulk RNA-seq | 20 | 212 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Yang, Y.; Han, X.; Hou, J. Novel AT2 Cell Subpopulations and Diagnostic Biomarkers in IPF: Integrating Machine Learning with Single-Cell Analysis. Int. J. Mol. Sci. 2024, 25, 7754. https://doi.org/10.3390/ijms25147754
Yang Z, Yang Y, Han X, Hou J. Novel AT2 Cell Subpopulations and Diagnostic Biomarkers in IPF: Integrating Machine Learning with Single-Cell Analysis. International Journal of Molecular Sciences. 2024; 25(14):7754. https://doi.org/10.3390/ijms25147754
Chicago/Turabian StyleYang, Zhuoying, Yanru Yang, Xin Han, and Jiwei Hou. 2024. "Novel AT2 Cell Subpopulations and Diagnostic Biomarkers in IPF: Integrating Machine Learning with Single-Cell Analysis" International Journal of Molecular Sciences 25, no. 14: 7754. https://doi.org/10.3390/ijms25147754
APA StyleYang, Z., Yang, Y., Han, X., & Hou, J. (2024). Novel AT2 Cell Subpopulations and Diagnostic Biomarkers in IPF: Integrating Machine Learning with Single-Cell Analysis. International Journal of Molecular Sciences, 25(14), 7754. https://doi.org/10.3390/ijms25147754