
Tumor Necrosis Factor-Alpha: Ally and Enemy in Protean Cutaneous Sceneries
 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
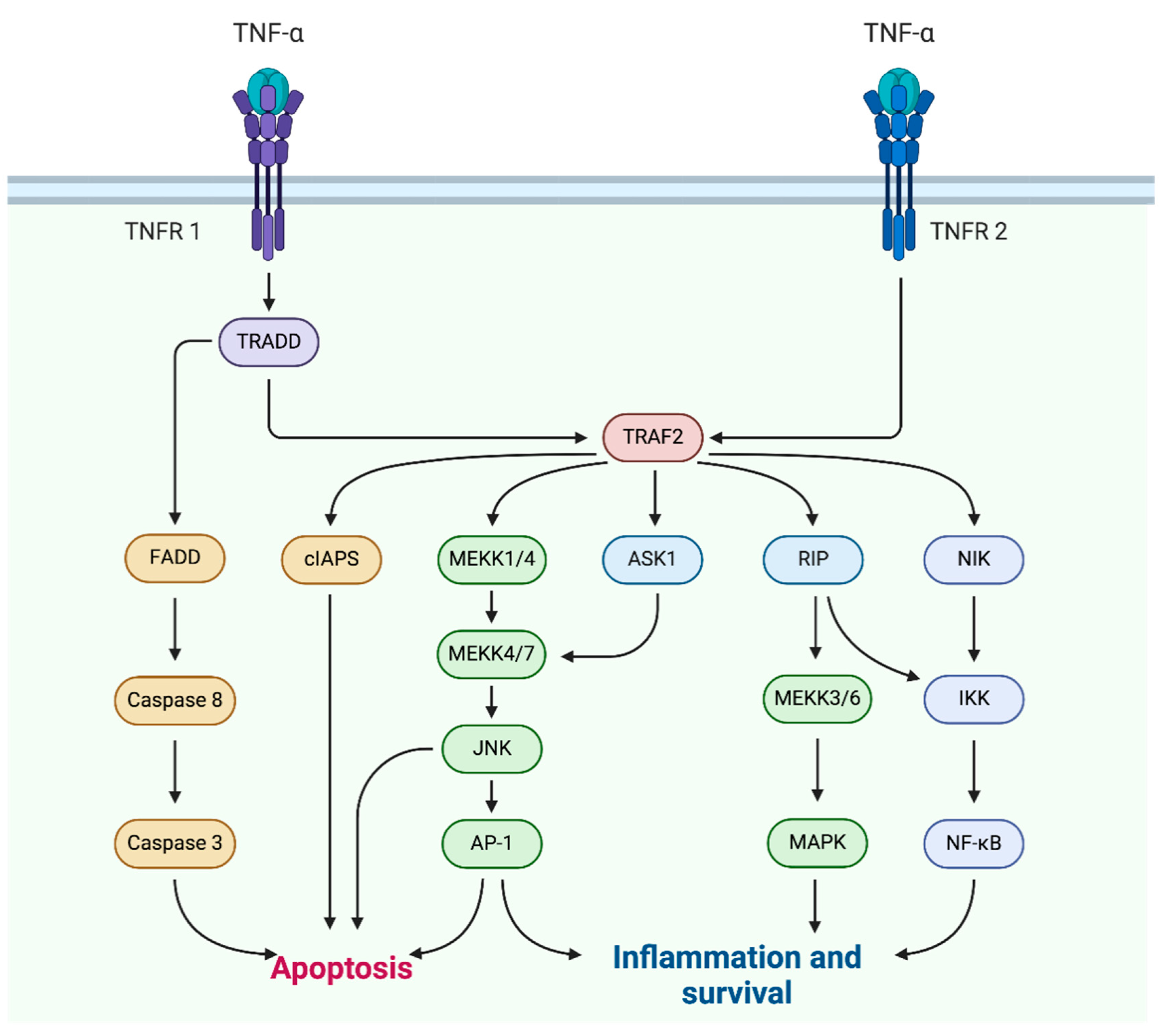
2. TNF-α in Skin Diseases
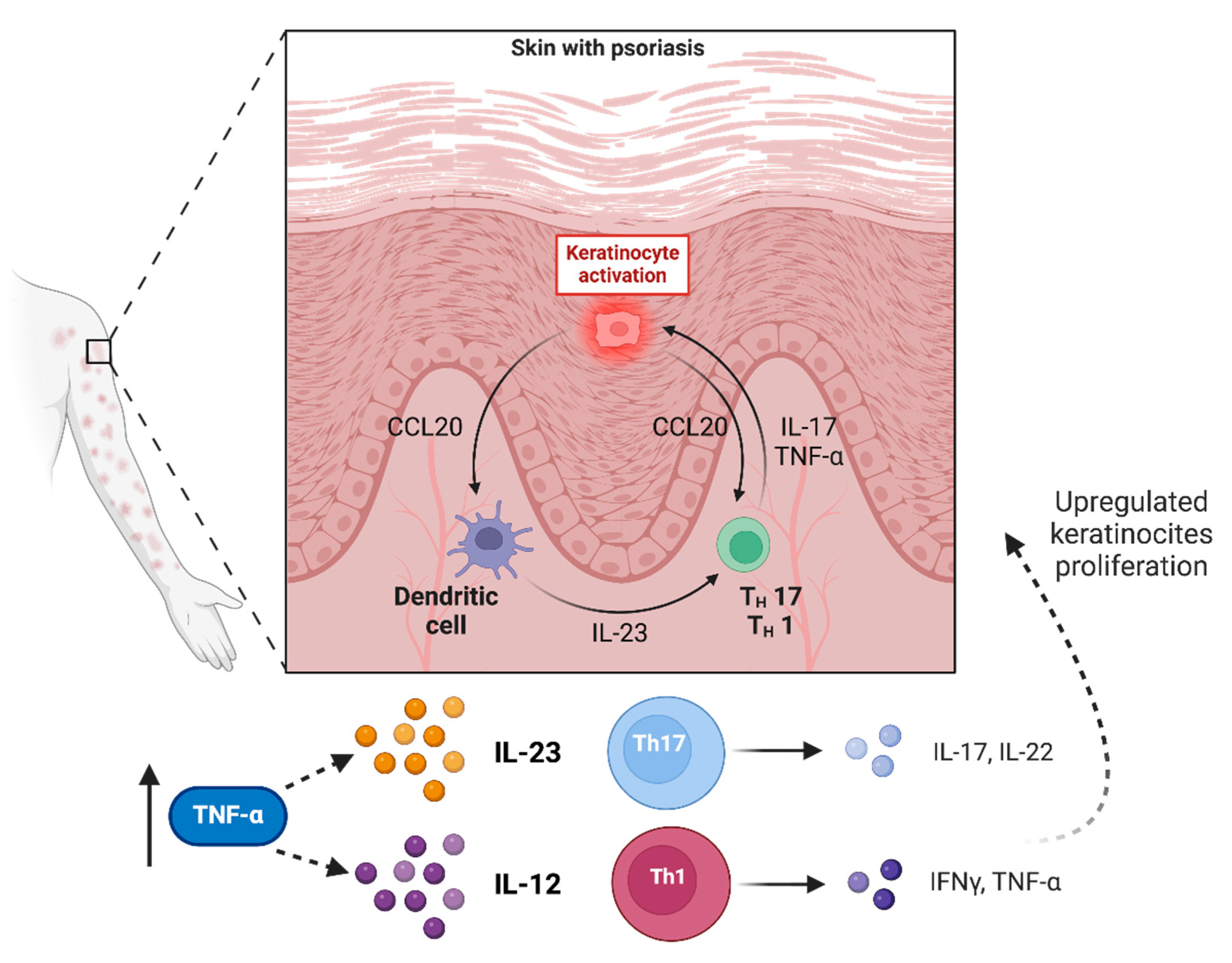
2.1. Psoriasis
2.2. Vitiligo
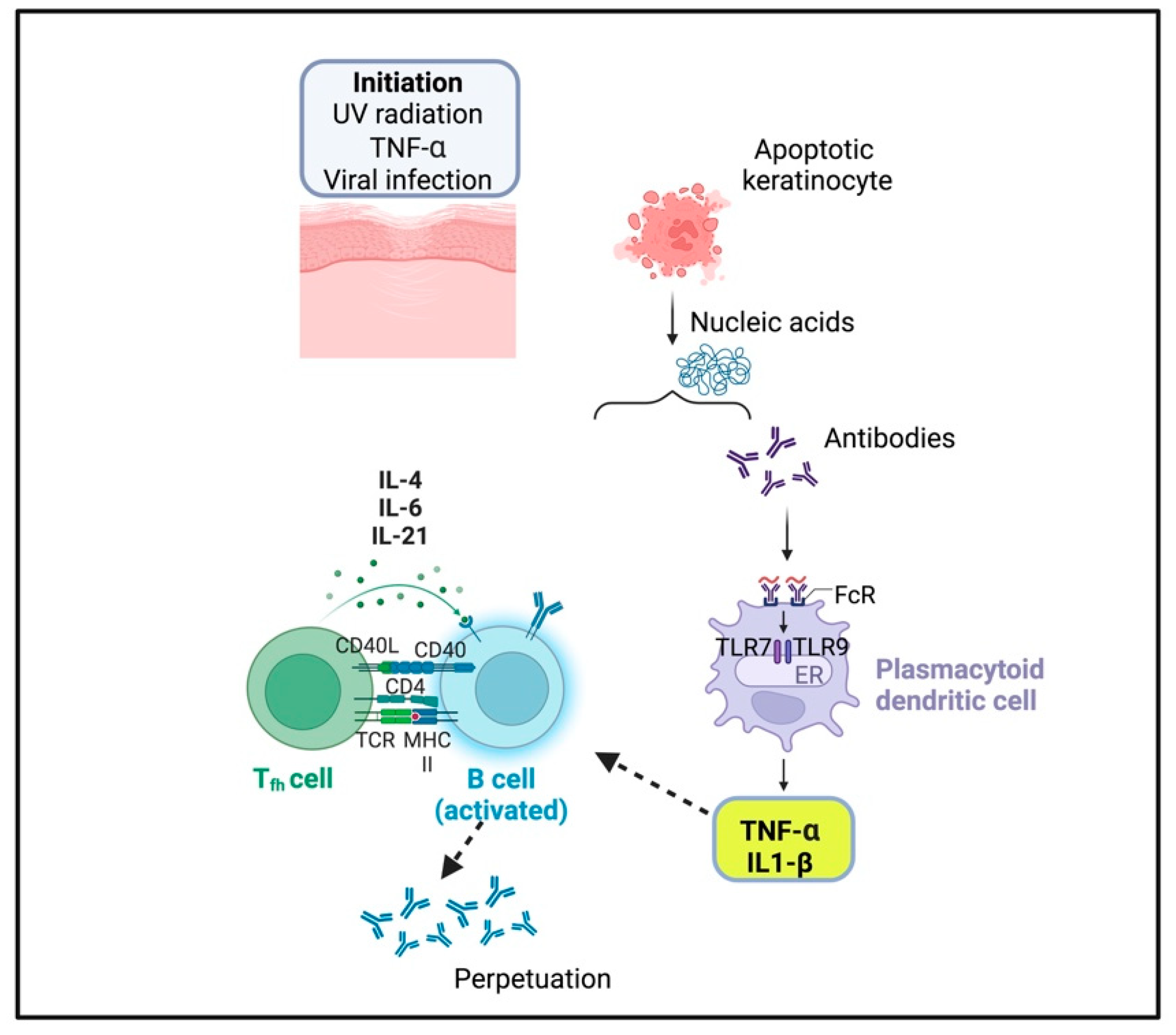
2.3. Cutaneous Lupus Erythematosus
2.4. Acne Vulgaris and Acne Inversa
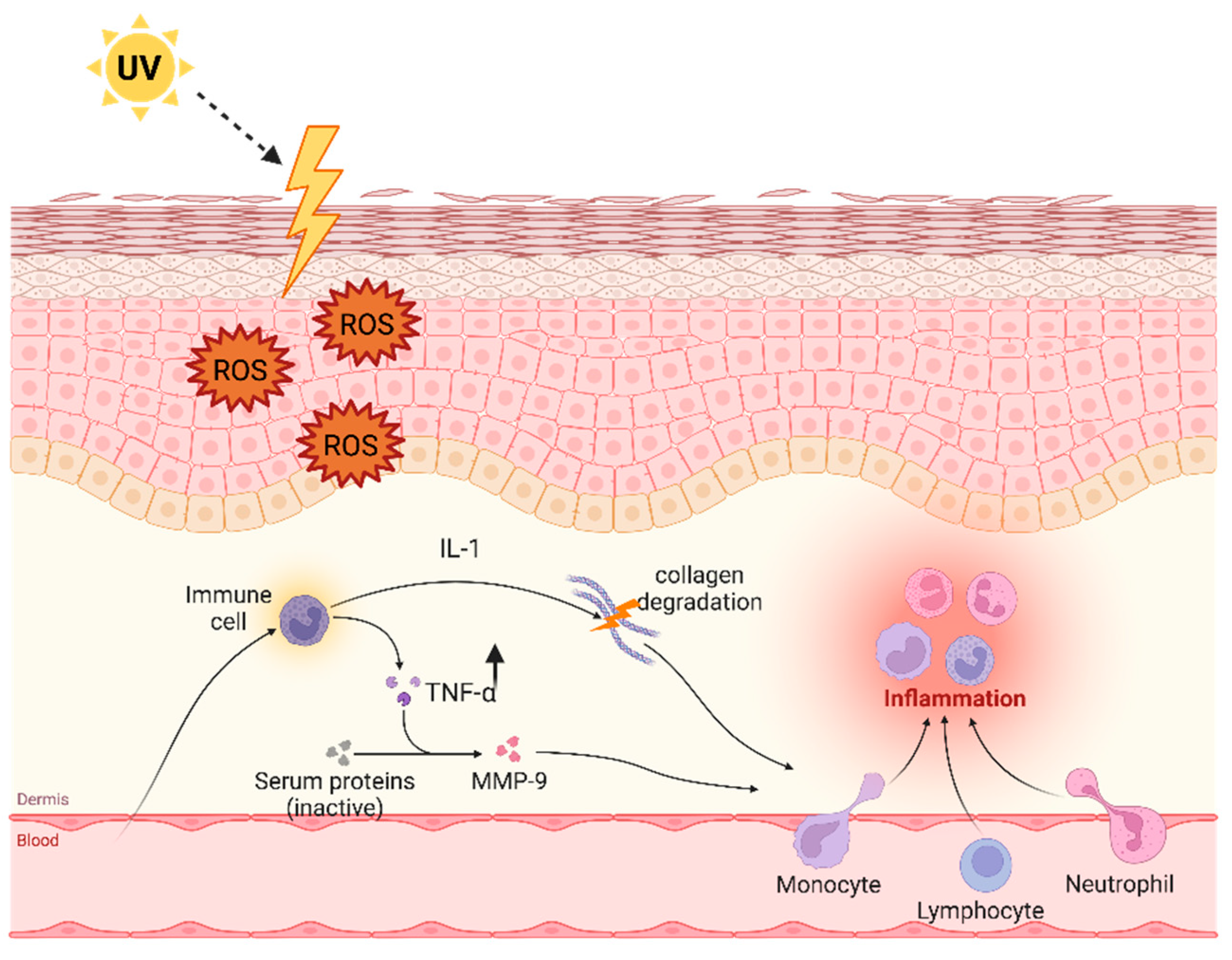
3. TNF-α after Exposition to Ultraviolet Light
Skin Aging
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Aggarwal, B.B.; Gupta, S.C.; Kim, J.H. Historical Perspectives on Tumor Necrosis Factor and Its Superfamily: 25 Years Later, a Golden Journey. Blood 2012, 119, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lu, J.; Liu, J.; Wu, J.; Zhang, X.; Meng, Y.; Wu, X.; Tai, Z.; Zhu, Q.; Chen, Z. Immune Cells in the Epithelial Immune Microenvironment of Psoriasis: Emerging Therapeutic Targets. Front. Immunol. 2023, 14, 1340677. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Guo, P.; Lv, N.; Huang, D. Lipopolysaccharide-Induced Tumor Necrosis Factor-α Factor Enhances Inflammation and Is Associated with Cancer. Mol. Med. Rep. 2015, 12, 6399–6404. [Google Scholar] [CrossRef] [PubMed]
- Zelová, H.; Hošek, J. TNF-α Signalling and Inflammation: Interactions between Old Acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Camussi, G.; Albano, E.; Tetta, C.; Bussolino, F. The Molecular Action of Tumor Necrosis Factor-Alpha. Eur. J. Biochem. 1991, 202, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Krochin, N.; Milsark, I.W.; Luedke, C.; Cerami, A. Control of Cachectin (Tumor Necrosis Factor) Synthesis: Mechanisms of Endotoxin Resistance. Science 1986, 232, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E. Tumor Necrosis Factor Has Positive and Negative Self Regulatory Feed Back Cycles Centered around cAMP. Int. J. Immunopharmacol. 2000, 22, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Idriss, H.T.; Naismith, J.H. TNF Alpha and the TNF Receptor Superfamily: Structure-Function Relationship(s). Microsc. Res. Tech. 2000, 50, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M. Tumour Necrosis Factor Signalling in Health and Disease. F1000Research 2019, 8, F1000 Faculty Rev-111. [Google Scholar] [CrossRef]
- Victor, F.C.; Gottlieb, A.B. TNF-Alpha and Apoptosis: Implications for the Pathogenesis and Treatment of Psoriasis. J. Drugs Dermatol. 2002, 1, 264–275. [Google Scholar]
- Terajima, S.; Higaki, M.; Igarashi, Y.; Nogita, T.; Kawashima, M. An Important Role of Tumor Necrosis Factor-Alpha in the Induction of Adhesion Molecules in Psoriasis. Arch. Dermatol. Res. 1998, 290, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Nicols, B.F.; Robledo-Pulido, J.J.; Alvarado-Navarro, A. Etiopathogenesis of Psoriasis: Integration of Proposed Theories. Immunol. Investig. 2024, 53, 348–415. [Google Scholar] [CrossRef] [PubMed]
- Harper, E.G.; Guo, C.; Rizzo, H.; Lillis, J.V.; Kurtz, S.E.; Skorcheva, I.; Purdy, D.; Fitch, E.; Iordanov, M.; Blauvelt, A. Th17 Cytokines Stimulate CCL20 Expression in Keratinocytes In Vitro and In Vivo: Implications for Psoriasis Pathogenesis. J. Investig. Dermatol. 2009, 129, 2175–2183. [Google Scholar] [CrossRef]
- Bernink, J.H.; Ohne, Y.; Teunissen, M.B.M.; Wang, J.; Wu, J.; Krabbendam, L.; Guntermann, C.; Volckmann, R.; Koster, J.; van Tol, S.; et al. C-Kit-Positive ILC2s Exhibit an ILC3-like Signature That May Contribute to IL-17-Mediated Pathologies. Nat. Immunol. 2019, 20, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.A. Traffic Signals for Lymphocyte Recirculation and Leukocyte Emigration: The Multistep Paradigm. Cell 1994, 76, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Harvima, I.T. Mast Cells of Psoriatic and Atopic Dermatitis Skin Are Positive for TNF-Alpha and Their Degranulation Is Associated with Expression of ICAM-1 in the Epidermis. Arch. Dermatol. Res. 1998, 290, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenberger, K.; Udey, M.C. Contact Allergens and Epidermal Proinflammatory Cytokines Modulate Langerhans Cell E-Cadherin Expression in Situ. J. Investig. Dermatol. 1996, 106, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, M.; Chu, C.Q.; Eedy, D.J.; Feldmann, M.; Brennan, F.M.; Breathnach, S.M. Localization of Tumour Necrosis Factor-Alpha (TNF-Alpha) and Its Receptors in Normal and Psoriatic Skin: Epidermal Cells Express the 55-kD but Not the 75-kD TNF Receptor. Clin. Exp. Immunol. 1993, 94, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Akcılar, R.; Dizen Namdar, N.; Yükcü, F.; Arslan Utku, S. TNF-α Gene -238G>A Polymorphism Is Associated with Psoriasis Patients. J. Cosmet. Dermatol. 2022, 21, 2662–2667. [Google Scholar] [CrossRef]
- Dapavo, P.; Siliquini, N.; Mastorino, L.; Avallone, G.; Merli, M.; Agostini, A.; Cariti, C.; Viola, R.; Stroppiana, E.; Verrone, A.; et al. Efficacy, Safety, and Drug Survival of IL-23, IL-17, and TNF-Alpha Inhibitors for Psoriasis Treatment: A Retrospective Study. J. Dermatol. Treat. 2022, 33, 2352–2357. [Google Scholar] [CrossRef]
- Camara-Lemarroy, C.R.; Salas-Alanis, J.C. The Role of Tumor Necrosis Factor-α in the Pathogenesis of Vitiligo. Am. J. Clin. Dermatol. 2013, 14, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Bergqvist, C.; Ezzedine, K. Vitiligo: A Review. Dermatology 2020, 236, 571–592. [Google Scholar] [CrossRef] [PubMed]
- Funasaka, Y.; Chakraborty, A.K.; Hayashi, Y.; Komoto, M.; Ohashi, A.; Nagahama, M.; Inoue, Y.; Pawelek, J.; Ichihashi, M. Modulation of Melanocyte-Stimulating Hormone Receptor Expression on Normal Human Melanocytes: Evidence for a Regulatory Role of Ultraviolet B, Interleukin-1alpha, Interleukin-1beta, Endothelin-1 and Tumour Necrosis Factor-Alpha. Br. J. Dermatol. 1998, 139, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Laddha, N.C.; Dwivedi, M.; Mansuri, M.S.; Gani, A.R.; Ansarullah, M.; Ramachandran, A.V.; Dalai, S.; Begum, R. Vitiligo: Interplay between Oxidative Stress and Immune System. Exp. Dermatol. 2013, 22, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; De Sarkar, S.; Pradhan, A.; Pati, A.K.; Pradhan, R.; Mondal, D.; Sen, S.; Ghosh, A.; Chatterjee, S.; Chatterjee, M. Levels of Oxidative Damage and Proinflammatory Cytokines Are Enhanced in Patients with Active Vitiligo. Free Radic. Res. 2017, 51, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Mansuri, M.S.; Kadam, A.; Palit, S.P.; Dwivedi, M.; Laddha, N.C.; Begum, R. Tumor Necrosis Factor-Alpha Affects Melanocyte Survival and Melanin Synthesis via Multiple Pathways in Vitiligo. Cytokine 2021, 140, 155432. [Google Scholar] [CrossRef] [PubMed]
- Al Badri, A.M.T.; Foulis, A.K.; Todd, P.M.; Garioch, J.J.; Gudgeon, J.E.; Stewart, D.G.; Gracie, J.A.; Goudie, R.B. Abnormal Expression of MHC Class II and ICAM-1 by Melanocytes in Vitiligo. J. Pathol. 1993, 169, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Wańkowicz-Kalińska, A.; van den Wijngaard, R.M.J.G.J.; Tigges, B.J.; Westerhof, W.; Ogg, G.S.; Cerundolo, V.; Storkus, W.J.; Das, P.K. Immunopolarization of CD4+ and CD8+ T Cells to Type-1-like Is Associated with Melanocyte Loss in Human Vitiligo. Lab. Investig. 2003, 83, 683–695. [Google Scholar] [CrossRef]
- Ranges, G.E.; Figari, I.S.; Espevik, T.; Palladino, M.A. Inhibition of Cytotoxic T Cell Development by Transforming Growth Factor Beta and Reversal by Recombinant Tumor Necrosis Factor Alpha. J. Exp. Med. 1987, 166, 991–998. [Google Scholar] [CrossRef]
- Liu, H.; Wang, Y.; Le, Q.; Tong, J.; Wang, H. The IFN-γ-CXCL9/CXCL10-CXCR3 Axis in Vitiligo: Pathological Mechanism and Treatment. Eur. J. Immunol. 2023, 54, e2250281. [Google Scholar] [CrossRef]
- Kim, N.H.; Torchia, D.; Rouhani, P.; Roberts, B.; Romanelli, P. Tumor Necrosis Factor-α in Vitiligo: Direct Correlation between Tissue Levels and Clinical Parameters. Cutan. Ocul. Toxicol. 2011, 30, 225–227. [Google Scholar] [CrossRef]
- Sushama, S.; Dixit, N.; Gautam, R.K.; Arora, P.; Khurana, A.; Anubhuti, A. Cytokine Profile (IL-2, IL-6, IL-17, IL-22, and TNF-α) in Vitiligo-New Insight into Pathogenesis of Disease. J. Cosmet. Dermatol. 2019, 18, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Webb, K.C.; Tung, R.; Winterfield, L.S.; Gottlieb, A.B.; Eby, J.M.; Henning, S.W.; Le Poole, I.C. Tumour Necrosis Factor-α Inhibition Can Stabilize Disease in Progressive Vitiligo. Br. J. Dermatol. 2015, 173, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Maruthappu, T.; Leandro, M.; Morris, S.D. Deterioration of Vitiligo and New Onset of Halo Naevi Observed in Two Patients Receiving Adalimumab. Dermatol. Ther. 2013, 26, 370–372. [Google Scholar] [CrossRef]
- Ramírez-Hernández, M.; Marras, C.; Martínez-Escribano, J.A. Infliximab-Induced Vitiligo. Dermatology 2005, 210, 79–80. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.M.; Lee, Y.J.; Won, C.H.; Chang, S.E.; Lee, M.W.; Choi, J.H.; Moon, K.C. Development of Vitiligo during Treatment with Adalimumab: A Plausible or Paradoxical Response? Ann. Dermatol. 2015, 27, 620–621. [Google Scholar] [CrossRef]
- Bae, J.M.; Kim, M.; Lee, H.H.; Kim, K.-J.; Shin, H.; Ju, H.J.; Kim, G.M.; Park, C.J.; Park, H.J. Increased Risk of Vitiligo Following Anti-Tumor Necrosis Factor Therapy: A 10-Year Population-Based Cohort Study. J. Investig. Dermatol. 2018, 138, 768–774. [Google Scholar] [CrossRef]
- Burlando, M.; Muracchioli, A.; Cozzani, E.; Parodi, A. Psoriasis, Vitiligo, and Biologic Therapy: Case Report and Narrative Review. Case Rep. Dermatol. 2021, 13, 372–378. [Google Scholar] [CrossRef]
- Biton, J.; Boissier, M.-C.; Bessis, N. TNFα: Activator or Inhibitor of Regulatory T Cells? Joint Bone Spine 2012, 79, 119–123. [Google Scholar] [CrossRef]
- Eby, J.M.; Kang, H.-K.; Tully, S.T.; Bindeman, W.E.; Peiffer, D.S.; Chatterjee, S.; Mehrotra, S.; Le Poole, I.C. CCL22 to Activate Treg Migration and Suppress Depigmentation in Vitiligo. J. Investig. Dermatol. 2015, 135, 1574–1580. [Google Scholar] [CrossRef]
- Vale, E.C.S.d.; Garcia, L.C. Cutaneous Lupus Erythematosus: A Review of Etiopathogenic, Clinical, Diagnostic and Therapeutic Aspects. An. Brasil Dermatol. 2023, 98, 355–372. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, J. Cutaneous Lupus Erythematosus: New Insights into Pathogenesis and Therapeutic Strategies. Nat. Rev. Rheumatol. 2019, 15, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Stannard, J.N.; Kahlenberg, J.M. Cutaneous Lupus Erythematosus: Updates on Pathogenesis and Associations with Systemic Lupus. Curr. Opin. Rheumatol. 2016, 28, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Achtman, J.C.; Werth, V.P. Pathophysiology of Cutaneous Lupus Erythematosus. Arthritis Res. Ther. 2015, 17, 182. [Google Scholar] [CrossRef] [PubMed]
- Petty, A.J.; Floyd, L.; Henderson, C.; Nicholas, M.W. Cutaneous Lupus Erythematosus: Progress and Challenges. Curr. Allergy Asthma Rep. 2020, 20, 12. [Google Scholar] [CrossRef] [PubMed]
- Nabatian, A.S.; Bashir, M.M.; Wysocka, M.; Sharma, M.; Werth, V.P. Tumor Necrosis Factor α Release in Peripheral Blood Mononuclear Cells of Cutaneous Lupus and Dermatomyositis Patients. Arthritis Res. Ther. 2012, 14, R1. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, F.; Itoh, T.; Wakita, H.; Yagi, H.; Tokura, Y.; Norris, D.A.; Takigawa, M. Keratinocytes from Patients with Lupus Erythematosus Show Enhanced Cytotoxicity to Ultraviolet Radiation and to Antibody-Mediated Cytotoxicity. Clin. Exp. Immunol. 2001, 118, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Zeidi, M.; Kim, H.J.; Werth, V.P. Increased Myeloid Dendritic Cells and TNF-α Expression Predicts Poor Response to Hydroxychloroquine in Cutaneous Lupus Erythematosus. J. Investig. Dermatol. 2019, 139, 324–332. [Google Scholar] [CrossRef]
- Walling, H.W.; Sontheimer, R.D. Cutaneous Lupus Erythematosus: Issues in Diagnosis and Treatment. Am. J. Clin. Dermatol. 2009, 10, 365–381. [Google Scholar] [CrossRef]
- Wahie, S.; Daly, A.K.; Cordell, H.J.; Goodfield, M.J.; Jones, S.K.; Lovell, C.R.; Carmichael, A.J.; Carr, M.M.; Drummond, A.; Natarajan, S.; et al. Clinical and Pharmacogenetic Influences on Response to Hydroxychloroquine in Discoid Lupus Erythematosus: A Retrospective Cohort Study. J. Investig. Dermatol. 2011, 131, 1981–1986. [Google Scholar] [CrossRef]
- Chang, A.Y. Response to Antimalarial Agents in Cutaneous Lupus Erythematosus: A Prospective Analysis. Arch. Dermatol. 2011, 147, 1261. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, E.P.; Sarno, E.N.; Galilly, R.; Cohn, Z.A.; Kaplan, G. Thalidomide Selectively Inhibits Tumor Necrosis Factor Alpha Production by Stimulated Human Monocytes. J. Exp. Med. 1991, 173, 699–703. [Google Scholar] [CrossRef]
- Domingo, S.; Solé, C.; Moliné, T.; Ferrer, B.; Ordi-Ros, J.; Cortés-Hernández, J. Efficacy of Thalidomide in Discoid Lupus Erythematosus: Insights into the Molecular Mechanisms. Dermatology 2020, 236, 467–476. [Google Scholar] [CrossRef]
- Cleaver, N.; Ramirez, J.; Gildenberg, S. Cutaneous Lupus Erythematosus in a Patient Undergoing Intravitreal Bevacizumab Injections: Case Report and Review of the Literature. J. Drugs Dermatol. 2013, 12, 1052–1055. [Google Scholar] [PubMed]
- Cemil, B.; Atas, H.; Canpolat, F.; Akca, Y.; Sasmaz, R. Infliximab-Induced Discoid Lupus Erythematosus. Lupus 2013, 22, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.; Switlyk, S.A.; Gottlieb, A. Cutaneous Lupus Erythematosus and Anti-TNF-Alpha Therapy: A Case Report with Review of the Literature. J. Drugs Dermatol. 2010, 9, 1283–1287. [Google Scholar] [PubMed]
- Suarez, A. Differential Effect of IL10 and TNF Genotypes on Determining Susceptibility to Discoid and Systemic Lupus Erythematosus. Ann. Rheum. Dis. 2005, 64, 1605–1610. [Google Scholar] [CrossRef] [PubMed]
- Solé, C.; Gimenez-Barcons, M.; Ferrer, B.; Ordi-Ros, J.; Cortés-Hernández, J. Microarray Study Reveals a Transforming Growth Factor-β-Dependent Mechanism of Fibrosis in Discoid Lupus Erythematosus. Br. J. Dermatol. 2016, 175, 302–313. [Google Scholar] [CrossRef]
- Werth, V.P.; Zhang, W.; Dortzbach, K.; Sullivan, K. Association of a Promoter Polymorphism of Tumor Necrosis Factor-α with Subacute Cutaneous Lupus Erythematosus and Distinct Photoregulation of Transcription. J. Investig. Dermatol. 2000, 115, 726–730. [Google Scholar] [CrossRef]
- Oge’, L.K.; Broussard, A.; Marshall, M.D. Acne Vulgaris: Diagnosis and Treatment. Am. Fam. Physician 2019, 100, 475–484. [Google Scholar]
- Dréno, B.; Pécastaings, S.; Corvec, S.; Veraldi, S.; Khammari, A.; Roques, C. Cutibacterium acnes (Propionibacterium acnes) and Acne Vulgaris: A Brief Look at the Latest Updates. Acad. Dermatol. Venereol. 2018, 32, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Chen, Y.; Shao, X.; Chen, J.; Liu, L.; Li, Y.; Pu, Y.; Chen, J. Hematological Parameters in Patients with Acnes. J. Cosmet. Dermatol. 2023, 22, 2099–2104. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, A.G.W.; Vaughn, L.T.; Huang, J.T.; Barbieri, J.S. Role of Tumor Necrosis Factor–α Inhibitors in the Treatment and Occurrence of Acne: A Systematic Review. JAMA Dermatol. 2023, 159, 504. [Google Scholar] [CrossRef] [PubMed]
- Rajaii, R.; Globerson, J.; Arnold, N.; Mahon, M. A Novel Treatment of Acne Fulminans with Adalimumab: A Case Report. Spartan Med. Res. J. 2018, 3, 7003. [Google Scholar] [CrossRef] [PubMed]
- Barry, K.K.; Neale, H.D.; Hawryluk, E.B. Tumor Necrosis Factor-Alpha Inhibitors and Acne Fulminans: Friend or Foe? Pediatr. Dermatol. 2023, 40, 678–680. [Google Scholar] [CrossRef] [PubMed]
- Taudorf, E.H.; Jensen, M.B.; Bouazzi, D.; Sand, C.; Thomsen, S.F.; Jemec, G.B.E.; Saunte, D.M.L. Tumor Necrosis Factor-α Inhibitor Treatment of Acne Fulminans—A Clinical and Literature Review. J. Dtsch. Dermatol. Ges. 2024, 22, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Vossen, A.R.J.V.; van der Zee, H.H.; Prens, E.P. Hidradenitis Suppurativa: A Systematic Review Integrating Inflammatory Pathways Into a Cohesive Pathogenic Model. Front. Immunol. 2018, 9, 2965. [Google Scholar] [CrossRef] [PubMed]
- Goldburg, S.R.; Strober, B.E.; Payette, M.J. Hidradenitis Suppurativa. J. Am. Acad. Dermatol. 2020, 82, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Moran, B.; Sweeney, C.M.; Hughes, R.; Malara, A.; Kirthi, S.; Tobin, A.-M.; Kirby, B.; Fletcher, J.M. Hidradenitis Suppurativa Is Characterized by Dysregulation of the Th17:Treg Cell Axis, Which Is Corrected by Anti-TNF Therapy. J. Investig. Dermatol. 2017, 137, 2389–2395. [Google Scholar] [CrossRef]
- Malara, A.; Hughes, R.; Jennings, L.; Sweeney, C.M.; Lynch, M.; Awdeh, F.; Timoney, I.; Tobin, A.M.; Lynam-Loane, K.; Tobin, L.; et al. Adipokines Are Dysregulated in Patients with Hidradenitis Suppurativa. Br. J. Dermatol. 2018, 178, 792–793. [Google Scholar] [CrossRef]
- Kadowaki, T. Adiponectin and Adiponectin Receptors in Insulin Resistance, Diabetes, and the Metabolic Syndrome. J. Clin. Investig. 2006, 116, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Bukvić Mokos, Z.; Miše, J.; Balić, A.; Marinović, B. Understanding the Relationship Between Smoking and Hidradenitis Suppurativa. Acta Dermatovenerol. Croat. 2020, 28, 9–13. [Google Scholar] [PubMed]
- Thomi, R.; Cazzaniga, S.; Seyed Jafari, S.M.; Schlapbach, C.; Hunger, R.E. Association of Hidradenitis Suppurativa With T Helper 1/T Helper 17 Phenotypes: A Semantic Map Analysis. JAMA Dermatol. 2018, 154, 592–595. [Google Scholar] [CrossRef]
- Sabat, R.; Jemec, G.B.E.; Matusiak, Ł.; Kimball, A.B.; Prens, E.; Wolk, K. Hidradenitis Suppurativa. Nat. Rev. Dis. Primers 2020, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Kimball, A.B.; Okun, M.M.; Williams, D.A.; Gottlieb, A.B.; Papp, K.A.; Zouboulis, C.C.; Armstrong, A.W.; Kerdel, F.; Gold, M.H.; Forman, S.B.; et al. Two Phase 3 Trials of Adalimumab for Hidradenitis Suppurativa. N. Engl. J. Med. 2016, 375, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Markota Čagalj, A.; Marinović, B.; Bukvić Mokos, Z. New and Emerging Targeted Therapies for Hidradenitis Suppurativa. Int. J. Mol. Sci. 2022, 23, 3753. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Zmijewski, M.A.; Zbytek, B.; Tobin, D.J.; Theoharides, T.C.; Rivier, J. Key Role of CRF in the Skin Stress Response System. Endocr. Rev. 2013, 34, 827–884. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Wortsman, J. Neuroendocrinology of the Skin. Endocr. Rev. 2000, 21, 457–487. [Google Scholar] [CrossRef]
- Slominski, A.T.; Slominski, R.M.; Raman, C.; Chen, J.Y.; Athar, M.; Elmets, C. Neuroendocrine Signaling in the Skin with a Special Focus on the Epidermal Neuropeptides. Am. J. Physiol. Cell Physiol. 2022, 323, C1757–C1776. [Google Scholar] [CrossRef]
- Slominski, A.T.; Zmijewski, M.A.; Plonka, P.M.; Szaflarski, J.P.; Paus, R. How UV Light Touches the Brain and Endocrine System Through Skin, and Why. Endocrinology 2018, 159, 1992–2007. [Google Scholar] [CrossRef]
- Bashir, M.M.; Sharma, M.R.; Werth, V.P. TNF-α Production in the Skin. Arch. Dermatol. Res. 2009, 301, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Suschek, C.V.; Mahotka, C.; Schnorr, O.; Kolb-Bachofen, V. UVB Radiation-Mediated Expression of Inducible Nitric Oxide Synthase Activity and the Augmenting Role of Co-Induced TNF-α in Human Skin Endothelial Cells. J. Investig. Dermatol. 2004, 123, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Wang, B.; Sauder, D.N. Review: Molecular Mechanism of Ultraviolet-Induced Keratinocyte Apoptosis. J. Interferon Cytokine Res. 2000, 20, 445–454. [Google Scholar] [CrossRef]
- Sugimoto, M.; Yamashita, R.; Ueda, M. Telomere Length of the Skin in Association with Chronological Aging and Photoaging. J. Dermatol. Sci. 2006, 43, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Thornton, M.J. Estrogens and Aging Skin. Dermato-Endocrinology 2013, 5, 264–270. [Google Scholar] [CrossRef]
- Pilkington, S.M.; Bulfone-Paus, S.; Griffiths, C.E.M.; Watson, R.E.B. Inflammaging and the Skin. J. Investig. Dermatol. 2021, 141, 1087–1095. [Google Scholar] [CrossRef]
- Khavkin, J.; Ellis, D.A.F. Aging Skin: Histology, Physiology, and Pathology. Facial Plast. Surg. Clin. N. Am. 2011, 19, 229–234. [Google Scholar] [CrossRef]
- Holvoet, S.; Vincent, C.; Schmitt, D.; Serres, M. The Inhibition of MAPK Pathway Is Correlated with Down-Regulation of MMP-9 Secretion Induced by TNF-Alpha in Human Keratinocytes. Exp. Cell Res. 2003, 290, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Rai, V.; Agrawal, D.K. Regulation of Collagen I and Collagen III in Tissue Injury and Regeneration. Cardiol. Cardiovasc. Med. 2023, 7, 5–16. [Google Scholar] [CrossRef]
- Verrecchia, F.; Mauviel, A. TGF-β and TNF-α: Antagonistic Cytokines Controlling Type I Collagen Gene Expression. Cell Signal 2004, 16, 873–880. [Google Scholar] [CrossRef]
- Agius, E.; Lacy, K.E.; Vukmanovic-Stejic, M.; Jagger, A.L.; Papageorgiou, A.-P.; Hall, S.; Reed, J.R.; Curnow, S.J.; Fuentes-Duculan, J.; Buckley, C.D.; et al. Decreased TNF-Alpha Synthesis by Macrophages Restricts Cutaneous Immunosurveillance by Memory CD4+ T Cells during Aging. J. Exp. Med. 2009, 206, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Tiemessen, M.M.; Jagger, A.L.; Evans, H.G.; van Herwijnen, M.J.C.; John, S.; Taams, L.S. CD4+CD25+Foxp3+ Regulatory T Cells Induce Alternative Activation of Human Monocytes/Macrophages. Proc. Natl. Acad. Sci. USA 2007, 104, 19446–19451. [Google Scholar] [CrossRef] [PubMed]
- Rigante, D. A Systematic Approach to Autoinflammatory Syndromes: A Spelling Booklet for the Beginner. Exp. Rev. Clin. Immunol. 2017, 13, 571–597. [Google Scholar] [CrossRef] [PubMed]
- Rigante, D. The Fresco of Autoinflammatory Diseases from the Pediatric Perspective. Autoimmun. Rev. 2012, 11, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Signalling Pathways of the TNF Superfamily: A Double-Edged Sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Blanco, P.; Palucka, A.K.; Pascual, V.; Banchereau, J. Dendritic Cells and Cytokines in Human Inflammatory and Autoimmune Diseases. Cytokine Growth Factor. Rev. 2008, 19, 41–52. [Google Scholar] [CrossRef]
- Rigante, D.; Cantarini, L.; Imazio, M.; Lucherini, O.M.; Sacco, E.; Galeazzi, M.; Brizi, M.G.; Brucato, A. Autoinflammatory Diseases and Cardiovascular Manifestations. Ann. Med. 2011, 43, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Cantarini, L.; Lopalco, G.; Selmi, C.; Napodano, S.; De Rosa, G.; Caso, F.; Costa, L.; Iannone, F.; Rigante, D. Autoimmunity and Autoinflammation as the Yin and Yang of Idiopathic Recurrent Acute Pericarditis. Autoimm Rev. 2015, 14, 90–97. [Google Scholar] [CrossRef]
- Jacobi, A.; Manger, B.; Schuler, G.; Hertl, M. Therapeutic application of TNF-alpha inhibitors infliximab and etanercept in inflammatory skin disorders. J. Dtsch. Dermatol. Ges. 2003, 1, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Cantarini, L.; Rigante, D.; Lucherini, O.M.; Cimaz, R.; Pasini, F.L.; Baldari, C.T.; Benucci, M.; Simonini, G.; Di Sabatino, V.; Brizi, M.G.; et al. Role of Etanercept in the Treatment of Tumor Necrosis Factor Receptor-Associated Periodic Syndrome: Personal Experience and Review of the Literature. Int. J. Immunopathol. Pharmacol. 2010, 23, 701–707. [Google Scholar] [CrossRef]
- De Luca, E.; Guerriero, C.; Capozio, G.; Peris, K.; Rigante, D. Cold-Induced Urticaria in Children. Skinmed 2021, 19, 339–348. [Google Scholar] [PubMed]
- Federico, G.; Rigante, D.; Pugliese, A.; Ranno, O.; Catania, S.; Stabile, A. Etanercept Induces Improvement of Arthropathy in Chronic Infantile Neurological Cutaneous Articular (CINCA) Syndrome. Scand. J. Rheumatol. 2003, 32, 312–314. [Google Scholar] [CrossRef] [PubMed]
- Stabile, A.; Bertoni, B.; Ansuini, V.; La Torraca, I.; Sallì, A.; Rigante, D. The Clinical Spectrum and Treatment Options of Macrophage Activation Syndrome in the Pediatric Age. Eur. Rev. Med. Pharmacol. Sci. 2006, 10, 53–59. [Google Scholar] [PubMed]
- De Rosa, G.; Pardeo, M.; Rigante, D. Current Recommendations for the Pharmacologic Therapy in Kawasaki Syndrome and Management of Its Cardiovascular Complications. Eur. Rev. Med. Pharmacol. Sci. 2007, 11, 301–308. [Google Scholar] [PubMed]
- Evangelatos, G.; Bamias, G.; Kitas, G.D.; Kollias, G.; Sfikakis, P.P. The Second Decade of Anti-TNF-a Therapy in Clinical Practice: New Lessons and Future Directions in the COVID-19 Era. Rheumatol. Int. 2022, 42, 1493–1511. [Google Scholar] [CrossRef] [PubMed]
- Palladino, M.A.; Bahjat, F.R.; Theodorakis, E.A.; Moldawer, L.L. Anti-TNF-α Therapies: The next Generation. Nat. Rev. Drug Discov. 2003, 2, 736–746. [Google Scholar] [CrossRef]
- Papadopoulou, D.; Drakopoulos, A.; Lagarias, P.; Melagraki, G.; Kollias, G.; Afantitis, A. In Silico Identification and Evaluation of Natural Products as Potential Tumor Necrosis Factor Function Inhibitors Using Advanced Enalos Asclepios KNIME Nodes. Int. J. Mol. Sci. 2021, 22, 10220. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Skin Disease | TNF-Alpha Effect |
|---|---|
| Psoriasis |
|
| Vitiligo | Melanogenesis alteration
|
| Cutaneous lupus erythematosus |
|
| Acne vulgaris and inversa |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pocino, K.; Carnazzo, V.; Stefanile, A.; Basile, V.; Guerriero, C.; Marino, M.; Rigante, D.; Basile, U. Tumor Necrosis Factor-Alpha: Ally and Enemy in Protean Cutaneous Sceneries. Int. J. Mol. Sci. 2024, 25, 7762. https://doi.org/10.3390/ijms25147762
Pocino K, Carnazzo V, Stefanile A, Basile V, Guerriero C, Marino M, Rigante D, Basile U. Tumor Necrosis Factor-Alpha: Ally and Enemy in Protean Cutaneous Sceneries. International Journal of Molecular Sciences. 2024; 25(14):7762. https://doi.org/10.3390/ijms25147762
Chicago/Turabian StylePocino, Krizia, Valeria Carnazzo, Annunziata Stefanile, Valerio Basile, Cristina Guerriero, Mariapaola Marino, Donato Rigante, and Umberto Basile. 2024. "Tumor Necrosis Factor-Alpha: Ally and Enemy in Protean Cutaneous Sceneries" International Journal of Molecular Sciences 25, no. 14: 7762. https://doi.org/10.3390/ijms25147762