
Behind the Curtain of Abnormal Placentation in Pre-Eclampsia: From Molecular Mechanisms to Histological Hallmarks


{kind=link}
{kind=link}
Abstract
:1. Introduction
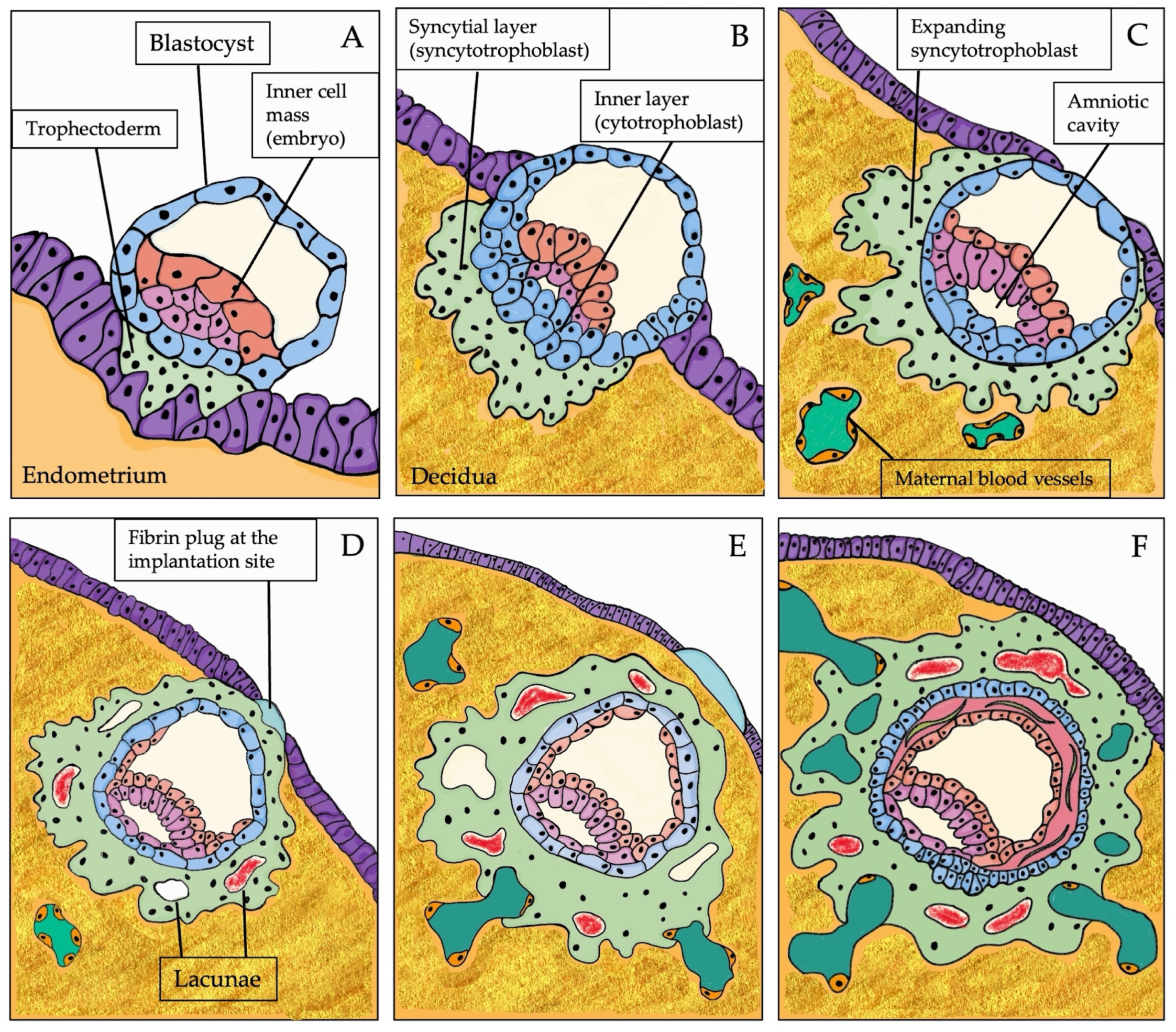
1.1. The Expected Path of Human Placentation
1.1.1. Physiology of Human Placentation
1.1.2. The Endometrium and the Molecular Crosstalk during Implantation
2. When the Placentation Goes Wrong
2.1. Molecular Markers of PE
2.1.1. Impaired Angiogenesis
2.1.2. Oxidative Stress
2.1.3. Inflammation
2.1.4. Immunological Imbalance and Autoimmunity
2.1.5. RAAS
2.2. Histological Hallmarks: The Elementary Lesions
Decidual Arteriopathy
2.3. Acute Atherosis and Fibrinoid Necrosis
2.4. Mural Hypertrophy and Chronic Perivasculitis
2.5. Incomplete/Absent Physiological Spiral Artery Remodeling
2.6. Arterial Thrombosis
2.7. Persistence of Intramural Endovascular Trophoblast
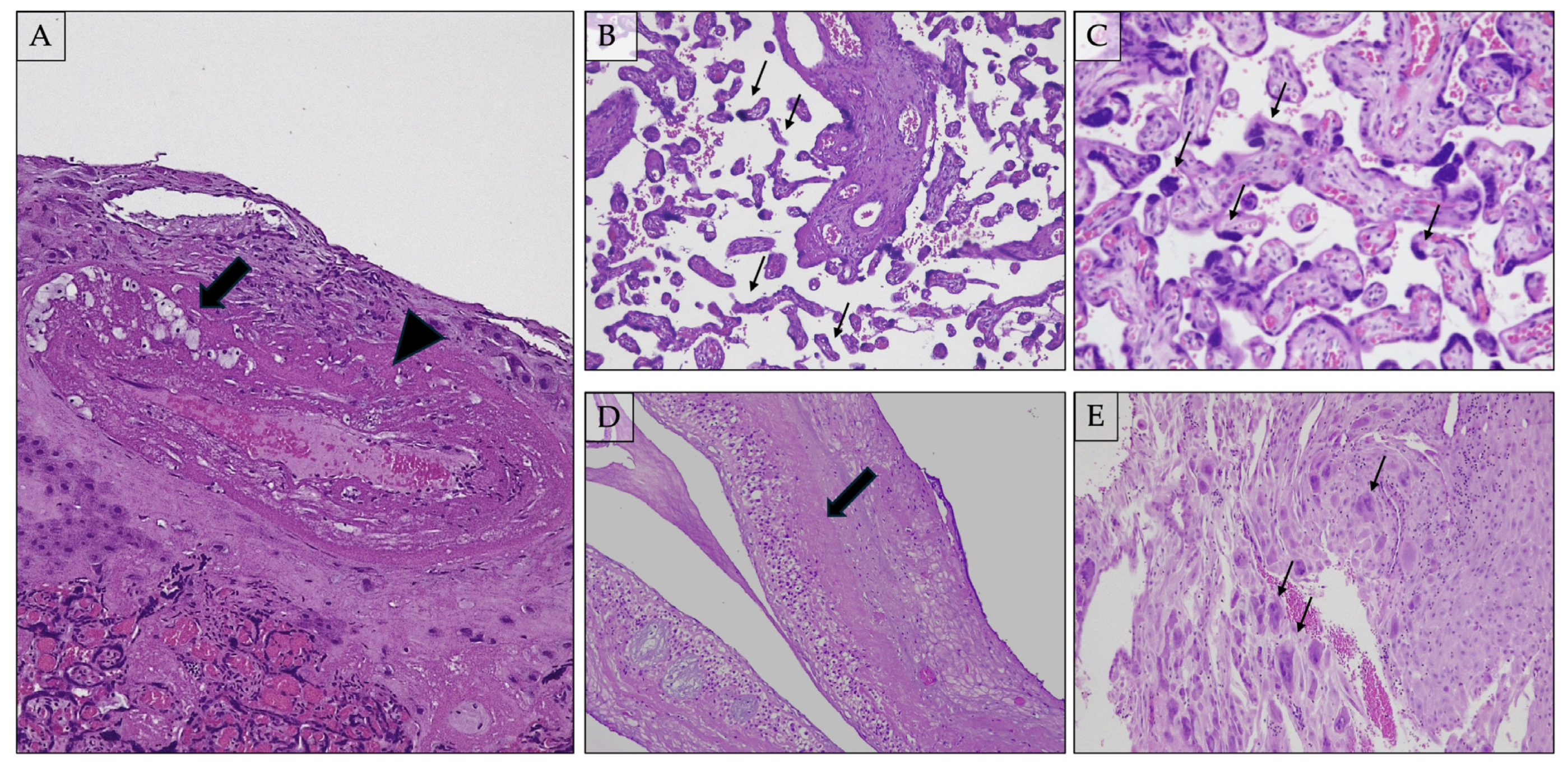
2.7.1. Distal Villous Hypoplasia (Figure 2B)
2.7.2. Accelerated Villous Maturation and Increased Syncytial Knots (i.e., Tenney–Parker Change) (Figure 2C)
2.7.3. Basal Plate Lamina Necrosis (Figure 2D)
2.7.4. Multinucleated Giant Cells in Decidua Basalis (Figure 2E)
2.7.5. Infarcts
2.7.6. Placental Abruptio
2.7.7. Increased Perivillous Fibrin
2.7.8. Extravillous Trophoblastic Cysts
3. Conclusions
4. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benirschke, K.; Burton, G.J.; Baergen, R.N. Pathology of the Human Placenta, 6th ed.; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Kim, S.M.; Kim, J.S. A Review of Mechanisms of Implantation. Dev. Reprod. 2017, 21, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Sadler, T.W. Langman’s Medical Embriology, 14th ed.; Wolters Kluwer Health: Philadelphia, PA, USA, 2018. [Google Scholar]
- Ng, S.W.; Norwitz, G.A.; Pavlicev, M.; Tilburgs, T.; Simon, C.; Norwitz, E.R. Endometrial Decidualization: The Primary Driver of Pregnancy Health. Int. J. Mol. Sci. 2020, 21, 4092. [Google Scholar] [CrossRef] [PubMed]
- Ramathal, C.Y.; Bagchi, I.C.; Taylor, R.N.; Bagchi, M.K. Endometrial decidualization: Of mice and men. Semin. Reprod. Med. 2010, 28, 17–26. [Google Scholar] [CrossRef]
- Cindrova-Davies, T.; Sferruzzi-Perri, A.N. Human placental development and function. Semin. Cell Dev. Biol. 2022, 131, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Hertig, A.T.; Rock, J.; Adams, E.C. A description of 34 human ova within the first 17 days of development. Am. J. Anat. 1956, 98, 435–493. [Google Scholar] [CrossRef] [PubMed]
- Enders, A.C.; Lantz, K.C.; Peterson, P.E.; Hendrickx, A.G. From blastocyst to placenta: The morphology of implantation in the baboon. Hum. Reprod. Update 1997, 3, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Turco, M.Y.; Gardner, L.; Hughes, J.; Cindrova-Davies, T.; Gomez, M.J.; Farrell, L.; Hollinshead, M.; Marsh, S.G.E.; Brosens, J.J.; Critchley, H.O.; et al. Long-term, hormone-responsive organoid cultures of human endometrium in a chemically defined medium. Nat. Cell Biol. 2017, 19, 568–577. [Google Scholar] [CrossRef]
- Gray, C.A.; Taylor, K.M.; Ramsey, W.S.; Hill, J.R.; Bazer, F.W.; Bartol, F.F.; Spencer, T.E. Endometrial glands are required for preimplantation conceptus elongation and survival. Biol. Reprod. 2001, 64, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- Filant, J.; Spencer, T.E. Endometrial glands are essential for blastocyst implantation and decidualization in the mouse uterus. Biol. Reprod. 2013, 88, 93. [Google Scholar] [CrossRef]
- Hempstock, J.; Cindrova-Davies, T.; Jauniaux, E.; Burton, G.J. Endometrial glands as a source of nutrients, growth factors and cytokines during the first trimester of human pregnancy: A morphological and immunohistochemical study. Reprod. Biol. Endocrinol. 2004, 2, 58. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E. The human placenta: New perspectives on its formation and function during early pregnancy. Proc. Biol. Sci. 2023, 290, 20230191. [Google Scholar] [CrossRef]
- Arias-Stella, J. The Arias-Stella reaction: Facts and fancies four decades after. Adv. Anat. Pathol. 2002, 9, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Douglas, N.C.; Thornton, M.H., 2nd; Nurudeen, S.K.; Bucur, M.; Lobo, R.A.; Sauer, M.V. Differential expression of serum glycodelin and insulin-like growth factor binding protein 1 in early pregnancy. Reprod. Sci. 2013, 20, 1376–1381. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Li, J.; Burton, G.J.; Koistinen, H.; Cheung, K.W.; Ng, E.H.Y.; Yao, Y.; Yeung, W.S.B.; Lee, C.L.; Chiu, P.C.N. The functional roles of protein glycosylation in human maternal-fetal crosstalk. Hum. Reprod. Update 2024, 30, 81–108. [Google Scholar] [CrossRef]
- Roberts, J.M.; August, P.A.; Bakris, G.; Barton, J.R.; Bernstein, I.M.; Druzin, M.; Gaiser, R.R.; Granger, J.P.; Jeyabalan, A.; Johnson, D.D.; et al. Hypertension in pregnancy: Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet. Gynecol. 2013, 122, 1122–1131. [Google Scholar] [CrossRef]
- Bastu, E.; Mutlu, M.F.; Yasa, C.; Dural, O.; Nehir Aytan, A.; Celik, C.; Buyru, F.; Yeh, J. Role of Mucin 1 and Glycodelin A in recurrent implantation failure. Fertil. Steril. 2015, 103, 1059–1064.e2. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.C.; Erikson, D.W.; McLendon, B.A.; Seo, H.; Hayashi, K.; Spencer, T.E.; Bazer, F.W.; Burghardt, R.C.; Johnson, G.A. SPP1 expression in the mouse uterus and placenta: Implications for implantationdagger. Biol. Reprod. 2021, 105, 892–904. [Google Scholar] [CrossRef]
- Moore, T.; Dveksler, G.S. Pregnancy-specific glycoproteins: Complex gene families regulating maternal-fetal interactions. Int. J. Dev. Biol. 2014, 58, 273–280. [Google Scholar] [CrossRef]
- Schlafke, S.; Enders, A.C. Cellular basis of interaction between trophoblast and uterus at implantation. Biol. Reprod. 1975, 12, 41–65. [Google Scholar] [CrossRef]
- Aplin, J.D.; Straszewski-Chavez, S.L.; Kalionis, B.; Dunk, C.; Morrish, D.; Forbes, K.; Baczyk, D.; Rote, N.; Malassine, A.; Knofler, M. Trophoblast differentiation: Progenitor cells, fusion and migration—A workshop report. Placenta 2006, 27 (Suppl. A), S141–S143. [Google Scholar] [CrossRef]
- Fritz, R.; Jain, C.; Armant, D.R. Cell signaling in trophoblast-uterine communication. Int. J. Dev. Biol. 2014, 58, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, E.B.; Abbondanzo, S.J.; Anderson, P.S.; Pollard, J.W.; Lessey, B.A.; Stewart, C.L. Leukemia inhibitory factor (LIF) and LIF receptor expression in human endometrium suggests a potential autocrine/paracrine function in regulating embryo implantation. Proc. Natl. Acad. Sci. USA 1996, 93, 3115–3120. [Google Scholar] [CrossRef] [PubMed]
- Suman, P.; Malhotra, S.S.; Gupta, S.K. LIF-STAT signaling and trophoblast biology. JAKSTAT 2013, 2, e25155. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Chen, L.H.; Chiu, W.J.; Tsai, C.L. LIF-STAT signaling in decidual cells: A possible role in embryo implantation and early pregnancy. J. Mol. Endocrinol. 2024, 73, e240006. [Google Scholar] [CrossRef] [PubMed]
- Aghajanova, L. Leukemia inhibitory factor and human embryo implantation. Ann. N. Y. Acad. Sci. 2004, 1034, 176–183. [Google Scholar] [CrossRef]
- Dominguez, F.; Gadea, B.; Mercader, A.; Esteban, F.J.; Pellicer, A.; Simon, C. Embryologic outcome and secretome profile of implanted blastocysts obtained after coculture in human endometrial epithelial cells versus the sequential system. Fertil. Steril. 2010, 93, 774–782.e1. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.; Ahmed Khan, T.; Gul, H.; Rehman, R. An interplay of Progesterone, Leukemia Inhibitor Factor and Interleukin-6 in the window of implantation; Impact on fertility. Cytokine 2023, 170, 156332. [Google Scholar] [CrossRef] [PubMed]
- Viti, J.; Feathers, A.; Phillips, J.; Lillien, L. Epidermal growth factor receptors control competence to interpret leukemia inhibitory factor as an astrocyte inducer in developing cortex. J. Neurosci. 2003, 23, 3385–3393. [Google Scholar] [CrossRef] [PubMed]
- Holbro, T.; Hynes, N.E. ErbB receptors: Directing key signaling networks throughout life. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 195–217. [Google Scholar] [CrossRef]
- Lim, H.; Das, S.K.; Dey, S.K. erbB genes in the mouse uterus: Cell-specific signaling by epidermal growth factor (EGF) family of growth factors during implantation. Dev. Biol. 1998, 204, 97–110. [Google Scholar] [CrossRef]
- Nishi, E.; Klagsbrun, M. Heparin-binding epidermal growth factor-like growth factor (HB-EGF) is a mediator of multiple physiological and pathological pathways. Growth Factors 2004, 22, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Poh, Q.H.; Rai, A.; Cross, J.; Greening, D.W. HB-EGF-loaded nanovesicles enhance trophectodermal spheroid attachment and invasion. Proteomics 2024, 24, e2200145. [Google Scholar] [CrossRef] [PubMed]
- Lessey, B.A.; Gui, Y.; Apparao, K.B.; Young, S.L.; Mulholland, J. Regulated expression of heparin-binding EGF-like growth factor (HB-EGF) in the human endometrium: A potential paracrine role during implantation. Mol. Reprod. Dev. 2002, 62, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.J.; Dey, S.K. HB-EGF: A unique mediator of embryo-uterine interactions during implantation. Exp. Cell Res. 2009, 315, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Chobotova, K.; Spyropoulou, I.; Carver, J.; Manek, S.; Heath, J.K.; Gullick, W.J.; Barlow, D.H.; Sargent, I.L.; Mardon, H.J. Heparin-binding epidermal growth factor and its receptor ErbB4 mediate implantation of the human blastocyst. Mech. Dev. 2002, 119, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Aplin, J.D.; Ruane, P.T. Embryo-epithelium interactions during implantation at a glance. J. Cell Sci. 2017, 130, 15–22. [Google Scholar] [CrossRef]
- Meinhardt, G.; Haider, S.; Kunihs, V.; Saleh, L.; Pollheimer, J.; Fiala, C.; Hetey, S.; Feher, Z.; Szilagyi, A.; Than, N.G.; et al. Pivotal role of the transcriptional co-activator YAP in trophoblast stemness of the developing human placenta. Proc. Natl. Acad. Sci. USA 2020, 117, 13562–13570. [Google Scholar] [CrossRef] [PubMed]
- Soncin, F.; Parast, M.M. Role of Hippo signaling pathway in early placental development. Proc. Natl. Acad. Sci. USA 2020, 117, 20354–20356. [Google Scholar] [CrossRef] [PubMed]
- Bruce, A.W. What is the role of maternally provided Cdx2 mRNA in early mouse embryogenesis? Reprod. Biomed. Online 2011, 22, 512–515. [Google Scholar] [CrossRef]
- Bacenkova, D.; Trebunova, M.; Cizkova, D.; Hudak, R.; Dosedla, E.; Findrik-Balogova, A.; Zivcak, J. In Vitro Model of Human Trophoblast in Early Placentation. Biomedicines 2022, 10, 904. [Google Scholar] [CrossRef]
- Knott, J.G.; Paul, S. Transcriptional regulators of the trophoblast lineage in mammals with hemochorial placentation. Reproduction 2014, 148, R121–R136. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Ganguly, A.; Home, P.; Bhattacharya, B.; Ray, S.; Ghosh, A.; Rumi, M.A.K.; Marsh, C.; French, V.A.; Gunewardena, S.; et al. TEAD4 ensures postimplantation development by promoting trophoblast self-renewal: An implication in early human pregnancy loss. Proc. Natl. Acad. Sci. USA 2020, 117, 17864–17875. [Google Scholar] [CrossRef] [PubMed]
- Lentjes, M.H.; Niessen, H.E.; Akiyama, Y.; de Bruine, A.P.; Melotte, V.; van Engeland, M. The emerging role of GATA transcription factors in development and disease. Expert. Rev. Mol. Med. 2016, 18, e3. [Google Scholar] [CrossRef] [PubMed]
- Niringiyumukiza, J.D.; Cai, H.; Xiang, W. Prostaglandin E2 involvement in mammalian female fertility: Ovulation, fertilization, embryo development and early implantation. Reprod. Biol. Endocrinol. 2018, 16, 43. [Google Scholar] [CrossRef] [PubMed]
- Malathy, P.V.; Cheng, H.C.; Dey, S.K. Production of leukotrienes and prostaglandins in the rat uterus during peri-implantation period. Prostaglandins 1986, 32, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Waclawik, A.; Kaczynski, P.; Jabbour, H.N. Autocrine and paracrine mechanisms of prostaglandin E(2) action on trophoblast/conceptus cells through the prostaglandin E(2) receptor (PTGER2) during implantation. Endocrinology 2013, 154, 3864–3876. [Google Scholar] [CrossRef] [PubMed]
- Waclawik, A. Novel insights into the mechanisms of pregnancy establishment: Regulation of prostaglandin synthesis and signaling in the pig. Reproduction 2011, 142, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Aplin, J.D. Adhesion molecules in endometrial epithelium: Tissue integrity and embryo implantation. J. Anat. 2009, 215, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Role of colony-stimulating factor-1 in reproduction and development. Mol. Reprod. Dev. 1997, 46, 54–60, discussion 60–51. [Google Scholar] [CrossRef]
- Robertson, S.A. GM-CSF regulation of embryo development and pregnancy. Cytokine Growth Factor. Rev. 2007, 18, 287–298. [Google Scholar] [CrossRef]
- Zhang, S.; Mesalam, A.; Joo, M.D.; Lee, K.L.; Hwang, J.Y.; Xu, L.; Song, S.H.; Koh, P.O.; Yuan, Y.G.; Lv, W.; et al. Matrix metalloproteinases improves trophoblast invasion and pregnancy potential in mice. Theriogenology 2020, 151, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Zhou, Y.; Li, J.; Zhang, D. MicroRNA-491-5p inhibits trophoblast cell migration and invasion through targeting matrix metalloproteinase-9 in preeclampsia. Mol. Med. Rep. 2020, 22, 5033–5040. [Google Scholar] [CrossRef] [PubMed]
- Brew, K.; Nagase, H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim. Biophys. Acta 2010, 1803, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, V.J.; Lane, D.A.; Beevers, D.G.; Lip, G.Y.; Blann, A.D. Matrix metalloproteinases and their tissue inhibitors in hypertension-related pregnancy complications. J. Hum. Hypertens. 2013, 27, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Hunkapiller, N.M.; Gasperowicz, M.; Kapidzic, M.; Plaks, V.; Maltepe, E.; Kitajewski, J.; Cross, J.C.; Fisher, S.J. A role for Notch signaling in trophoblast endovascular invasion and in the pathogenesis of pre-eclampsia. Development 2011, 138, 2987–2998. [Google Scholar] [CrossRef] [PubMed]
- Jauniaux, E.; Hempstock, J.; Greenwold, N.; Burton, G.J. Trophoblastic oxidative stress in relation to temporal and regional differences in maternal placental blood flow in normal and abnormal early pregnancies. Am. J. Pathol. 2003, 162, 115–125. [Google Scholar] [CrossRef] [PubMed]
- American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins—Obstetrics and the Society for Maternal-FetalMedicin. ACOG Practice Bulletin No. 204: Fetal Growth Restriction. Obstet. Gynecol. 2019, 133, e97–e109. [Google Scholar] [CrossRef] [PubMed]
- Ives, C.W.; Sinkey, R.; Rajapreyar, I.; Tita, A.T.N.; Oparil, S. Preeclampsia-Pathophysiology and Clinical Presentations: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 76, 1690–1702. [Google Scholar] [CrossRef] [PubMed]
- de Ganzo Suarez, T.; de Paco Matallana, C.; Plasencia, W. Spiral, uterine artery doppler and placental ultrasound in relation to preeclampsia. Best. Pract. Res. Clin. Obstet. Gynaecol. 2024, 92, 102426. [Google Scholar] [CrossRef]
- Williams, P.J.; Broughton Pipkin, F. The genetics of pre-eclampsia and other hypertensive disorders of pregnancy. Best. Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 405–417. [Google Scholar] [CrossRef]
- Yang, M.; Wang, M.; Li, N. Advances in pathogenesis of preeclampsia. Arch. Gynecol. Obstet. 2024, 309, 1815–1823. [Google Scholar] [CrossRef] [PubMed]
- Vitoratos, N.; Vrachnis, N.; Iavazzo, C.; Kyrgiou, M. Preeclampsia: Molecular mechanisms, predisposition, and treatment. J. Pregnancy 2012, 2012, 145487. [Google Scholar] [CrossRef] [PubMed]
- Vitoratos, N.; Hassiakos, D.; Iavazzo, C. Molecular mechanisms of preeclampsia. J. Pregnancy 2012, 2012, 298343. [Google Scholar] [CrossRef] [PubMed]
- Hod, T.; Cerdeira, A.S.; Karumanchi, S.A. Molecular Mechanisms of Preeclampsia. Cold Spring Harb. Perspect. Med. 2015, 5, a023473. [Google Scholar] [CrossRef] [PubMed]
- Furuya, M.; Ishida, J.; Aoki, I.; Fukamizu, A. Pathophysiology of placentation abnormalities in pregnancy-induced hypertension. Vasc. Health Risk Manag. 2008, 4, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Fantone, S.; Tossetta, G.; Di Simone, N.; Tersigni, C.; Scambia, G.; Marcheggiani, F.; Giannubilo, S.R.; Marzioni, D. CD93 a potential player in cytotrophoblast and endothelial cell migration. Cell Tissue Res. 2022, 387, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Piani, F.; Tossetta, G.; Fantone, S.; Agostinis, C.; Di Simone, N.; Mandala, M.; Bulla, R.; Marzioni, D.; Borghi, C. First Trimester CD93 as a Novel Marker of Preeclampsia and Its Complications: A Pilot Study. High. Blood Press. Cardiovasc. Prev. 2023, 30, 591–594. [Google Scholar] [CrossRef]
- Denny, K.J.; Woodruff, T.M.; Taylor, S.M.; Callaway, L.K. Complement in pregnancy: A delicate balance. Am. J. Reprod. Immunol. 2013, 69, 3–11. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E. Placental oxidative stress: From miscarriage to preeclampsia. J. Soc. Gynecol. Investig. 2004, 11, 342–352. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E. Oxidative stress. Best. Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 287–299. [Google Scholar] [CrossRef]
- Chiarello, D.I.; Abad, C.; Rojas, D.; Toledo, F.; Vazquez, C.M.; Mate, A.; Sobrevia, L.; Marin, R. Oxidative stress: Normal pregnancy versus preeclampsia. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165354. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, B. Infarctions. In Pathology of the Placenta; Khong, T.Y., Ed.; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.H.; Yuan, H.T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Robillard, P.Y.; Dekker, G.; Chaouat, G.; Hulsey, T.C.; Saftlas, A. Epidemiological studies on primipaternity and immunology in preeclampsia--a statement after twelve years of workshops. J. Reprod. Immunol. 2011, 89, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Daher, S.; Fonseca, F.; Ribeiro, O.G.; Musatti, C.C.; Gerbase-DeLima, M. Tumor necrosis factor during pregnancy and at the onset of labor and spontaneous abortion. Eur. J. Obstet. Gynecol. Reprod. Biol. 1999, 83, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yip, K.C.; He, A.; Tang, J.; Liu, S.; Yan, R.; Zhang, Q.; Li, R. Plasma Olink Proteomics Identifies CCL20 as a Novel Predictive and Diagnostic Inflammatory Marker for Preeclampsia. J. Proteome Res. 2022, 21, 2998–3006. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.J.; Chen, C.P.; Schatz, F.; Rahman, M.; Abrahams, V.M.; Lockwood, C.J. Pre-eclampsia is associated with dendritic cell recruitment into the uterine decidua. J. Pathol. 2008, 214, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.W.; Zhang, Y.C.; Wu, F.; Tian, F.J.; Lin, Y. The role of extravillous trophoblasts and uterine NK cells in vascular remodeling during pregnancy. Front. Immunol. 2022, 13, 951482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Dunk, C.E.; Shynlova, O.; Caniggia, I.; Lye, S.J. TGFb1 suppresses the activation of distinct dNK subpopulations in preeclampsia. EBioMedicine 2019, 39, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Yang, X. The central role of natural killer cells in preeclampsia. Front. Immunol. 2023, 14, 1009867. [Google Scholar] [CrossRef]
- Miller, D.; Motomura, K.; Galaz, J.; Gershater, M.; Lee, E.D.; Romero, R.; Gomez-Lopez, N. Cellular immune responses in the pathophysiology of preeclampsia. J. Leukoc. Biol. 2022, 111, 237–260. [Google Scholar] [CrossRef]
- Reeve, K.E.; Deer, E.; Amaral, L.M.; Cornelius, D.C.; Herrock, O.; Harmon, A.C.; Campbell, N.; Fitzgerald, S.; Ibrahim, T.; Wallukat, G.; et al. Placental CD4(+) T cells from preeclamptic patients cause autoantibodies to the angiotensin II type I receptor and hypertension in a pregnant rat model of preeclampsia. Explor. Med. 2022, 3, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Irani, R.A.; Xia, Y. The functional role of the renin-angiotensin system in pregnancy and preeclampsia. Placenta 2008, 29, 763–771. [Google Scholar] [CrossRef]
- Moghaddas Sani, H.; Zununi Vahed, S.; Ardalan, M. Preeclampsia: A close look at renal dysfunction. Biomed. Pharmacother. 2019, 109, 408–416. [Google Scholar] [CrossRef] [PubMed]
- ACOG Practice Bulletin No. 202: Gestational Hypertension and Preeclampsia. Obstet. Gynecol. 2019, 133, 1. [CrossRef]
- Khong, T.Y.; Mooney, E.E.; Ariel, I.; Balmus, N.C.; Boyd, T.K.; Brundler, M.A.; Derricott, H.; Evans, M.J.; Faye-Petersen, O.M.; Gillan, J.E.; et al. Sampling and Definitions of Placental Lesions: Amsterdam Placental Workshop Group Consensus Statement. Arch. Pathol. Lab. Med. 2016, 140, 698–713. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Woods, A.W.; Jauniaux, E.; Kingdom, J.C. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta 2009, 30, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.; Audette, M.C.; Ye, X.Y.; Keating, S.; Hoffman, B.; Lye, S.J.; Shah, P.S.; Kingdom, J.C. Maternal Vascular Malperfusion and Adverse Perinatal Outcomes in Low-Risk Nulliparous Women. Obstet. Gynecol. 2017, 130, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Ernst, L.M. Maternal vascular malperfusion of the placental bed. APMIS 2018, 126, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Z.; Sheehan, P.M.; Brennecke, S.P.; Keogh, R.J. Vessel remodelling, pregnancy hormones and extravillous trophoblast function. Mol. Cell Endocrinol. 2012, 349, 138–144. [Google Scholar] [CrossRef]
- Zhou, Y.; Damsky, C.H.; Fisher, S.J. Preeclampsia is associated with failure of human cytotrophoblasts to mimic a vascular adhesion phenotype. One cause of defective endovascular invasion in this syndrome? J. Clin. Investig. 1997, 99, 2152–2164. [Google Scholar] [CrossRef]
- Rana, S.; Lemoine, E.; Granger, J.P.; Karumanchi, S.A. Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ. Res. 2019, 124, 1094–1112. [Google Scholar] [CrossRef] [PubMed]
- Hertig, A.T. On the development of the amnion and exoccelomic membrane in the previllous human ovum. Yale J. Biol. Med. 1945, 18, 107–115. [Google Scholar] [PubMed]
- Pitz Jacobsen, D.; Fjeldstad, H.E.; Johnsen, G.M.; Fosheim, I.K.; Moe, K.; Alnaes-Katjavivi, P.; Dechend, R.; Sugulle, M.; Staff, A.C. Acute Atherosis Lesions at the Fetal-Maternal Border: Current Knowledge and Implications for Maternal Cardiovascular Health. Front. Immunol. 2021, 12, 791606. [Google Scholar] [CrossRef] [PubMed]
- Back, M.; Yurdagul, A., Jr.; Tabas, I.; Oorni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Subbotin, V.M. Neovascularization of coronary tunica intima (DIT) is the cause of coronary atherosclerosis. Lipoproteins invade coronary intima via neovascularization from adventitial vasa vasorum, but not from the arterial lumen: A hypothesis. Theor. Biol. Med. Model. 2012, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Staff, A.C.; Fjeldstad, H.E.; Fosheim, I.K.; Moe, K.; Turowski, G.; Johnsen, G.M.; Alnaes-Katjavivi, P.; Sugulle, M. Failure of physiological transformation and spiral artery atherosis: Their roles in preeclampsia. Am. J. Obstet. Gynecol. 2022, 226, S895–S906. [Google Scholar] [CrossRef] [PubMed]
- Robertson, W.B.; Brosens, I.; Dixon, G. Uteroplacental vascular pathology. Eur. J. Obstet. Gynecol. Reprod. Biol. 1975, 5, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Redline, R.W.; Boyd, T.; Campbell, V.; Hyde, S.; Kaplan, C.; Khong, T.Y.; Prashner, H.R.; Waters, B.L.; Society for Pediatric Pathology, P.S.M.V.P.N.C. Maternal vascular underperfusion: Nosology and reproducibility of placental reaction patterns. Pediatr. Dev. Pathol. 2004, 7, 237–249. [Google Scholar] [CrossRef]
- Olaya, C.M.; Franco Zuluaga, J.A. More Tools for Evaluating Decidual Artery Disease. Int. J. Surg. Pathol. 2023, 31, 1217–1224. [Google Scholar] [CrossRef]
- Kos, M.; Czernobilsky, B.; Hlupic, L.; Kunjko, K. Pathological changes in placentas from pregnancies with preeclampsia and eclampsia with emphasis on persistence of endovascular trophoblastic plugs. Croat. Med. J. 2005, 46, 404–409. [Google Scholar]
- Nikkels, P.G.; Severens-Rijvers, C.A.H. Distal Villous Hypoplasia; Khong, T.Y., Ed.; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Parks, W.T. Manifestations of Hypoxia in the Second and Third Trimester Placenta. Birth Defects Res. 2017, 109, 1345–1357. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Chan, A.D.; Keating, S.; Redline, R.W.; Fritsch, M.K.; Machin, G.A.; Cornejo-Palma, D.; de Nanassy, J.; El-Demellawy, D.; von Dadelszen, P.; et al. The Placental Distal Villous Hypoplasia Pattern: Interobserver Agreement and Automated Fractal Dimension as an Objective Metric. Pediatr. Dev. Pathol. 2016, 19, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Jaiman, S.; Romero, R.; Pacora, P.; Jung, E.; Bhatti, G.; Yeo, L.; Kim, Y.M.; Kim, B.; Kim, C.J.; Kim, J.S.; et al. Disorders of placental villous maturation in fetal death. J. Perinat. Med. 2020, 48, 345–368. [Google Scholar] [CrossRef] [PubMed]
- Brosens, I.; Pijnenborg, R.; Vercruysse, L.; Romero, R. The “Great Obstetrical Syndromes” are associated with disorders of deep placentation. Am. J. Obstet. Gynecol. 2011, 204, 193–201. [Google Scholar] [CrossRef]
- Fogarty, N.M.; Ferguson-Smith, A.C.; Burton, G.J. Syncytial knots (Tenney-Parker changes) in the human placenta: Evidence of loss of transcriptional activity and oxidative damage. Am. J. Pathol. 2013, 183, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.A.; Mayhew, T.M.; Barnes, P.R. From 13 weeks to term, the trophoblast of human placenta grows by the continuous recruitment of new proliferative units: A study of nuclear number using the disector. Placenta 1992, 13, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Fox, H. The Significance of Villous Syncytial Knots in the Human Placenta. J. Obstet. Gynaecol. Br. Commonw. 1965, 72, 347–355. [Google Scholar] [CrossRef]
- Tenney, B.; Parker, F. The placenta in toxemia of pregnancy. Am. J. Obstet. Gynecol. 1940, 39, 1000–1005. [Google Scholar] [CrossRef]
- Salafia, C.M.; Pezzullo, J.C.; Lopez-Zeno, J.A.; Simmens, S.; Minior, V.K.; Vintzileos, A.M. Placental pathologic features of preterm preeclampsia. Am. J. Obstet. Gynecol. 1995, 173, 1097–1105. [Google Scholar] [CrossRef]
- Stanek, J.; Al-Ahmadie, H.A. Laminar necrosis of placental membranes: A histologic sign of uteroplacental hypoxia. Pediatr. Dev. Pathol. 2005, 8, 34–42. [Google Scholar] [CrossRef]
- Stanek, J.; Biesiada, J. Sensitivity and specificity of finding of multinucleate trophoblastic giant cells in decidua in placentas from high-risk pregnancies. Hum. Pathol. 2012, 43, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Little, W.A. Placental infarction. Obstet. Gynecol. 1960, 15, 109–130. [Google Scholar] [PubMed]
- Redline, R.W.; Patterson, P. Pre-eclampsia is associated with an excess of proliferative immature intermediate trophoblast. Hum. Pathol. 1995, 26, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, V.G.; Sunilkumar, K.B.; Nagaraj, T.S.; Uddin, Z.; Ahmed, I.; Hwang, K.; Goudar, S.S.; Guruprasad, G.; Saleem, S.; Tikmani, S.S.; et al. Maternal and fetal vascular lesions of malperfusion in the placentas associated with fetal and neonatal death: Results of a prospective observational study. Am. J. Obstet. Gynecol. 2021, 225, 660.e1–660.e12. [Google Scholar] [CrossRef] [PubMed]
- Becroft, D.M.; Thompson, J.M.; Mitchell, E.A. The epidemiology of placental infarction at term. Placenta 2002, 23, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Heerema-McKenney, A.; De Paepe, M.E.; Popek, E.J. Placenta; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Ananth, C.V.; Berkowitz, G.S.; Savitz, D.A.; Lapinski, R.H. Placental abruption and adverse perinatal outcomes. JAMA 1999, 282, 1646–1651. [Google Scholar] [CrossRef] [PubMed]
- Oyelese, Y.; Ananth, C.V. Placental abruption. Obstet. Gynecol. 2006, 108, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Dommisse, J.; Tiltman, A.J. Placental bed biopsies in placental abruption. Br. J. Obstet. Gynaecol. 1992, 99, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.; Cooper, D.; Lees, C.; Hecher, K.; Campbell, S. Doppler ultrasound of the uterine arteries: The importance of bilateral notching in the prediction of pre-eclampsia, placental abruption or delivery of a small-for-gestational-age baby. Ultrasound Obstet. Gynecol. 1996, 7, 182–188. [Google Scholar] [CrossRef]
- Chen, A.; Roberts, D.J. Placental pathologic lesions with a significant recurrence risk-what not to miss! APMIS 2018, 126, 589–601. [Google Scholar] [CrossRef]
- Huang, Z.; Cheng, S.; Jash, S.; Fierce, J.; Agudelo, A.; Higashiyama, T.; Hanna, N.; Nakashima, A.; Saito, S.; Padbury, J.; et al. Exploiting sweet relief for preeclampsia by targeting autophagy-lysosomal machinery and proteinopathy. Exp. Mol. Med. 2024, 56, 1206–1220. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gusella, A.; Martignoni, G.; Giacometti, C. Behind the Curtain of Abnormal Placentation in Pre-Eclampsia: From Molecular Mechanisms to Histological Hallmarks. Int. J. Mol. Sci. 2024, 25, 7886. https://doi.org/10.3390/ijms25147886
Gusella A, Martignoni G, Giacometti C. Behind the Curtain of Abnormal Placentation in Pre-Eclampsia: From Molecular Mechanisms to Histological Hallmarks. International Journal of Molecular Sciences. 2024; 25(14):7886. https://doi.org/10.3390/ijms25147886
Chicago/Turabian StyleGusella, Anna, Guido Martignoni, and Cinzia Giacometti. 2024. "Behind the Curtain of Abnormal Placentation in Pre-Eclampsia: From Molecular Mechanisms to Histological Hallmarks" International Journal of Molecular Sciences 25, no. 14: 7886. https://doi.org/10.3390/ijms25147886