
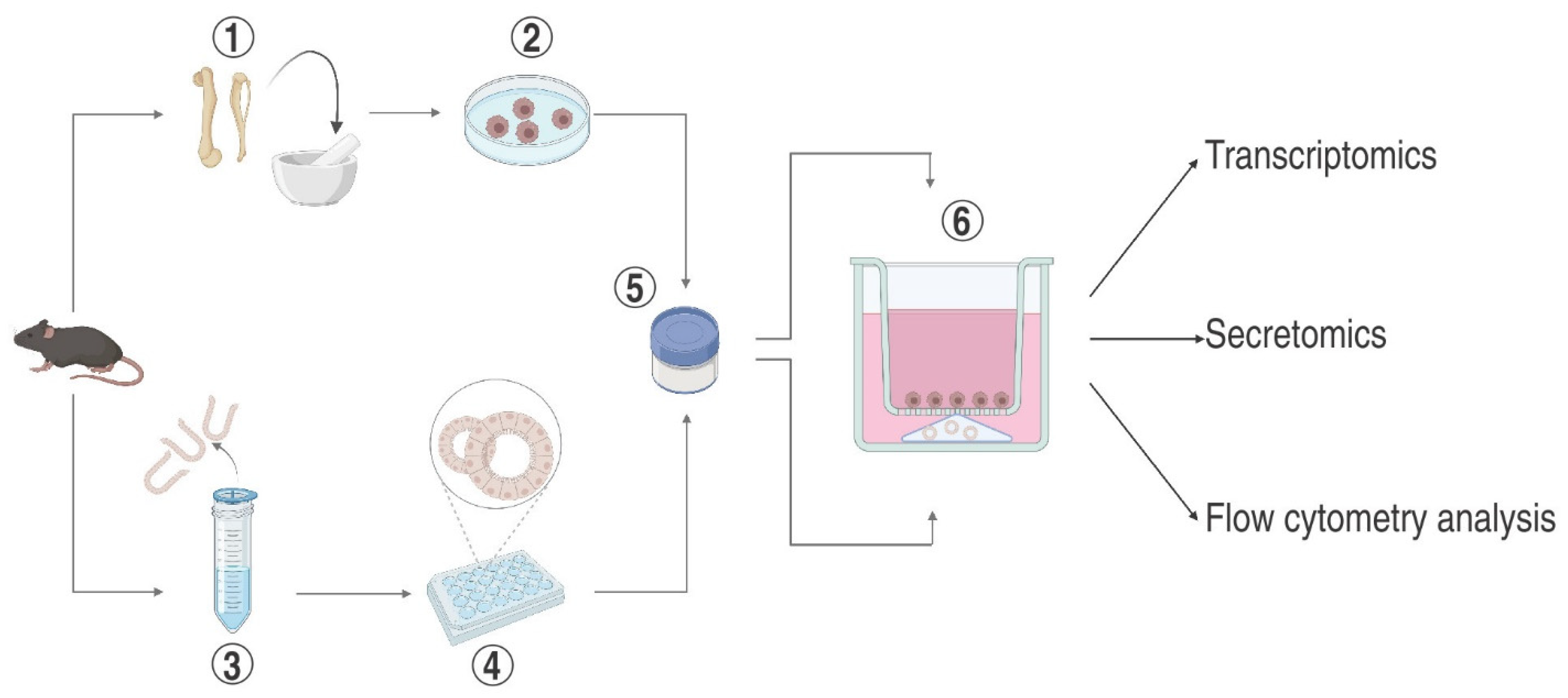
An Adaptable Protocol to Generate a Murine Enteroid–Macrophage Co-Culture System


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials
2.1. Animals and Cell Lines
- C57BL/6j mice were bred and housed under specific pathogen conditions at the University of Ulm’s animal research facility. Breeding pairs were initially obtained from the Jackson Laboratory. Five mice were housed per SPF cage at 21–24 °C room temperature, air humidity 42–44%, light/dark cycle of 14:10 h.
- L-WNR cells, stably expressing Wnt3a, Noggin, and R-Spondin 3, #S0011002, AddexBio, San Diego, Ca, USA.
2.2. Reagents and Chemicals
2.2.1. General Requirements
- Fetal bovine serum (FBS), heat-inactivated, # F7524, Sigma-Aldrich, Taufkirchen, Germany.
- Dulbecco’s Phosphate Buffered Saline (DPBS), ‘# 14190-094, Gibco, Paisley, UK.
- Penicillin/Streptomycin, 10,000 units/mL/10 mg/mL, # 15140-122, Gibco, Paisley, UK.
- Dimethyl Sulfoxide (DMSO), # A3672, PanReac AppliChem, Darmstadt, Germany.
- Ethylenediaminetetraacetic acid disodium (EDTA), # E7889-100ML, Sigma-Aldrich, Darmstadt, Germany.
- Trypsin-EDTA (0.5%), # 15400054, Gibco, Paisley, UK.
2.2.2. Isolation and Differentiation of Bone Marrow-Derived Macrophages
- Dulbecco’s Modified Eagle Medium (DMEM), supplemented with D-Glucose, L-Glutamine, Pyruvate, # 41966-029, Gibco, Paisley, UK.
- Recombinant mouse Macrophage Colony Stimulating Factor (M-CSF), # 315-02, PeproTech (part of Thermo Fisher, Weil am Rhein, Germany) (lot no. 0518245 A0220).
- o
- Reconstitute lyophilized recombinant M-CSF with sterile distilled H20 (e.g., Ampuwa Plastipur) to obtain a stock solution of 100 µg/mL, aliquot at appropriate amounts (e.g., 10 µL), and store at −20 °C.
- Ethanol, # 32205, Sigma-Aldrich, Darmstadt, Germany.
- NH4Cl, # K298.2, Roth, Karlsruhe, Germany.
- Trizma-Base (Tris-Base), # 93362, Sigma-Aldrich, Darmstadt, Germany.
- Ampuwa Plastipur (sterile distilled H2O), # B230673, Fresenius Kabi, Bad Homburg, Germany.
- Lipopolysaccharide from Escherichia coli O55:B5, # L6529, Sigma-Aldrich, Darmstadt, Germany.
- RNeasy Mini Kit, # 74106, Qiagen, Hamburg, Germany.
- Primers: Mm_Tnf_1_SG QuantiTect Primer Assay, # QT00104006; Primer Assay for Mouse GAPDH primer, # PPM02946E-200, Hamburg, Qiagen, Germany.
2.3. Enteroid Culture
- L-WNR-conditioned medium. Prepared according to [16]). For steps of production please refer to “Media and Solutions Formulations”.
- Antibiotic-Antimycotic 100X, # 15240096, Gibco, Paisley, UK.
- Advanced DMEM/F12 (1x), # 12634-010, Gibco, Paisley, UK.
- GlutaMAX (100X), # 35050061, Gibco, Paisley, UK.
- B-27 Supplement (50X), # 12587010, Gibco, Paisley, UK (lot. no. 2584583).
- N-2 Supplement (100X), # 17502048, Gibco, Paisley, UK (lot. no. 2584679).
- Murine Epidermal Growth Factor (EGF), # 315-09, PeproTech (part of Thermo Fisher, Weil am Rhein, Germany) (lot. no. 0519179 L1219).
- R-Spondin 1, # 3474-RS-050, R&D Systems, Mineapolis, MN, USA (lot. no. OHO5420011).
- Murine Noggin, # 250-38, PeproTech (part of Thermo Fisher, Weil am Rhein, Germany) (lot. no. 0618407 J0318).
- Collagenase type I, # 17100-017, Invitrogen (part of Thermo Fisher, Weil am Rhein, Germany) (lot. no. 2445202).
- Gentamicin 50 mg/mL, # 15750060, Invitrogen (part of Thermo Fisher, Weil am Rhein, Germany).
- HEPES, 1 M, # 7365-45-9, Sigma-Aldrich, Darmstadt, Germany.
- Y-27632, # Y0503-1MG, Sigma-Aldrich, Darmstadt, Germany (lot. no. 0000310878).
- o
- Reconstitute lyophilized Y-27632 with sterile distilled H2O (e.g., Ampuwa Plastipur) to obtain a stock solution of 1 mM, aliquot at appropriate amounts (e.g., 100 µL), and store at −20 °C.
- Basement Membrane Matrix, # 354234, Corning, Bedford, MA, USA (lot. no. 3116001).
- CryoStor CS10, #07930, Stem Cell Technologies, Cologne, Germany.
2.4. Equipment
- Water bath: Bender & Hobein GmbH, München, Germany (set to 37 °C).
- Cell culture incubator: Heracell 150, Thermo Scientific, Weil am Rhein, Germany (set to 37 °C 5% CO2).
- Biosafety cabinet (S1): Herasafe 2025 1.8, Thermo Scientific, Weil am Rhein, Germany.
- Refrigerated centrifuge: Multifuge 3 S-R, Heraeus, Thermo Scientific, Weil am Rhein, Germany.
- CO2 rodent euthanasia chamber: # 51370, Otto Environmental, Greenfield, WI, USA.
- Hemocytometer: Neubauer improved, depth 0.1 mm, Marienfeld, Lauda-Königshofen, Germany.
- Stainless steel blunt-end, strait scissors, ball scissors, tweezers, and gavaging needles: Fine Science Tools GmbH, Heidelberg, Germany.
- TC-coated dishes of 10 mm and 100 mm: #150288 and #150350, Nunclon Delta Surface, ThermoFisher Scientific, Weil am Rhein, Germany.
- Polystyrene conical tubes measuring 15 and 50 mL: Falcon 15 mL and 50 mL, #352099 and #352070, Corning, Bedford, MA, USA.
- Cell strainers measuring 70 µm and 100 µm: Falcon cell strainer 70 µm and 100 µm, #352350 and #352360, Corning, Bedford, MA, USA.
- Disposable safety scalpels, feather-shaped: # BA810SU, Braun, Tuttlingen, Germany.
- Freezing container: Mr. Frosty, ThermoFisher, Weil am Rhein, Germany.
- Mortar and pestle (sterile): # GZ-63100-56 (capacity 125 mL), Cole-Parmer, Cambridgeshire, UK.
- Non-TC-treated Petri dishes: 100 × 20 mm Style dish, # 430591, Corning, Bedford, MA, USA.
- Plunger of a 5 mL syringe (sterile): # 4606051V, B. Braun Medical AG, Switzerland.
- Twenty-four-well plates: CellStar, #662160, Greiner bio-one, Darmstadt, Germany.
- Permeable supports: Transwell pore size 0.4 µm, # 3470, Corning, Bedford, MA, USA.
2.5. Media and Solutions Formulations
- DPBS-0: Add Antimycotic-Antibiotic to DPBS to obtain a final 1% concentration. Can be stored at 4° C for up to 4 weeks.
- Coating Solution: Add FBS to DPBS to obtain a final concentration of 1%. NOTE: Must be prepared fresh and used on the same day.
- Macrophage Basic Culture Medium: Add penicillin/streptomycin and FBS to DMEM medium to obtain a final 1% and 10% concentration, respectively. Can be stored at 4 °C for up to 4 weeks.
- Macrophage Complete Culture Medium: Add recombinant M-CSF to Macrophage Basic Culture Medium to obtain a final concentration of 20 ng/mL. Can be stored at 4 °C for up to 1 week.
- Red Blood Cell (RBC) Lysis Buffer: Dissolve NH4Cl (final concentration 144 mM) and Tris-Base (final concentration 17 mM) in distilled H2O. Adjust the pH to 7.2. Can be stored at 4 °C for up to 4 weeks.
- Macrophages Freezing Medium: Add FBS and DMSO to DMEM to achieve a final concentration of 20% and 10%, respectively.
- Enteroid Proliferation Medium (adapted from Stappenbeck et al. [16]): Briefly, L-WNR cells are maintained under dual antibiotic selection with hygromycin B and geneticin until 100% confluence is reached. Then, media is replaced and the conditioned medium is harvested on 4 consecutive days. Eventually, all batches are pooled together and sterile-filtered through a syringe filtration unit (pore size 0.2 µm) before storing at −20 °C. For detailed steps and additional reagents required please refer to the original protocol [16].
- CRITICAL: It is strongly recommended that the L-WNR-conditioned medium be supplemented with Y-27632 (final concentration 10 µM) when performing critical steps such as starting cultivation, passing, and thawing of enteroids.
- Enteroid Basic Medium: Add penicillin/streptomycin and GlutaMAX to Advanced DMEM/F12 to obtain a final concentration of 1% per supplement.
- Enteroid Differentiation Medium (adapted from Sato et al. [15]): On the day of experiment, mix Enteroid Basic Medium with B27 (final concentration 1x), N2 (final concentration 1x), Noggin (final concentration 100 ng/mL), EGF (final concentration 20 ng/mL), and R-Spondin 1 (final concentration 500 ng/mL). For the purpose of co-culture with BMDM, the Enteroid Differentiation Medium must additionally be supplemented with M-CSF (final concentration 20 ng/mL).
3. Experimental Procedures
3.1. Phase I
3.1.1. Isolation and Differentiation of Bone Marrow-Derived Macrophages
- Euthanize the mouse and place it onto the surgical pad in the supine position. Sterilize the skin of the abdomen and hindlimbs with skin antiseptic.
- Make a longitudinal incision along the medial hind limb to expose the muscles. Dissect and remove the muscles surrounding the femur with sharp scissors. Extend skin incision and tissue preparation proximally towards the pelvic hip joint and distally towards the ankle. Separate the femur from the pelvic girdle by exarticulating the hip joint. Separate the distal portion of the hind limb from the femur with a twisting–tugging movement to exarticulate the knee joint. Expose the ankle joint and perform the same twisting–tugging movement to separate the paw from the tibia.
- Remove residual muscles and soft tissues from the femur and tibia by thoroughly rubbing the bone with a dry paper towel, avoiding inadvertently damaging the corticalis or breaking the bone.
- CRITICAL: The articular surface should remain intact. Leaving the corticalis undamaged preserves a sterile environment inside the medullary cavity, decreasing the risk of contamination of the bone marrow cell culture.
- Dip the femur and tibiae in a 15 mL conical tube filled with 70% ethanol. Transfer to the sterile culture safety cabinet and continue processing in a sterile environment. Remove the bones with flame-scarved tweezers and place them in a 10 cm TC-treated dish with ice-cold DPBS to remove residual ethanol.
- Transfer bones with a tweezer into a sterile mortar, add 1–2 mL DPBS, and grind until the bone marrow cell extract’s colour turns red to white.
- Mount a 70 µm cell strainer on top of a 50 mL polystyrene conical tube and pour bone marrow cell suspension, including bone fragments, onto the membrane.
- Squeeze the bone marrow cells through the cell strainer with a sterile plunger of a 5 mL syringe, then rinse the strainer with 5 mL ice-cold DPBS.
- Centrifuge bone marrow cells (400× g), 7 min, 4 °C and aspirate the supernatant.
- Resuspend the bone marrow cell pellet with 1 mL RBC lysis buffer and incubate for 5 min at room temperature to lyse red blood cells. Stop the lysis reaction by adding 5 mL FBS.
- Pelletize RBC-depleted bone marrow cells (400× g, 7 min, 4 °C) and remove the supernatant. Repeat steps 9 and 10 at least once to ensure near-complete removal of RBC.
- Resuspend the cell pellet with 5 mL of Basic Macrophage Culture Medium and leave it on ice.
- Count the bone marrow cells with a hemocytometer (optionally: check for cell viability with Trypan Blue staining) and adjust concentration for a final density of 5 × 106 cells/mL in Macrophage Complete Culture Medium.
- Seed bone marrow cells in non-TC-treated Petri dishes at a concentration of 5 × 106 cells/mL Macrophage Complete Culture Medium to induce monocyte differentiation to macrophages.
- CRITICAL: Tissue culture-treated dishes should be avoided for seeding bone marrow cells, as macrophages tend to adhere firmly to coated surfaces and can only be detached by applying mechanical forces, e.g., scraping them off with a rubber policeman.
- Replace the Macrophage Complete Culture Medium every other day. Unlike other leukocyte fractions, monocytes remain attached to the dish while transforming into macrophages.
- BMDM differentiation is expected to be completed by day 7. To release the BMDM, add 5–10 mL of DPBS supplemented with 10 mM EDTA to the dish and incubate for 10 min in the biosafety cabinet. Swirl the dish gently every 2–3 min to promote BMDM detachment.
- Rinse the dish with a 10 mL pipet several times to release residual BMDM. As BMDM are accumulating, the EDTA/DPBS solution should adopt a turbid appearance. Collect the suspension in a 15 mL conical tube.
- Centrifuge the BMDM suspension (400× g, 5 min, 4 °C). Resuspend the pellet in 250–500 µL Macrophage Complete Culture medium, dispense in aliquots, and freeze, or immediately use for co-culture experiments.
3.1.2. Freezing of Mouse BMDM
- Count the BMDM using a hemocytometer and centrifuge (400× g, 5 min, 4 °C).
- Gently resuspend the cell pellet in freezing media to obtain a concentration of 5 × 106 cells/mL.
- Dispense 1 mL of BMDM suspension per cryovial and place it in a freezing container for gradual freezing. After 24 h, transfer to a liquid nitrogen tank for long-term storage.
- Centrifuge (400× g, 5 min, room temperature) and remove the supernatant. Resuspend the BMDM pellet in the desired volume of Macrophage Complete Culture Medium or immediately use it to establish enteroid–BMDM co-culture.
3.1.3. Thawing of Mouse BMDM
- 5.
- Thaw BMDM by immersing a cryovial in a 37 °C water bath until only an ice chunk is left, then immediately resuspend in a 15 mL conical tube filled with 10 mL pre-warmed Macrophage Basic Culture Medium to dilute residual DMSO.
- 6.
- Centrifuge (400× g, 5 min, room temperature) and remove the supernatant. Resuspend the BMDM pellet in the desired volume of Macrophage Complete Culture Medium or immediately use it for establishing enteroid–BMDM co-culture (continue with section “Establishing co-culture of enteroids and BMDM”).
3.1.4. Cultivation of Mouse Enteroids
- Euthanize the mouse and immobilize it in the supine position on a surgical pad. Sterilize the abdomen and hindlimbs with skin antiseptic.
- Make a longitudinal incision with pointed scissors to open the peritoneal cavity. Remove the pancreas and spleen. Separate the duodenum from the stomach at the gastric-intestinal junction. Stretch the small intestine to its full length and separate at the transition to the cecum. Remove as much mesenterial tissue from the intestine as possible.
- Place the isolated small intestine in a 10 cm dish with an ice-cold DPBS-0 and flush it using a 20 mL syringe fitted with a stainless-steel gavaging needle to remove large fecal matter.
- Slice the intestine longitudinally using a ball scissor and cut it into 3–4 pieces.
- Transfer the intestinal fragments to a new 10 cm dish filled with ice-cold DPBS-0 and wash it vigorously. Repeat the washing step.
- Transfer the intestinal fragments to a new 10 cm dish filled with ice-cold DPBS-0 and incubate for 15 min at 4 °C with gentle agitation on a rotating shaker (set speed to 100 rpm). Repeat this step once more.
- CRITICAL: From that point, work must be carried out under laminar flow to maintain sterile conditions.
- Place the intestinal fragments in an empty 10 cm dish and mince them finely into a mash using two scalpels.
- CRITICAL: From now on, all dishes and pipettes that come in contact with the minced tissues must be pre-coated with Coating Solution to avoid sticking and tissue loss.
- Transfer the mash to a pre-coated 50 mL conical tube filled with 20 mL DPBS-0 and invert 20 times. Allow the tissue pieces to settle to the bottom of the tube before the supernatant is aspirated. Then, replenish with 10 mL cold DPBS-0 and repeat Step 10 at least 5 times until the supernatant has become clear.
- Aspirate supernatant and resuspend the pellet in 30 mL of 2 mM EDTA/DPBS-0. Incubate at 4 °C for 30 min on a rotating shaker.
- Let the pellet settle at the bottom of the tube and remove the supernatant.
- Add 10 mL DPBS-0 and, using a Pipet boy, forcefully aspirate the tissue fragments at maximum pipetting speed and release slowly by gravity dispense (repeat this procedure 10 times). Let the pellet settle and discard the supernatant (fraction I: contains villi).
- Repeat step 11 and discard the supernatant (fraction II: contains both villi and crypts).
- Repeat step 11 to obtain fraction III enriched in crypts. Immediately filter the supernatant through a 100 µm cell pre-coated strainer mounted on a 50 mL pre-coated conical tube. Keep the flow-through.
- Add another 10 mL DPBS-0 to the remaining tissue fragments and shake vigorously to release residual crypts. Pour the entire suspension through the same 100 µm cell strainer as indicated in step 13.
- Centrifuge the crypt suspension (400× g, 5 min, 4 °C) and remove the supernatant.
- Carefully resuspend the crypt pellet in 10 mL Advanced DMEM/F12 medium and filter through a 70 µm cell strainer.
- Centrifuge the crypt-enriched flow-through (400× g, 5 min, 4 °C), remove the supernatant, and place the tube on ice.
- Resuspend the crypt pellet in an appropriate amount of Matrigel aiming at 15–20 µL of Matrigel per well, and mix gently.
- Place a droplet of 15–20 µL crypt–Matrigel suspension in the centre of a well of a 24-well plate and let polymerize in the incubator (20 min, 37 °C, 5% CO2).
- Overlay each crypt–Matrigel dome with either 500 µL Enteroid Proliferation Medium or Enteroid Differentiation Medium supplemented with Y-27632 and cultivate at 37 °C, 5% CO2.
- Change medium after 2 days.
- NOTE: On day 5, intestinal crypts are expected to have developed into cyst-like (Enteroid Proliferation Medium) or crypt-like structures (Enteroid Differentiation Medium). At this stage, both types of enteroids require routine passaging (continue with section “Passaging of mouse enteroids”). Additionally, enteroids with a cyst-like phenotype can be cryopreserved for future use (continue with section “Freezing of enteroids”).
3.1.5. Passaging of Mouse Enteroids
- Aspirate the medium and add 500 µL DPBS-0 to rinse the wells.
- Add 200 µL Trypsin-EDTA per well and keep the plate in the incubator at 37 °C for 3 min.
- Add 500 µL washing medium and lift off the enteroid–Matrigel domes by scratching horizontally and vertically with the tip of a 10 mL pipet.
- Dissociate enteroids by vigorous pipetting 5 times up and down with a 1000-µL pipet tip.
- Collect enteroid suspension from all wells with a 10 mL pipet and transfer it to a 50 mL conical tube.
- Attach a 1000 µL pipet tip to a 10 mL tip and pipet up and down 5 times to break down enteroids into small fragments and single cells.
- Centrifuge the resulting suspension (400× g, 5 min, 4 °C).
- Aspirate the supernatant and resuspend the pellet with 5 mL washing medium. Transfer the suspension to the 15 mL conical tube.
- Centrifuge the enteroid suspension (400× g, 5 min, 4 °C).
- Aspirate the supernatant and place the enteroid pellet on the ice.
- Resuspend the enteroid pellet in an appropriate volume of Matrigel aiming at 15–20 µL of Matrigel per well, and mix gently.
- Plate a droplet of 15–20 µL of crypt–Matrigel suspension in the centre of a well of a 24-well plate and let polymerize in the incubator (20 min, 37 °C, 5% CO2).
- Overlay each crypt–Matrigel dome with 500 µL Enteroid Proliferation Medium or Enteroid Differentiation Medium supplemented with Y-27632 and cultivate at 37 °C, 5 CO2.
- Change medium after 2 days or freeze enteroids (continue with section “Freezing of enteroids”).
3.1.6. Freezing of Mouse Enteroids
- Depending on the size of the enteroid pellet, add an appropriate volume of CryoStor freezing medium and mix thoroughly.
- Dispense 1 mL of the enteroid suspension per cryovial and place it in a freezing container for gradual cooling down to −80 °C overnight. Then, transfer to a liquid nitrogen tank for long-term cryo-storage.
3.1.7. Thawing of Mouse Enteroids
- Thaw enteroids by immersing a cryovial in a 37 °C water bath until only a few ice chunks are left, then immediately resuspend in 10 mL pre-warmed Advanced DMEM/F12.
- Centrifuge (400× g, 5 min, room temperature) and remove supernatant
- Resuspend the enteroid pellet in the desired amount of Matrigel (continue with the section “Establishing co-culture of enteroids and BMDM”).
3.2. Phase II
- Follow the instructions in the section “Thawing of mouse enteroids”.
- Resuspend the enteroid pellet in an appropriate volume of Matrigel aiming at 15–20 µL of Matrigel per well, and mix gently.
- Plate a droplet of 15–20 µL of crypt–Matrigel suspension in the centre of a well of a 24-well plate and let polymerize in the incubator (20 min, 37 °C, 5% CO2).
- Cover each crypt–Matrigel dome with 500 µL Enteroid Proliferation Medium supplemented with 10 µM Y-27632 and incubate for 3 h at 37 °C, 5 CO2.
- CRITICAL Immediately after thawing, enteroids tend to be fragile and thus should first be exposed to Enteroid Proliferation Medium for a minimum of 3 h to speed up their recovery from liquid nitrogen. Subsequent differentiation/crypt budding of the enteroids is promoted by replacing Enteroid Proliferation Medium with Enteroid Differentiation Medium devoid of Wnt3a.
- Meanwhile, defrost BMDM following the instruction in the section “Thawing of BMDM”, then immediately place on ice.
- Determine the concentration of viable BMDM by counting Trypan blue-negative cells using a Neubauer hemocytometer. Suitable BMDM numbers for co-culture are ranging between 105–106 cells per Matrigel dome.
- Centrifuge the BMDM suspension containing the required absolute number of cells (400× g, 5 min, 4 °C) and aspirate the supernatant.
- Resuspend the BMDM pellet with the appropriate volume of Enteroid Differentiation Medium supplemented with M-CSF (20 ng/mL) and mix by gently pipetting up and down.
- NOTE: For a Transwell insert compatible with a 24-well plate, calculate a volume of 200 µL.
- Dispense 200 µL of BMDM suspension to each Transwell insert and position above the already solidified Matrigel dome containing the enteroids to enable close contact between the cell populations. Preferably cultivate at 37 °C, 5 CO2 overnight before co-culture experiments can be carried out.
4. Results and Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
Abbreviations
| FBS | Fetal Bovine Serum |
| BMDM | Bone Marrow-Derived Macrophages |
| DPBS | Dulbecco’s Phosphate Buffered Saline |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | Dimethyl Sulfoxide |
| EDTA | Ethylenediaminetetraacetic Acid Disodium |
| EGF | Epidermal Growth Factor |
| M-CSF | Mouse Macrophage Colony Stimulating Factor |
| RBC | Red Blood Cell |
| ROCK inhibitor | Rho-Associated Protein Kinase Inhibitor |
| SPF | Specific Pathogen Free |
References
- Santana, P.T.; Rosas, S.L.B.; Ribeiro, B.E.; Marinho, Y.; de Souza, H.S.P. Dysbiosis in Inflammatory Bowel Disease: Pathogenic Role and Potential Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 3464. [Google Scholar] [CrossRef]
- Delfini, M.; Stakenborg, N.; Viola, M.F.; Boeckxstaens, G. Macrophages in the gut: Masters in multitasking. Immunity 2022, 55, 1530–1548. [Google Scholar] [CrossRef]
- Mass, E.; Nimmerjahn, F.; Kierdorf, K.; Schlitzer, A. Tissue-specific macrophages: How they develop and choreograph tissue biology. Nat. Rev. Immunol. 2023, 23, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Karve, S.S.; Pradhan, S.; Ward, D.V.; Weiss, A.A. Intestinal organoids model human responses to infection by commensal and Shiga toxin producing Escherichia coli. PLoS ONE 2017, 12, e0178966. [Google Scholar] [CrossRef]
- Staab, J.F.; Lemme-Dumit, J.M.; Latanich, R.; Pasetti, M.F.; Zachos, N.C. Co-Culture System of Human Enteroids/Colonoids with Innate Immune Cells. Curr. Protoc. Immunol. 2020, 131, e113. [Google Scholar] [CrossRef] [PubMed]
- Noel, G.; Baetz, N.W.; Staab, J.F.; Donowitz, M.; Kovbasnjuk, O.; Pasetti, M.F.; Zachos, N.C. Erratum: A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions. Sci. Rep. 2017, 7, 46790. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, S.; Kawasaki, T.; Machida, M.; Iwatsuki, K.; Inaba, A.; Shibata, S.; Shindo, T.; Nakabayashi, K.; Hakamada, K.; Umezawa, A.; et al. Development of Human Gut Organoids With Resident Tissue Macrophages as a Model of Intestinal Immune Responses. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 726–729.e725. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Ye, L.; Liu, H.; Huang, L.; Yang, Q.; Turner, J.R.; Yu, Q. Correction: Lactobacillus accelerates ISCs regeneration to protect the integrity of intestinal mucosa through activation of STAT3 signaling pathway induced by LPLs secretion of IL-22. Cell Death Differ. 2021, 28, 2025–2027. [Google Scholar] [CrossRef]
- Nozaki, K.; Mochizuki, W.; Matsumoto, Y.; Matsumoto, T.; Fukuda, M.; Mizutani, T.; Watanabe, M.; Nakamura, T. Co-culture with intestinal epithelial organoids allows efficient expansion and motility analysis of intraepithelial lymphocytes. J. Gastroenterol. 2016, 51, 206–213. [Google Scholar] [CrossRef]
- Belaid, M.; Javorovic, J.; Pastorin, G.; Vllasaliu, D. Development of an in vitro co-culture model using Caco-2 and J774A.1 cells to mimic intestinal inflammation. Eur. J. Pharm. Biopharm. Off. J. Arbeitsgemeinschaft Fur Pharm. Verfahrenstechnik 2024, 197, 114243. [Google Scholar] [CrossRef] [PubMed]
- Kämpfer, A.A.M.; Urbán, P.; Gioria, S.; Kanase, N.; Stone, V.; Kinsner-Ovaskainen, A. Development of an in vitro co-culture model to mimic the human intestine in healthy and diseased state. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2017, 45, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.K.; Faulk, D.M.; Sundaram, N.; Mahe, M.M.; Stout, K.M.; von Furstenberg, R.J.; Smith, B.J.; McNaughton, K.K.; Shroyer, N.F.; Helmrath, M.A.; et al. Intestinal stem cells remain viable after prolonged tissue storage. Cell Tissue Res. 2013, 354, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.J.; Cui, J.; Wang, Z.Q.; Liu, R.D. Normal mouse intestinal epithelial cells as a model for the in vitro invasion of Trichinella spiralis infective larvae. PLoS ONE 2011, 6, e27010. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, H.; Stappenbeck, T.S. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc. 2013, 8, 2471–2482. [Google Scholar] [CrossRef]
- Wilson, S.S.; Mayo, M.; Melim, T.; Knight, H.; Patnaude, L.; Wu, X.; Phillips, L.; Westmoreland, S.; Dunstan, R.; Fiebiger, E.; et al. Optimized Culture Conditions for Improved Growth and Functional Differentiation of Mouse and Human Colon Organoids. Front. Immunol 2020, 11, 547102. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Lee, H.J.; Gu, N.Y.; Park, Y.R.; Kim, E.J.; Kang, S.J.; Hyun, B.H.; Yang, D.K. Evaluation of porcine intestinal organoids as an in vitro model for mammalian orthoreovirus 3 infection. J. Vet. Sci. 2023, 24, e53. [Google Scholar] [CrossRef] [PubMed]
- Oost, M.J.; Ijaz, A.; van Haarlem, D.A.; van Summeren, K.; Velkers, F.C.; Kraneveld, A.D.; Venema, K.; Jansen, C.A.; Pieters, R.H.H.; Ten Klooster, J.P. Chicken-derived RSPO1 and WNT3 contribute to maintaining longevity of chicken intestinal organoid cultures. Sci. Rep. 2022, 12, 10563. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Zhou, J.; Li, C.; Liu, X.; Chiu, M.C.; Zhao, X.; Wang, D.; Wei, Y.; Lee, A.; Zhang, A.J.; Chu, H.; et al. Infection of bat and human intestinal organoids by SARS-CoV-2. Nat. Med. 2020, 26, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Verhulsel, M.; Simon, A.; Bernheim-Dennery, M.; Gannavarapu, V.R.; Gérémie, L.; Ferraro, D.; Krndija, D.; Talini, L.; Viovy, J.L.; Vignjevic, D.M.; et al. Developing an advanced gut on chip model enabling the study of epithelial cell/fibroblast interactions. Lab Chip 2021, 21, 365–377. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hentschel, V.; Govindarajan, D.; Seufferlein, T.; Armacki, M. An Adaptable Protocol to Generate a Murine Enteroid–Macrophage Co-Culture System. Int. J. Mol. Sci. 2024, 25, 7944. https://doi.org/10.3390/ijms25147944
Hentschel V, Govindarajan D, Seufferlein T, Armacki M. An Adaptable Protocol to Generate a Murine Enteroid–Macrophage Co-Culture System. International Journal of Molecular Sciences. 2024; 25(14):7944. https://doi.org/10.3390/ijms25147944
Chicago/Turabian StyleHentschel, Viktoria, Deepalakshmi Govindarajan, Thomas Seufferlein, and Milena Armacki. 2024. "An Adaptable Protocol to Generate a Murine Enteroid–Macrophage Co-Culture System" International Journal of Molecular Sciences 25, no. 14: 7944. https://doi.org/10.3390/ijms25147944