
Martini 3 Coarse-Grained Model for the Cofactors Involved in Photosynthesis


Abstract
:1. Introduction
2. Results and Discussion
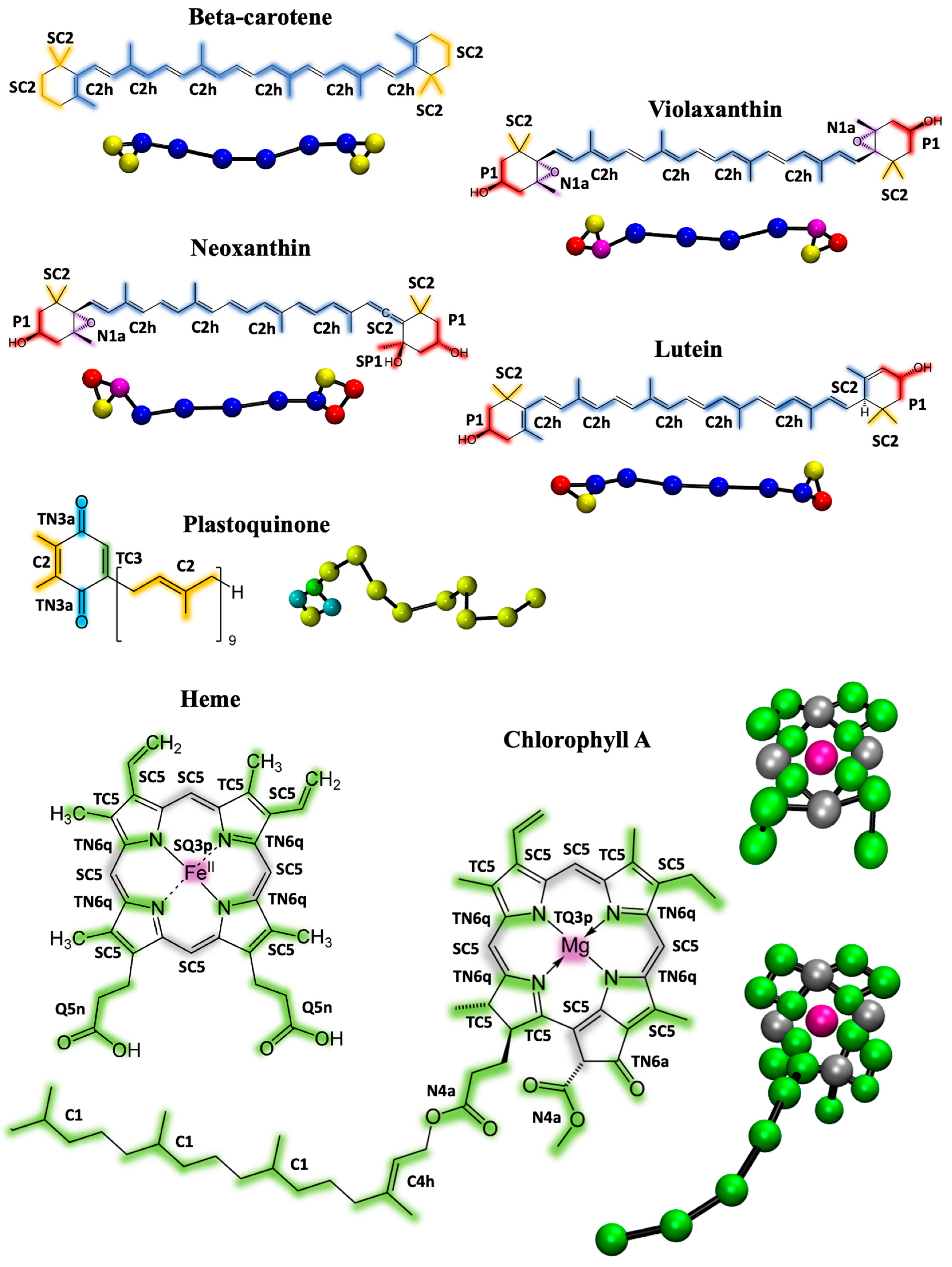
2.1. Parametrization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Martini 3 | Literature |
|---|---|---|
| Beta-carotene | 17.6 ± 0.05 | 17.6 1 |
| Violaxanthin | 12.0 ± 0.04 | 12.0 1 |
| Neoxanthin | 10.9 ± 0.04 | 11.9 1 |
| Lutein | 12.9 ± 0.04 | 14.8 1 |
| Plastoquinone 3 | 19.8 ± 0.11 | |
| Plastoquinol | 18.6 ± 0.10 | |
| Plastoquinone (head) | 2.6 ± 0.03 | 2.5 4 |
| Plastoquinol (head) | 1.4 ± 0.04 | 1.8 4 |
| Chlorophyll A | 11.9 ± 0.10 | 2.21 2 |
| Chlorophyll B | 11.2 ± 0.10 | |
| Heme | −0.31 ± 0.04 | 0.95 2 |
| Chlorophyll A (aromatic core) | 1.9 ± 0.07 | |
| Chlorophyll B (aromatic core) | 1.4 ± 0.05 |
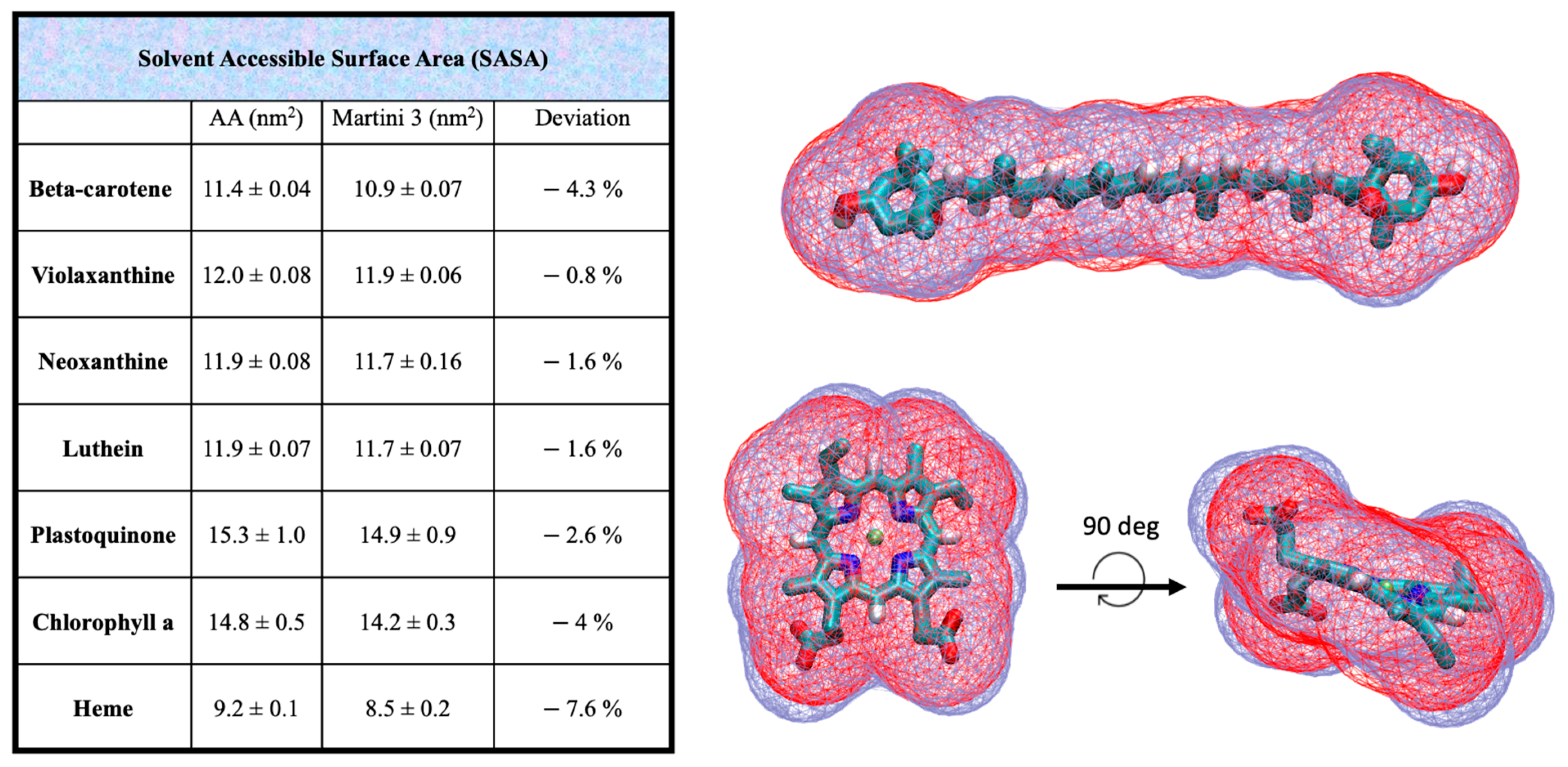
2.2. Validation in Membrane Environment
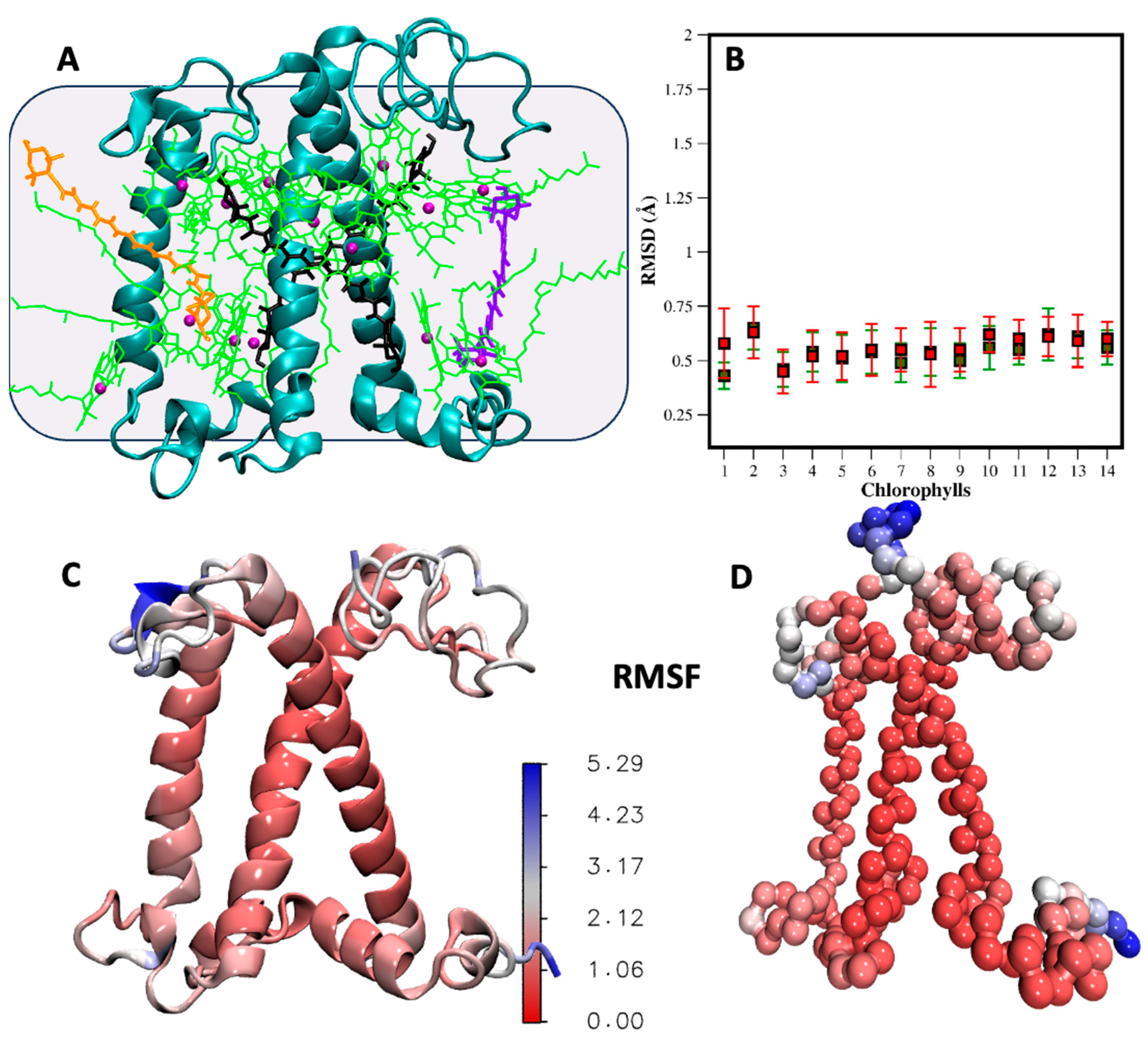
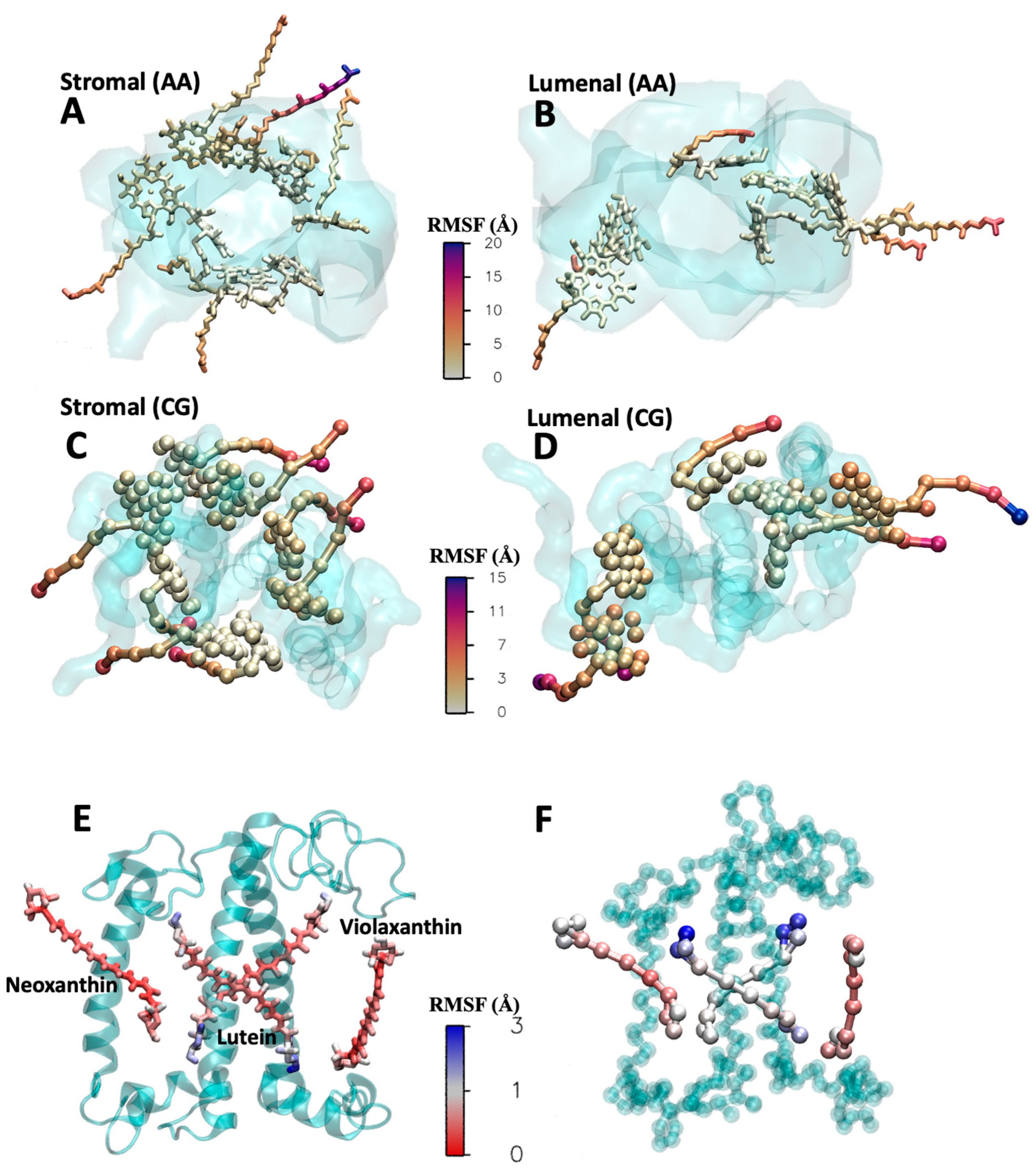
2.3. Validation in Protein Environment
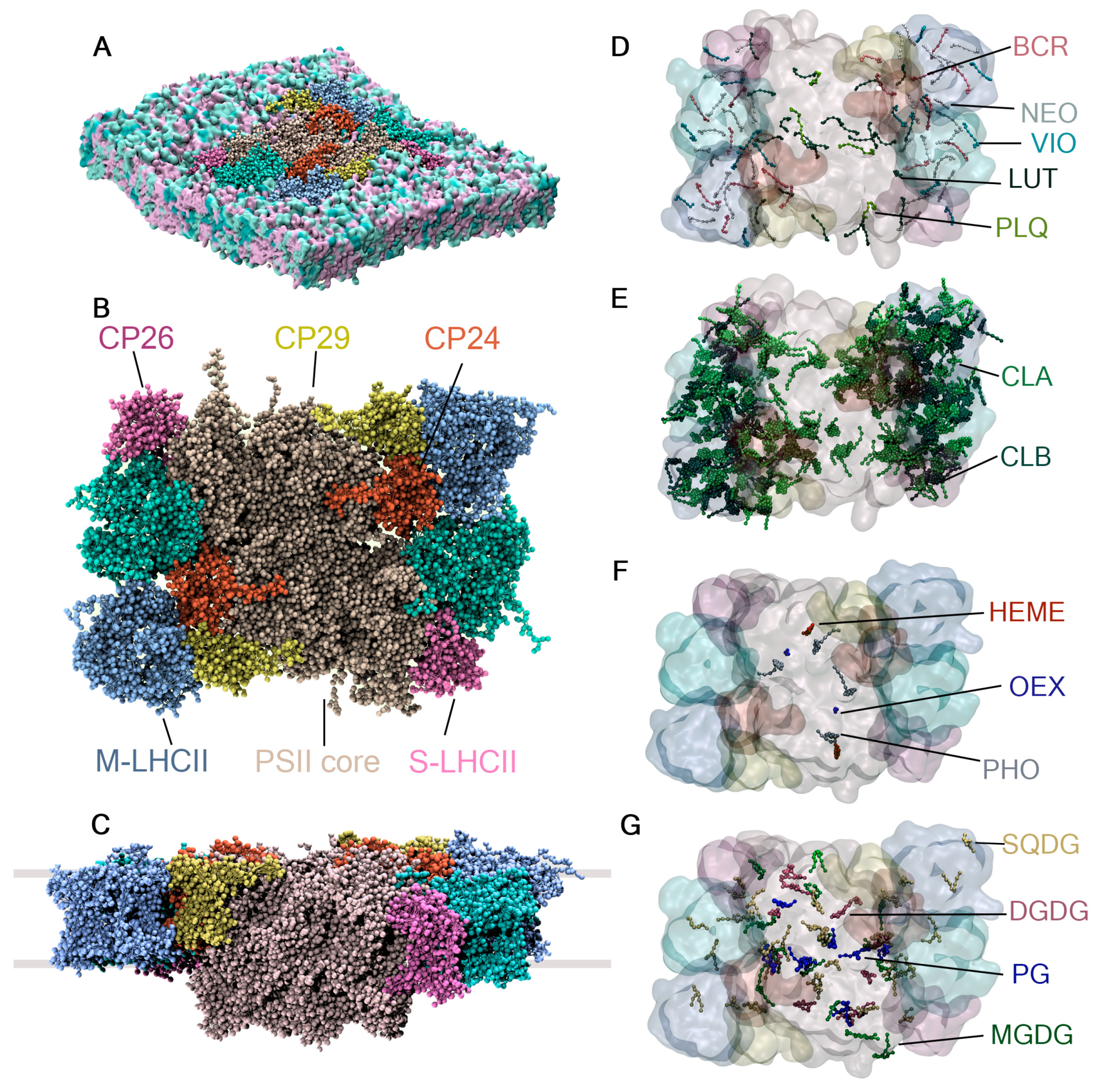
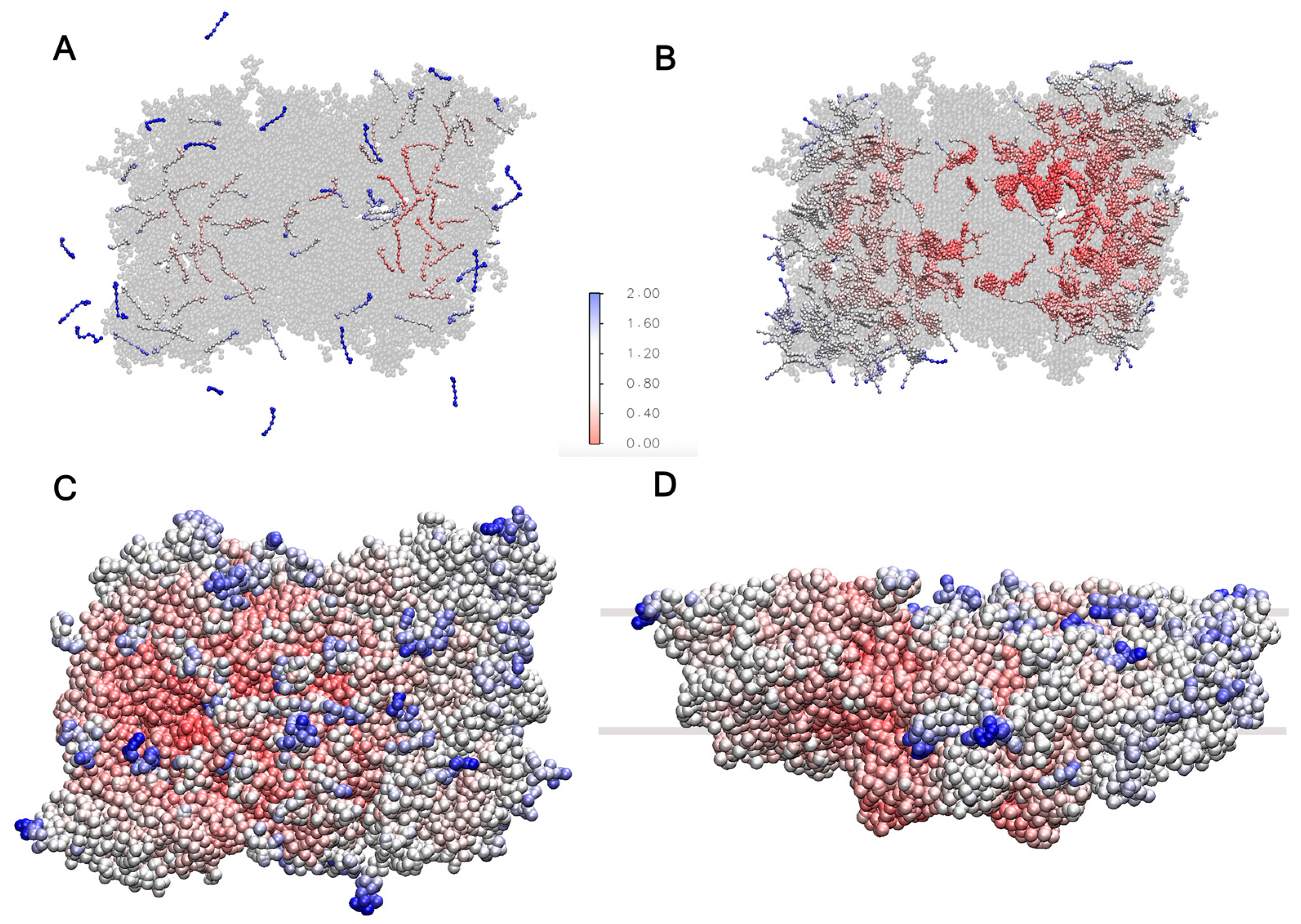
2.4. Cofactors Embedded in the LHCII-PSII Super-Complex
3. Methods
3.1. Parametrization
3.2. Free Energy and Partition Coefficient Calculations
3.3. Cofactors in Membrane
3.4. LHCII
- (1)
- NVT equilibration. Here, no pressure coupling was applied. Long-range interactions were evaluated using particle mesh Ewald summation with a 12 Å cut-off in real space [62]. Lennard–Jones (LJ) interactions were truncated at 12 Å with an atom-based force switching function, which starts to be effective at 10 Å. The integration time step was set at 2 fs for 200 ps. Bonds involving hydrogen atoms were constrained using the LINCS algorithm [63]. A velocity rescale thermostat [56] was used to maintain the temperature at 300 K. Position restraints were applied on the protein.
- (2)
- NPT equilibration. Here, A semi-isotropic Parrinello–Rahman [57] barostat with a reference pressure of 1 bar and isothermal compressibility of 4.5 × 10−5 bar−1 was used to maintain the pressure of the system, while a Nosé–Hoover chain thermostat was used to maintain the temperature at 300 K [64]. The simulation was run for 1 ns.
3.5. PSII-LHCII
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Suga, M.; Akita, F.; Hirata, K.; Ueno, G.; Murakami, H.; Nakajima, Y.; Shimizu, T.; Yamashita, K.; Yamamoto, M.; Ago, H.; et al. Native structure of photosystem II at 1.95 Å resolution viewed by femtosecond X-ray pulses. Nature 2015, 517, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Vinyard, D.J.; Ananyev, G.M.; Charles Dismukes, G. Photosystem II: The Reaction Center of Oxygenic Photosynthesis. Annu. Rev. Biochem. 2013, 82, 577–606. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, H.; Yuan, Q.; Feng, Y. Structure and Function of the Photosystem Supercomplexes. Front. Plant Sci. 2018, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, J.P.; Brudvig, G.W. Water-Splitting Chemistry of Photosystem II. Chem. Rev. 2006, 106, 4455–4483. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.-R. The Structure of Photosystem II and the Mechanism of Water Oxidation in Photosynthesis. Annu. Rev. Plant Biol. 2015, 66, 23–48. [Google Scholar] [CrossRef] [PubMed]
- Lubitz, W.; Chrysina, M.; Cox, N. Water oxidation in photosystem II. Photosynth. Res. 2019, 142, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Rochaix, J.-D. Regulation of photosynthetic electron transport. Biochim. Biophys. Acta (BBA)—Bioenerg. 2011, 1807, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Nikkanen, L.; Solymosi, D.; Jokel, M.; Allahverdiyeva, Y. Regulatory electron transport pathways of photosynthesis in cyanobacteria and microalgae: Recent advances and biotechnological prospects. Physiol. Plant. 2021, 173, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Croce, R.; van Amerongen, H. Natural Strategies for Photosynthetic Light Harvesting. Nat. Chem. Biol. 2014, 10, 492–501. [Google Scholar] [CrossRef]
- Van Amerongen, H.; Valkunas, L.; Van Grondelle, R. Photosynthetic Excitons; World Scientific: Singapore, 2000. [Google Scholar]
- Liu, Z.; Yan, H.; Wang, K.; Kuang, T.; Zhang, J.; Gui, L.; An, X.; Chang, W. Crystal Structure of Spinach Major Light-Harvesting Complex at 2.72 Å Resolution. Nature 2004, 428, 287–292. [Google Scholar] [CrossRef]
- Ruban, A.V.; Johnson, M.P.; Duffy, C.D.P. The Photoprotective Molecular Switch in the Photosystem II Antenna. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.P.; Goral, T.K.; Duffy, C.D.P.; Brain, A.P.R.; Mullineaux, C.W.; Ruban, A.V. Photoprotective Energy Dissipation Involves the Reorganization of Photosystem II Light-Harvesting Complexes in the Grana Membranes of Spinach Chloroplasts. Plant Cell 2011, 23, 1468–1479. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Ma, J.; Wei, X.; Cao, P.; Zhu, D.; Chang, W.; Liu, Z.; Zhang, X.; Li, M. Structure and Assembly Mechanism of plant C2S2M2-type PSII-LHCII Supercomplex. Science 2017, 357, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Huang, Z.; Chang, S.; Wang, W.; Wang, J.; Kuang, T.; Han, G.; Shen, J.-R.; Zhang, X. Structure of a C2S2M2N2-type PSII–LHCII supercomplex from the green alga Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA 2019, 116, 21246–21255. [Google Scholar] [CrossRef] [PubMed]
- Liguori, N.; Campos, S.R.R.; Baptista, A.M.; Croce, R. Molecular Anatomy of Plant Photoprotective Switches: The Sensitivity of PsbS to the Environment, Residue by Residue. J. Phys. Chem. Lett. 2019, 10, 1737–1742. [Google Scholar] [CrossRef] [PubMed]
- Liguori, N.; Croce, R.; Marrink, S.J.; Thallmair, S. Molecular dynamics simulations in photosynthesis. Photosynth. Res. 2020, 144, 273–295. [Google Scholar] [CrossRef]
- Liguori, N.; Periole, X.; Marrink, S.J.; Croce, R. From light-harvesting to photoprotection: Structural basis of the dynamic switch of the major antenna complex of plants (LHCII). Sci. Rep. 2015, 5, 15661. [Google Scholar] [CrossRef] [PubMed]
- Segatta, F.; Cupellini, L.; Garavelli, M.; Mennucci, B. Quantum Chemical Modeling of the Photoinduced Activity of Multichromophoric Biosystems. Chem. Rev. 2019, 119, 9361–9380. [Google Scholar] [CrossRef]
- Cupellini, L.; Bondanza, M.; Nottoli, M.; Mennucci, B. Successes & challenges in the atomistic modeling of light-harvesting and its photoregulation. Biochim. Biophys. Acta (BBA)—Bioenerg. 2020, 1861, 148049. [Google Scholar] [CrossRef]
- Daskalakis, V.; Papadatos, S.; Kleinekathöfer, U. Fine tuning of the photosystem II major antenna mobility within the thylakoid membrane of higher plants. Biochim. Biophys. Acta (BBA)—Biomembr. 2019, 1861, 183059. [Google Scholar] [CrossRef]
- Ogata, K.; Yuki, T.; Hatakeyama, M.; Uchida, W.; Nakamura, S. All-Atom Molecular Dynamics Simulation of Photosystem II Embedded in Thylakoid Membrane. J. Am. Chem. Soc. 2013, 135, 15670–15673. [Google Scholar] [CrossRef] [PubMed]
- van Eerden, F.J.; de Jong, D.H.; de Vries, A.H.; Wassenaar, T.A.; Marrink, S.J. Characterization of thylakoid lipid membranes from cyanobacteria and higher plants by molecular dynamics simulations. Biochim. Biophys. Acta (BBA)—Biomembr. 2015, 1848, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Van Eerden, F.J.; Melo, M.N.; Frederix, P.W.J.M.; Marrink, S.J. Prediction of Thylakoid Lipid Binding Sites on Photosystem II. Biophys. J. 2017, 113, 2669–2681. [Google Scholar] [CrossRef] [PubMed]
- Van Eerden, F.J.; Melo, M.N.; Frederix, P.W.J.M.; Periole, X.; Marrink, S.J. Exchange pathways of plastoquinone and plastoquinol in the photosystem II complex. Nat. Commun. 2017, 8, 15214. [Google Scholar] [CrossRef] [PubMed]
- van Eerden, F.J.; van den Berg, T.; Frederix, P.W.J.M.; de Jong, D.H.; Periole, X.; Marrink, S.J. Molecular Dynamics of Photosystem II Embedded in the Thylakoid Membrane. J. Phys. Chem. B 2017, 121, 3237–3249. [Google Scholar] [CrossRef] [PubMed]
- Thallmair, S.; Vainikka, P.A.; Marrink, S.J. Lipid Fingerprints and Cofactor Dynamics of Light-Harvesting Complex II in Different Membranes. Biophys. J. 2019, 116, 1446–1455. [Google Scholar] [CrossRef]
- Souza, P.C.T.; Alessandri, R.; Barnoud, J.; Thallmair, S.; Faustino, I.; Grünewald, F.; Patmanidis, I.; Abdizadeh, H.; Bruininks, B.M.H.; Wassenaar, T.A.; et al. Martini 3: A General Purpose Force Field for Coarse-Grained Molecular Dynamics. Nat. Methods 2021, 18, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.C.T.; Thallmair, S.; Conflitti, P.; Ramírez-Palacios, C.; Alessandri, R.; Raniolo, S.; Limongelli, V.; Marrink, S.J. Protein–ligand binding with the coarse-grained Martini model. Nat. Commun. 2020, 11, 3714. [Google Scholar] [CrossRef]
- Chiariello, M.G.; Grünewald, F.; Zarmiento-Garcia, R.; Marrink, S.J. pH-Dependent Conformational Switch Impacts Stability of the PsbS Dimer. J. Phys. Chem. Lett. 2023, 14, 905–911. [Google Scholar] [CrossRef]
- de Jong, D.H.; Liguori, N.; van den Berg, T.; Arnarez, C.; Periole, X.; Marrink, S.J. Atomistic and Coarse Grain Topologies for the Cofactors Associated with the Photosystem II Core Complex. J. Phys. Chem. B 2015, 119, 7791–7803. [Google Scholar] [CrossRef]
- Alessandri, R.; Barnoud, J.; Gertsen, A.S.; Patmanidis, I.; de Vries, A.H.; Souza, P.C.T.; Marrink, S.J. Martini 3 Coarse-Grained Force Field: Small Molecules. Adv. Theory Simul. 2022, 5, 2100391. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.-J. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Monticelli, L.; Melo, M.N.; Alessandri, R.; Tieleman, D.P.; Souza, P.C.T. Two Decades of Martini: Better Beads, Broader Scope. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 13, e1620. [Google Scholar] [CrossRef]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of Octanol−Water Partition Coefficients by Guiding an Additive Model with Knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.A.; Webb, D.R.; Peters, J.C. Evaluation of the Potential for Olestra to Affect the Availability of Dietary Phytochemicals1,2. J. Nutr. 1997, 127, 1699S–1709S. [Google Scholar] [CrossRef]
- Rich, P.R.; Harper, R. Partition coefficients of quinones and hydroquinones and their relation to biochemical reactivity. FEBS Lett. 1990, 269, 139–144. [Google Scholar] [CrossRef]
- de Vogel, J.; Jonker-Termont, D.S.M.L.; Katan, M.B.; van der Meer, R. Natural Chlorophyll but Not Chlorophyllin Prevents Heme-Induced Cytotoxic and Hyperproliferative Effects in Rat Colon12. J. Nutr. 2005, 135, 1995–2000. [Google Scholar] [CrossRef] [PubMed]
- Guskov, A.; Kern, J.; Gabdulkhakov, A.; Broser, M.; Zouni, A.; Saenger, W. Cyanobacterial photosystem II at 2.9-Å resolution and the role of quinones, lipids, channels and chloride. Nat. Struct. Mol. Biol. 2009, 16, 334–342. [Google Scholar] [CrossRef]
- Socaciu, C.; Jessel, R.; Diehl, H.A. Competitive carotenoid and cholesterol incorporation into liposomes: Effects on membrane phase transition, fluidity, polarity and anisotropy. Chem. Phys. Lipids 2000, 106, 79–88. [Google Scholar] [CrossRef]
- Socaciu, C.; Lausch, C.; Diehl, H.A. Carotenoids in DPPC vesicles: Membrane dynamics. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1999, 55, 2289–2297. [Google Scholar] [CrossRef]
- Gruszecki, W.I. Carotenoids in Membranes. In The Photochemistry of Carotenoids; Frank, H.A., Young, A.J., Britton, G., Cogdell, R.J., Eds.; Springer: Dordrecht, The Netherlands, 1999; pp. 363–379. [Google Scholar] [CrossRef]
- Jemioła-Rzemińska, M.; Pasenkiewicz-Gierula, M.; Strzałka, K. The behaviour of β-carotene in the phosphatidylcholine bilayer as revealed by a molecular simulation study. Chem. Phys. Lipids 2005, 135, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Mostofian, B.; Johnson, Q.R.; Smith, J.C.; Cheng, X. Carotenoids promote lateral packing and condensation of lipid membranes. Phys. Chem. Chem. Phys. 2020, 22, 12281–12293. [Google Scholar] [CrossRef] [PubMed]
- Sujak, A.; Okulski, W.; Gruszecki, W.I. Organisation of xanthophyll pigments lutein and zeaxanthin in lipid membranes formed with dipalmitoylphosphatidylcholine. Biochim. Biophys. Acta (BBA)—Biomembr. 2000, 1509, 255–263. [Google Scholar] [CrossRef]
- Gruszecki, W.I.; Strzałka, K. Carotenoids as modulators of lipid membrane physical properties. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2005, 1740, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Li, M.; Wan, T.; Wang, L.; Jia, C.; Hou, Z.; Zhao, X.; Zhang, J.; Chang, W. Structural Insights into Energy Regulation of Light-Harvesting Complex CP29 from Spinach. Nat. Struct. Mol. Biol. 2011, 18, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Janik, E.; Bednarska, J.; Zubik, M.; Sowinski, K.; Luchowski, R.; Grudzinski, W.; Gruszecki, W.I. Is It Beneficial for the Major Photosynthetic Antenna Complex of Plants to form Trimers? J. Phys. Chem. B 2015, 119, 8501–8508. [Google Scholar] [CrossRef] [PubMed]
- Garab, G.; Cseh, Z.; Kovács, L.; Rajagopal, S.; Várkonyi, Z.; Wentworth, M.; Mustárdy, L.; Dér, A.; Ruban, A.V.; Papp, E.; et al. Light-Induced Trimer to Monomer Transition in the Main Light-Harvesting Antenna Complex of Plants: Thermo-Optic Mechanism. Biochemistry 2002, 41, 15121–15129. [Google Scholar] [CrossRef]
- Poma, A.B.; Cieplak, M.; Theodorakis, P.E. Combining the MARTINI and Structure-Based Coarse-Grained Approaches for the Molecular Dynamics Studies of Conformational Transitions in Proteins. J. Chem. Theory Comput. 2017, 13, 1366–1374. [Google Scholar] [CrossRef]
- Souza, P.C.T.; Borges-Araújo, L.; Brasnett, C.; Moreira, R.A.; Grünewald, F.; Park, P.; Wang, L.; Razmazma, H.; Borges-Araújo, A.C.; Cofas-Vargas, L.F.; et al. GōMartini 3: From large conformational changes in proteins to environmental bias corrections. bioRxiv 2024, 2024.04.15.589479. [Google Scholar] [CrossRef]
- Mao, R.; Zhang, H.; Bie, L.; Liu, L.-N.; Gao, J. Million-atom molecular dynamics simulations reveal the interfacial interactions and assembly of plant PSII-LHCII supercomplex. RSC Adv. 2023, 13, 6699–6712. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Li, M.; Liu, Z.; Cao, P.; Pan, X.; Zhang, H.; Zhao, X.; Zhang, J.; Chang, W. Crystal Structures of the PsbS Protein Essential for Photoprotection in Plants. Nat. Struct. Mol. Bio. 2015, 22, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Sarewicz, M.; Szwalec, M.; Pintscher, S.; Indyka, P.; Rawski, M.; Pietras, R.; Mielecki, B.; Koziej, Ł.; Jaciuk, M.; Glatt, S.; et al. High-resolution cryo-EM structures of plant cytochrome b6f at work. Sci. Adv. 2023, 9, eadd9688. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling Through velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations Through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Bennett, C.H. Efficient estimation of free energy differences from Monte Carlo data. J. Comput. Phys. 1976, 22, 245–268. [Google Scholar] [CrossRef]
- Wassenaar, T.A.; Ingólfsson, H.I.; Böckmann, R.A.; Tieleman, D.P.; Marrink, S.J. Computational Lipidomics with insane: A Versatile Tool for Generating Custom Membranes for Molecular Simulations. J. Chem. Theory Comput. 2015, 11, 2144–2155. [Google Scholar] [CrossRef] [PubMed]
- de Jong, D.H.; Baoukina, S.; Ingólfsson, H.I.; Marrink, S.J. Martini Straight: Boosting Performance Using a Shorter Cutoff and GPUs. Comput. Phys. Commun. 2016, 199, 1–7. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Nosé, S. A unified Formulation of the Constant Temperature Molecular Dynamics Methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Schrödinger, L.; DeLano, W. PyMOL. 2020. Available online: http://www.pymol.org/pymol (accessed on 15 January 2024).
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Kroon, P.C.; Grünewald, F.; Barnoud, J.; van Tilburg, M.; Souza, P.C.T.; Wassenaar, T.A.; Marrink, S.-J. Martinize2 and Vermouth: Unified Framework for Topology Generation. eLife 2024, 12, RP90627. [Google Scholar]
- Graham, J.A.; Essex, J.W.; Khalid, S. PyCGTOOL: Automated Generation of Coarse-Grained Molecular Dynamics Models from Atomistic Trajectories. J. Chem. Inf. Model. 2017, 57, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Sakurai, I.; Shen, J.-R.; Leng, J.; Ohashi, S.; Kobayashi, M.; Wada, H. Lipids in Oxygen-Evolving Photosystem II Complexes of Cyanobacteria and Higher Plants. J. Biochem. 2006, 140, 201–209. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Kim, H.; Fábián, B.; Hummer, G. Neighbor List Artifacts in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2023, 19, 8919–8929. [Google Scholar] [CrossRef] [PubMed]
- Bruininks, B.M.H.; Wassenaar, T.A.; Vattulainen, I. Unbreaking Assemblies in Molecular Simulations with Periodic Boundaries. J. Chem. Inf. Model. 2023, 63, 3448–3452. [Google Scholar] [CrossRef] [PubMed]
- Cignoni, E.; Lapillo, M.; Cupellini, L.; Acosta-Gutiérrez, S.; Gervasio, F.L.; Mennucci, B. A Different Perspective for NonPhotochemical Quenching in Plant Antenna Complexes. Nat. Commun. 2021, 12, 7152. [Google Scholar] [CrossRef] [PubMed]
- Sacharz, J.; Giovagnetti, V.; Ungerer, P.; Mastroianni, G.; Ruban, A.V. The Xanthophyll Cycle Affects Reversible Interactions between PsbS and light-harvesting Complex II to Control Non-Photochemical Quenching. Nat. Plants 2017, 3, 16225. [Google Scholar] [CrossRef] [PubMed]
- Correa-Galvis, V.; Poschmann, G.; Melzer, M.; Stühler, K.; Jahns, P. PsbS Interactions Involved in the activation of Energy Dissipation in Arabidopsis. Nat. Plants 2016, 2, 15225. [Google Scholar] [CrossRef]
- Hilpert, C.; Beranger, L.; Souza, P.C.T.; Vainikka, P.A.; Nieto, V.; Marrink, S.J.; Monticelli, L.; Launay, G. Facilitating CG Simulations with MAD: The MArtini Database Server. J. Chem. Inf. Model. 2023, 63, 702. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiariello, M.G.; Zarmiento-Garcia, R.; Marrink, S.-J. Martini 3 Coarse-Grained Model for the Cofactors Involved in Photosynthesis. Int. J. Mol. Sci. 2024, 25, 7947. https://doi.org/10.3390/ijms25147947
Chiariello MG, Zarmiento-Garcia R, Marrink S-J. Martini 3 Coarse-Grained Model for the Cofactors Involved in Photosynthesis. International Journal of Molecular Sciences. 2024; 25(14):7947. https://doi.org/10.3390/ijms25147947
Chicago/Turabian StyleChiariello, Maria Gabriella, Rubi Zarmiento-Garcia, and Siewert-Jan Marrink. 2024. "Martini 3 Coarse-Grained Model for the Cofactors Involved in Photosynthesis" International Journal of Molecular Sciences 25, no. 14: 7947. https://doi.org/10.3390/ijms25147947