
Development of Bivalent Aptamer-DNA Carrier-Doxorubicin Conjugates for Targeted Killing of Esophageal Squamous Cell Carcinoma Cells


Abstract
:1. Introduction
2. Results and Discussion
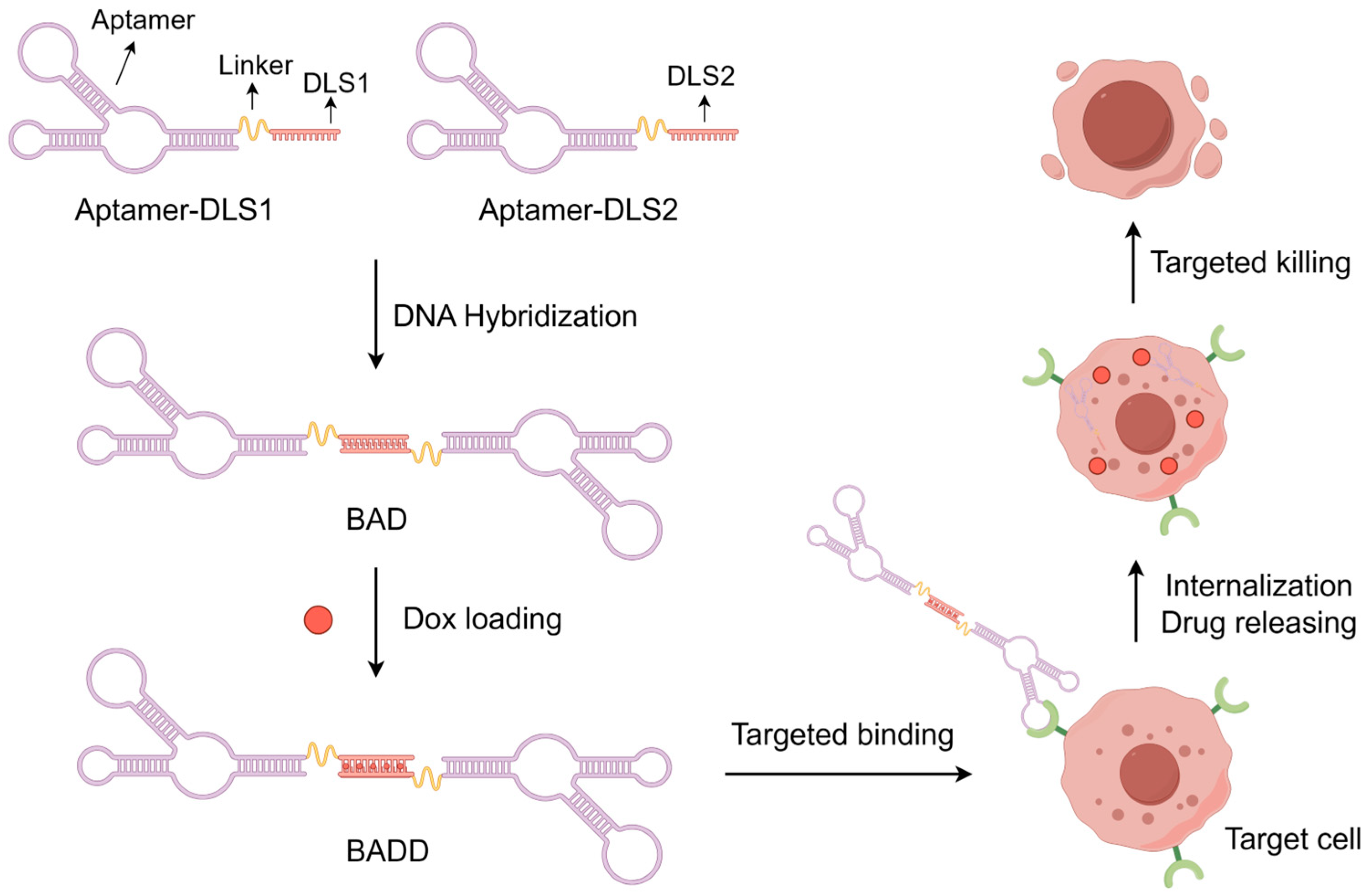
2.1. Design Principle of the BADD
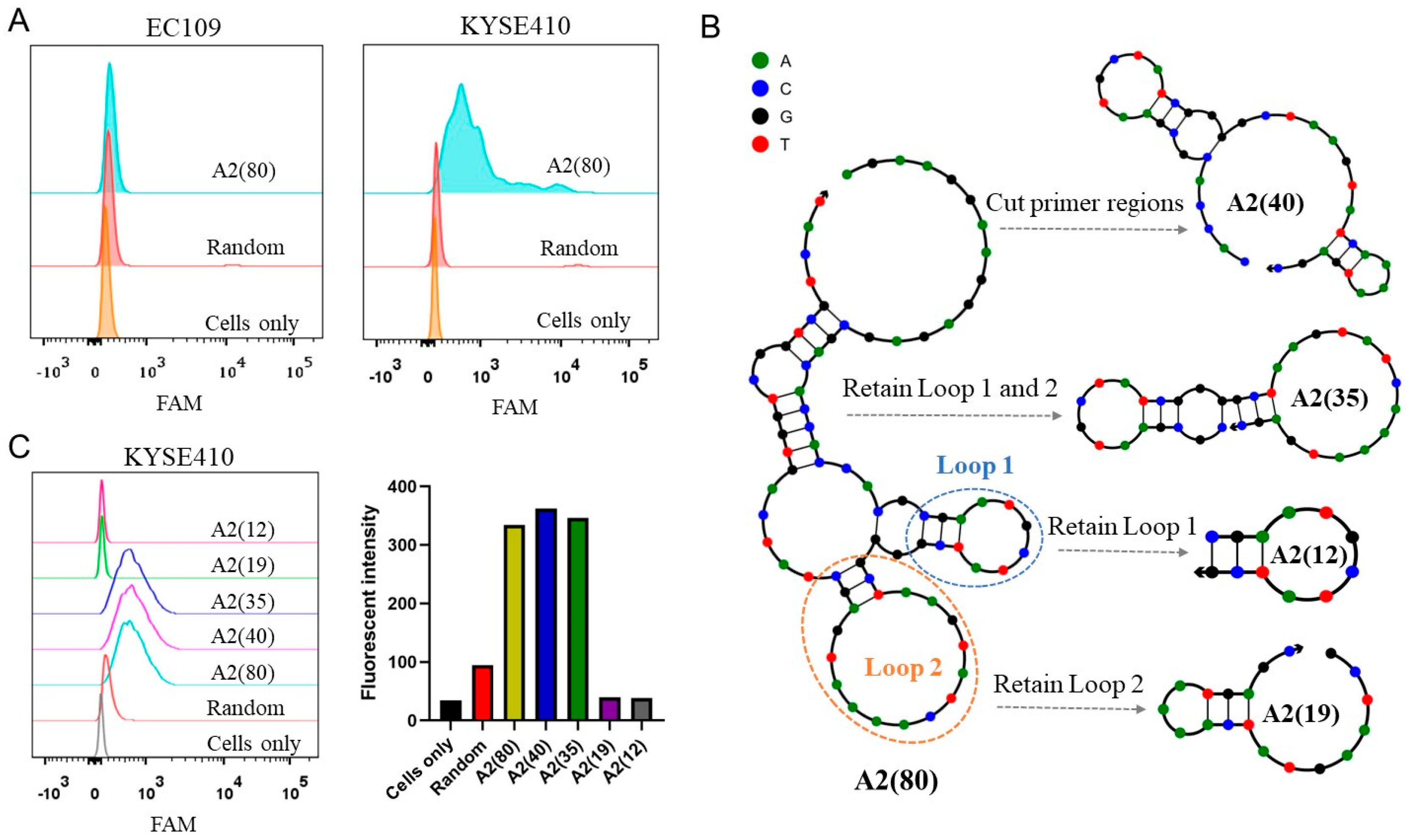
2.2. Optimization of the Aptamer
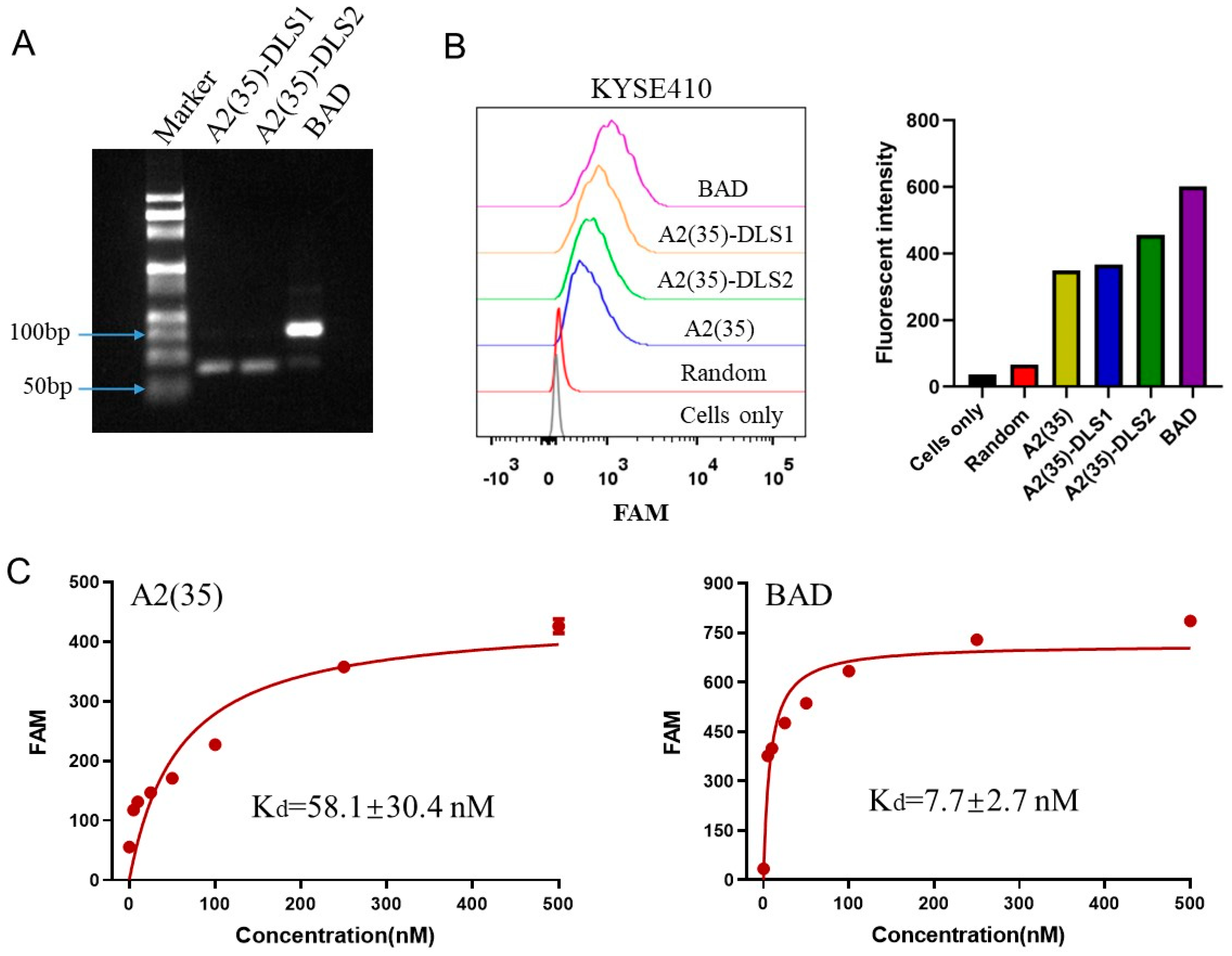
2.3. Construction of the BAD
2.4. Characterization of the BAD
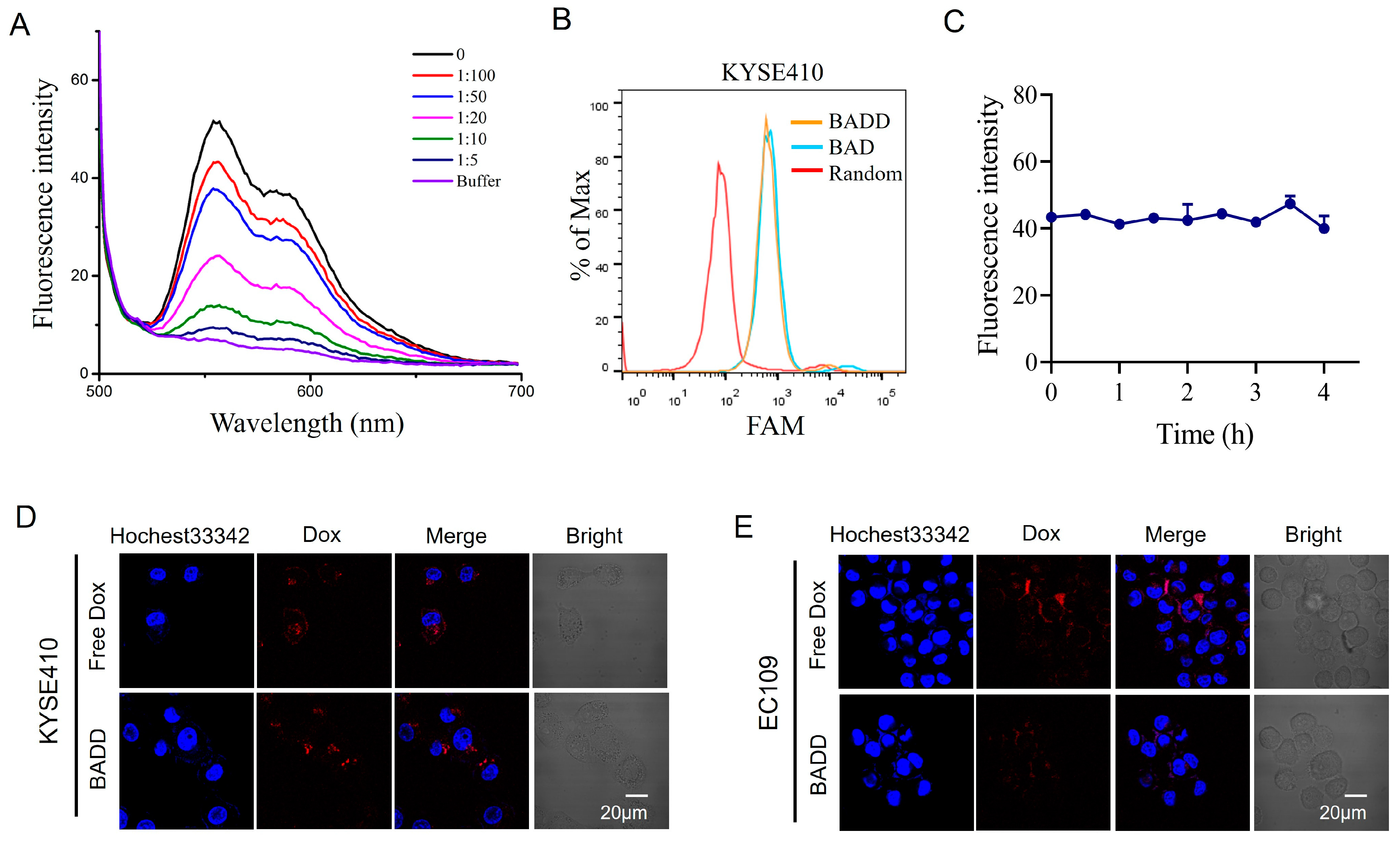
2.5. Construction of the BADD
2.6. Selective Internalization of the BADD
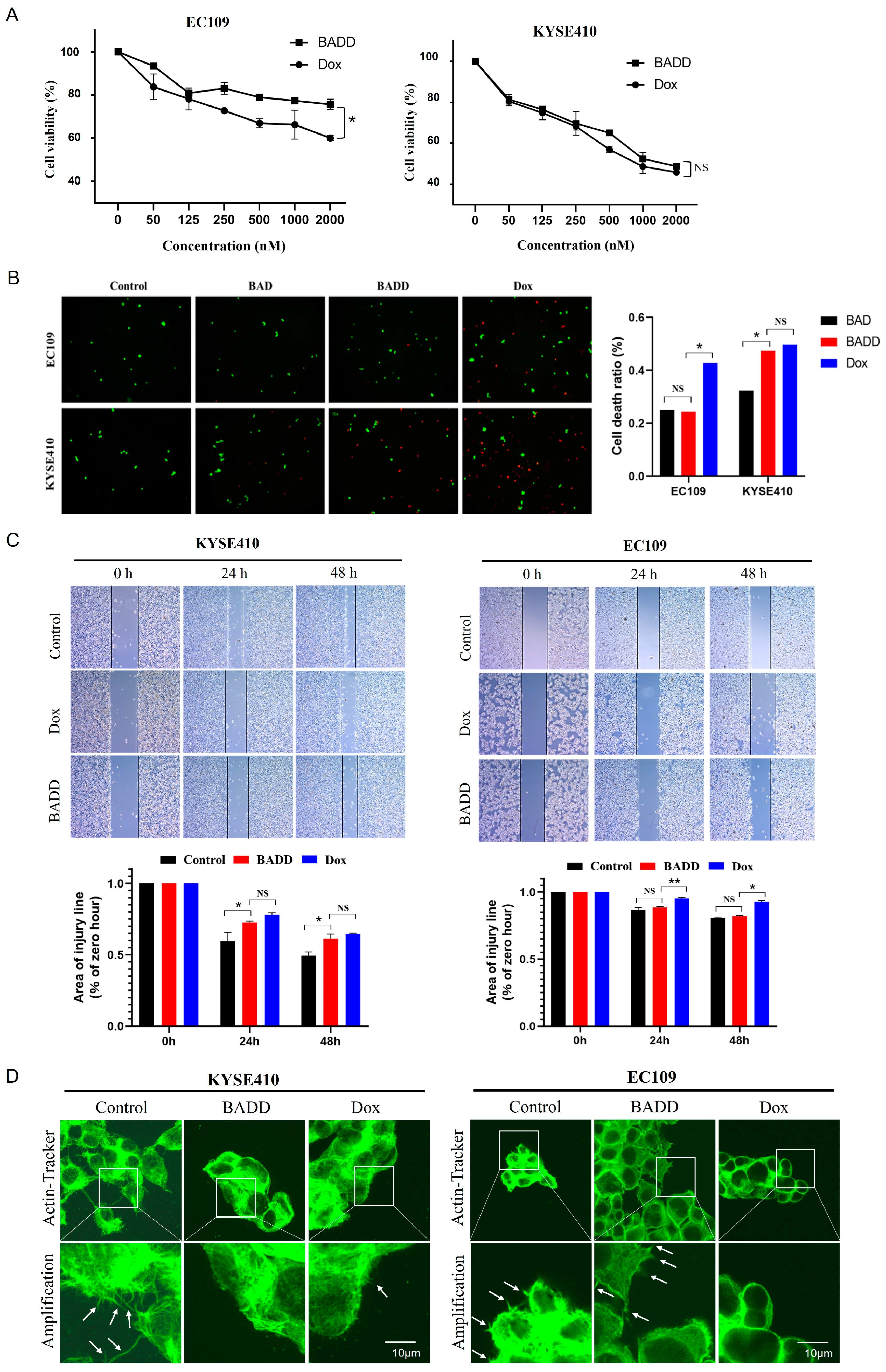
2.7. BADD-Induced Selective Killing of Target Cells
3. Conclusion
4. Materials and Methods
4.1. Chemicals and Materials
4.2. Cells and Culturing
4.3. Preparation and Characterization of the BAD and the BADD
4.4. Flow Cytometry Analysis
4.5. Confocal Imaging
4.6. CCK-8 Analysis
4.7. Calcein AM/PI Staining
4.8. Wound Healing Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Abnet, C.C.; Arnold, M.; Wei, W.Q. Epidemiology of Esophageal Squamous Cell Carcinoma. Gastroenterology 2018, 154, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Lagergren, J.; Fitzgerald, R.C.; Lordick, F.; Shah, M.A.; Lagergren, P.; Cunningham, D. Oesophageal cancer. Nat. Rev. Dis. Primers 2017, 3, 17048. [Google Scholar] [CrossRef] [PubMed]
- Lagergren, J.; Smyth, E.; Cunningham, D.; Lagergren, P. Oesophageal cancer. Lancet 2017, 390, 2383–2396. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, S.; Miyamoto, S.; Kikuchi, O.; Goto, T.; Amanuma, Y.; Muto, M. Recent Advances from Basic and Clinical Studies of Esophageal Squamous Cell Carcinoma. Gastroenterology 2015, 149, 1700–1715. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Hong, P.; Xu, W.W.; He, Q.Y.; Li, B. Advances in targeted therapy for esophageal cancer. Signal Transduct. Target Ther. 2020, 5, 229. [Google Scholar] [CrossRef]
- He, S.; Xu, J.; Liu, X.; Zhen, Y. Advances and challenges in the treatment of esophageal cancer. Acta Pharm. Sin. B 2021, 11, 3379–3392. [Google Scholar] [CrossRef] [PubMed]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Kołat, D.; Kałuzińska-Kołat, Ż.; Celik, I.; Kontek, R. Doxorubicin-An Agent with Multiple Mechanisms of Anticancer Activity. Cells 2023, 12, 659. [Google Scholar] [CrossRef]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The Good, the Bad and the Ugly Effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef]
- Kong, C.Y.; Guo, Z.; Song, P.; Zhang, X.; Yuan, Y.P.; Teng, T.; Yan, L.; Tang, Q.Z. Underlying the Mechanisms of Doxorubicin-Induced Acute Cardiotoxicity: Oxidative Stress and Cell Death. Int. J. Biol. Sci. 2022, 18, 760–770. [Google Scholar] [CrossRef]
- Pugazhendhi, A.; Edison, T.; Velmurugan, B.K.; Jacob, J.A.; Karuppusamy, I. Toxicity of Doxorubicin (Dox) to different experimental organ systems. Life Sci. 2018, 200, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Zou, X.; Wang, Y.; Dong, Z.; Weng, X.; Pei, Z.; Song, S.; Zhao, Y.; Wei, Z.; Gao, R.; et al. Critical Role of the cGAS-STING Pathway in Doxorubicin-Induced Cardiotoxicity. Circ. Res. 2023, 132, e223–e242. [Google Scholar] [CrossRef]
- Ijäs, H.; Shen, B.; Heuer-Jungemann, A.; Keller, A.; Kostiainen, M.A.; Liedl, T.; Ihalainen, J.A.; Linko, V. Unraveling the interaction between doxorubicin and DNA origami nanostructures for customizable chemotherapeutic drug release. Nucleic Acids Res. 2021, 49, 3048–3062. [Google Scholar] [CrossRef]
- Yuan, B.; Xi, Y.; Qi, C.; Zhao, M.; Zhu, X.; Tang, J. A sequentially triggered DNA nanocapsule for targeted drug delivery based on pH-responsive i-motif and tumor cell-specific aptamer. Front. Bioeng. Biotechnol. 2022, 10, 965337. [Google Scholar] [CrossRef]
- Ma, W.; Sun, H.; Chen, B.; Jia, R.; Huang, J.; Cheng, H.; He, X.; Huang, M.; Wang, K. Engineering a Facile Aptamer “Molecule-Doctor” with Hairpin-Contained I-Motif Enables Accurate Imaging and Killing of Cancer Cells. Anal. Chem. 2021, 93, 14552–14559. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- DeRosa, M.C.; Lin, A.; Mallikaratchy, P.; McConnell, E.M.; McKeague, M.; Patel, R.; Shigdar, S. In vitro selection of aptamers and their applications. Nat. Rev. Methods Primers 2023, 3, 55. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu, S.; Yan, H.; Li, X.; Yazd, H.S.; Li, X.; Huang, T.; Cui, C.; Jiang, J.; Tan, W. Nucleic Acid Aptamers for Molecular Diagnostics and Therapeutics: Advances and Perspectives. Angew. Chem. Int. Ed. 2021, 60, 2221–2231. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef]
- Tang, J.; Li, B.; Qi, C.; Wang, Z.; Yin, K.; Guo, L.; Zhang, W.; Yuan, B. Imaging specific cell-surface sialylation using DNA dendrimer-assisted FRET. Talanta 2022, 243, 123399. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Qi, C.; Bai, X.; Ji, M.; Wang, Z.; Luo, Y.; Ni, S.; Zhang, T.; Liu, K.; Yuan, B. Cell Membrane-Anchored DNA Nanoinhibitor for Inhibition of Receptor Tyrosine Kinase Signaling Pathways via Steric Hindrance and Lysosome-Induced Protein Degradation. ACS Pharmacol. Transl. Sci. 2024, 7, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yuan, B.; Lin, X.; Meng, X.; Wen, X.; Guo, Q.; Li, L.; Jiang, H.; Wang, K. Intramolecular trigger remodeling-induced HCR for amplified detection of protein-specific glycosylation. Talanta 2020, 215, 120889. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tam, W.W.; Yu, Y.; Zhuo, Z.; Xue, Z.; Tsang, C.; Qiao, X.; Wang, X.; Wang, W.; Li, Y.; et al. The application of Aptamer in biomarker discovery. Biomark. Res. 2023, 11, 70. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Liu, L.; Liu, X.; Jiang, H.; Wang, X. Aptamer-Based Immune Drug Systems (AptIDCs) Potentiating Cancer Immunotherapy. Chemistry 2023, 5, 1656–1680. [Google Scholar] [CrossRef]
- Shangguan, D.; Li, Y.; Tang, Z.; Cao, Z.C.; Chen, H.W.; Mallikaratchy, P.; Sefah, K.; Yang, C.J.; Tan, W. Aptamers evolved from live cells as effective molecular probes for cancer study. Proc. Natl. Acad. Sci. USA 2006, 103, 11838–11843. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, X.; Qiao, Y.; Yang, S.; Wang, Z.; Ji, M.; Yin, K.; Zhao, J.; Liu, K.; Yuan, B. DNA Aptamer Selected against Esophageal Squamous Cell Carcinoma for Tissue Imaging and Targeted Therapy with Integrin beta1 as a Molecular Target. Anal. Chem. 2022, 94, 17212–17222. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, Y.; Shi, Y.; Niu, T.; Li, B.; Guo, L.; Qiao, Y.; Zhao, J.; Yuan, B.; Liu, K. Evolution of DNA aptamers against esophageal squamous cell carcinoma using cell-SELEX. Analyst 2021, 146, 4180–4187. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Shi, Y.; Ji, M.; Wang, Z.; Bai, X.; Zhang, K.; Yin, K.; Zhang, Y.; Chen, X.; Zhang, Y.; et al. Selection and identification of a prohibitin 2-binding DNA aptamer for tumor tissue imaging and targeted chemotherapy. Int. J. Biol. Macromol. 2024, 259, 129002. [Google Scholar] [CrossRef]
- Zhu, L.; Yang, J.; Ma, Y.; Zhu, X.; Zhang, C. Aptamers Entirely Built from Therapeutic Nucleoside Analogues for Targeted Cancer Therapy. J. Am. Chem. Soc. 2022, 144, 1493–1497. [Google Scholar] [CrossRef]
- Li, F.; Lu, J.; Liu, J.; Liang, C.; Wang, M.; Wang, L.; Li, D.; Yao, H.; Zhang, Q.; Wen, J.; et al. A water-soluble nucleolin aptamer-paclitaxel conjugate for tumor-specific targeting in ovarian cancer. Nat. Commun. 2017, 8, 1390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jin, C.; Zhang, L.; Peng, B.; Zhang, Y.; Liu, Y.; Li, L.; Ye, M.; Xiong, W.; Tan, W. CD71-Specific Aptamer Conjugated with Monomethyl Auristatin E for the Treatment of Uveal Melanoma. ACS Appl. Mater. Interfaces 2022, 14, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Peng, Y.; Tan, Y.; Xuan, W.; Fu, T.; Wang, X.Q.; Tan, W. A new paradigm for artesunate anticancer function: Considerably enhancing the cytotoxicity via conjugating artesunate with aptamer. Signal Transduct. Target Ther. 2021, 6, 327. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Tang, L.; Zheng, M.; Liu, J.; Zhang, Z.; Li, Z.; Yang, Q.; Xiang, S.; Fang, L.; Ren, Q.; et al. Structure-Guided Designing Pre-Organization in Bivalent Aptamers. J. Am. Chem. Soc. 2022, 144, 4507–4514. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, X.; Mao, M.; Peng, C.; Wang, Z. Accelerated CRISPR/Cas12a-based small molecule detection using bivalent aptamer. Biosens. Bioelectron. 2022, 217, 114725. [Google Scholar] [CrossRef]
- Riccardi, C.; Napolitano, E.; Musumeci, D.; Montesarchio, D. Dimeric and Multimeric DNA Aptamers for Highly Effective Protein Recognition. Molecules 2020, 25, 5227. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ma, R.; Li, L.; Wang, L.; Li, J.; Sun, J.; Mao, X.; Tan, W. Engineering Robust Aptamers with High Affinity by Key Fragment Evolution and Terminal Fixation. Anal. Chem. 2022, 94, 16282–16289. [Google Scholar] [CrossRef]
- Gao, S.; Zheng, X.; Jiao, B.; Wang, L. Post-SELEX optimization of aptamers. Anal. Bioanal. Chem. 2016, 408, 4567–4573. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Zhou, Y.; Guo, Q.; Wang, K.; Yang, X.; Meng, X.; Wan, J.; Tan, Y.; Huang, Z.; Xie, Q.; et al. A signal-on split aptasensor for highly sensitive and specific detection of tumor cells based on FRET. Chem. Commun. 2016, 52, 1590–1593. [Google Scholar] [CrossRef]
- Xue, L.; Maihle, N.J.; Yu, X.; Tang, S.C.; Liu, H.Y. Synergistic Targeting HER2 and EGFR with Bivalent Aptamer-siRNA Chimera Efficiently Inhibits HER2-Positive Tumor Growth. Mol. Pharm. 2018, 15, 4801–4813. [Google Scholar] [CrossRef]
- Yang, F.; Li, S.; Yuan, R.; Xiang, Y. A bivalent aptamer and terminus-free siRNA junction nanostructure for targeted gene silencing in cancer cells. J. Mater. Chem. B 2022, 10, 8315–8321. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Liu, K.; Huang, Z.; Lu, J.; Wang, H.-H.; Hong, Y.; Xie, Z.; Li, H. A Bivalent Aptamer-Based DNA Agonist for EGFR Signaling Effectively Alleviates Ulcerative Colitis In Vivo. ACS Chem. Biol. 2024, 19, 1280–1290. [Google Scholar] [CrossRef]
- Kuai, H.; Zhao, Z.; Mo, L.; Liu, H.; Hu, X.; Fu, T.; Zhang, X.; Tan, W. Circular Bivalent Aptamers Enable in Vivo Stability and Recognition. J. Am. Chem. Soc. 2017, 139, 9128–9131. [Google Scholar] [CrossRef]
- Jiang, Y.; Pan, X.; Chang, J.; Niu, W.; Hou, W.; Kuai, H.; Zhao, Z.; Liu, J.; Wang, M.; Tan, W. Supramolecularly Engineered Circular Bivalent Aptamer for Enhanced Functional Protein Delivery. J. Am. Chem. Soc. 2018, 140, 6780–6784. [Google Scholar] [CrossRef]
- Zhou, F.; Wang, P.; Peng, Y.; Zhang, P.; Huang, Q.; Sun, W.; He, N.; Fu, T.; Zhao, Z.; Fang, X.; et al. Molecular Engineering-Based Aptamer-Drug Conjugates with Accurate Tunability of Drug Ratios for Drug Combination Targeted Cancer Therapy. Angew. Chem. Int. Ed. 2019, 58, 11661–11665. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yu, J.; Zhang, R.; Chen, P.; Jiang, H.; Yu, W. Doxorubicin prodrug-based nanomedicines for the treatment of cancer. Eur. J. Med. Chem. 2023, 258, 115612. [Google Scholar] [CrossRef]
- Agudelo, D.; Bourassa, P.; Bérubé, G.; Tajmir-Riahi, H.A. Intercalation of antitumor drug doxorubicin and its analogue by DNA duplex: Structural features and biological implications. Int. J. Biol. Macromol. 2014, 66, 144–150. [Google Scholar] [CrossRef]
- Shen, L.Y.; Wang, H.; Dong, B.; Yan, W.P.; Lin, Y.; Shi, Q.; Chen, K.N. Possible prediction of the response of esophageal squamous cell carcinoma to neoadjuvant chemotherapy based on gene expression profiling. Oncotarget 2016, 7, 4531–4541. [Google Scholar] [CrossRef] [PubMed]
- Egli, M.; Manoharan, M. Chemistry, structure and function of approved oligonucleotide therapeutics. Nucleic Acids Res. 2023, 51, 2529–2573. [Google Scholar] [CrossRef]
- Wang, R.E.; Wu, H.; Niu, Y.; Cai, J. Improving the stability of aptamers by chemical modification. Curr. Med. Chem. 2011, 18, 4126–4138. [Google Scholar] [CrossRef]
- Ni, S.; Yao, H.; Wang, L.; Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Chemical Modifications of Nucleic Acid Aptamers for Therapeutic Purposes. Int. J. Mol. Sci. 2017, 18, 1683. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, A. R, Nuclease resistance of DNA nanostructures. Nat. Rev. Chem. 2021, 5, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Li, W.; Luo, Y.; Ni, S.; Ji, M.; Wang, Z.; Zhang, T.; Bai, X.; Tang, J.; Yuan, B.; et al. Selective inhibition of c-Met signaling pathways with a bispecific DNA nanoconnector for the targeted therapy of cancer. Int. J. Biol. Macromol. 2024, 273, 133134. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequences (5′–3′) |
|---|---|
| A2(80) | AGAAGGAAGGAGAGCGACACCACCACGCGAATGCTATCGGGGCTAAGTATCAAAATGAGCTATCAGTGGTCGGTCGTCAT |
| A2(40) | CACCACGCGAATGCTATCGGGGCTAAGTATCAAAATGAGC |
| A2(35) | CGCGAATGCTATCGGGGCTAAGTATCAAAATGAGC |
| A2(19) | GCTAAGTATCAAAATGAGC |
| A2(12) | CGAATGCTATCG |
| A2(35)-DLS1 | CGTCGTCGTCGTCGTCGTTTTCGCGAATGCTATCGGGGCTAAGTATCAAAATGAGC |
| A2(35)-DLS2 | ACGACGACGACGACGACGTTTCGCGAATGCTATCGGGGCTAAGTATCAAAATGAGC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Yin, K.; Niu, X.; Bai, X.; Wang, Z.; Ji, M.; Yuan, B. Development of Bivalent Aptamer-DNA Carrier-Doxorubicin Conjugates for Targeted Killing of Esophageal Squamous Cell Carcinoma Cells. Int. J. Mol. Sci. 2024, 25, 7959. https://doi.org/10.3390/ijms25147959
Zhang T, Yin K, Niu X, Bai X, Wang Z, Ji M, Yuan B. Development of Bivalent Aptamer-DNA Carrier-Doxorubicin Conjugates for Targeted Killing of Esophageal Squamous Cell Carcinoma Cells. International Journal of Molecular Sciences. 2024; 25(14):7959. https://doi.org/10.3390/ijms25147959
Chicago/Turabian StyleZhang, Tianlu, Kai Yin, Xidong Niu, Xue Bai, Zhaoting Wang, Mengmeng Ji, and Baoyin Yuan. 2024. "Development of Bivalent Aptamer-DNA Carrier-Doxorubicin Conjugates for Targeted Killing of Esophageal Squamous Cell Carcinoma Cells" International Journal of Molecular Sciences 25, no. 14: 7959. https://doi.org/10.3390/ijms25147959