1. Introduction
Invasive species are a leading cause of biodiversity loss and ecosystem disruption worldwide, necessitating an in-depth understanding of the mechanisms underlying successful invasions [
1]. The dynamics of species dominance within ecological niches are complex, with population genetics playing a crucial role in determining the outcome of species interactions [
2]. For nonnative species, genetic diversity is often a key determinant of their invasiveness and potential to spread and establish in novel habitats [
3,
4]. High levels of genetic variation can facilitate rapid adaptation to novel environments, predisposing some populations to becoming invasive [
5]. Therefore, understanding the genetic underpinnings of successful invasion and competitive displacement of populations is critical to advancing our understanding of invasion mechanisms.
The whitefly
Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae), also known as the silverleaf whitefly or sweet potato whitefly, provides an interesting case study for dissecting the genetic factors that contribute to invasive success and population displacement. It is one of the most widespread and insidious pests plaguing agriculture and horticulture worldwide [
6] and is notorious for its ability to transmit plant viruses, cause direct damage through sap feeding, and induce the growth of sooty mold on plants, causing severe economic losses globally [
7,
8]. This pest is generally considered to consist of a complex of more than 44 cryptic species that are morphologically identical but exhibit significant variation in biological traits, such as the ecological niche, plant host range, endosymbionts, insecticide resistance, and ability to transmit viruses [
9,
10,
11,
12,
13,
14].
The invasive nature of two members within this complex, namely, Middle East-Asia Minor 1 (MEAM1, formerly known as the B biotype) and Mediterranean (MED, formerly known as the Q biotype), has led to widespread concern among both researchers and agricultural practitioners [
15,
16,
17]. In China, these two invasive whiteflies were successively discovered in the mid-1990s and early 21st century (approximately 2003) [
18,
19]. Following a succession of events in which MEAM1 replaced native cryptic species populations and was subsequently displaced by MED populations, the latter invader MED became the dominant cryptic species in most Chinese field regions by 2020 [
20]. Despite extensive research aimed at elucidating the ecological and biological mechanisms contributing to this shift, such as insecticide application, host plants, feeding behavior, and environmental suitability [
21,
22,
23], the specific role of population genetics in driving the dynamics of invasion and dominance between these two species has not been fully explored.
Exploring the molecular variability within species is a useful approach for population genetic studies [
24]. Currently, molecular markers such as mitochondrial DNA and microsatellites are widely used for species identification and population genetics studies [
25]. Owing to their high resolution in revealing genetic diversity, mitochondrial DNA markers, specifically the cytochrome
c oxidase subunit I (
mtCOI) gene, have been widely applied in DNA barcoding [
26,
27]. Microsatellites, another type of molecular marker, exhibit high polymorphism and codominance, making them valuable for evaluating genetic diversity, population structure, and gene flow patterns both within and among populations [
28,
29]. By integrating both types of markers, a comprehensive analysis of intra- and interspecific variation can be performed [
30,
31,
32].
In this study, we used mitochondrial DNA sequencing and microsatellite genotyping techniques to analyze the genetic diversity and population structure of 22 MED and 8 MEAM1 populations collected from different regions of China. We not only investigated the levels of genetic differentiation and gene flow between and among populations of both species, shedding light on the geographical structuring of genetic variation but also compared their genetic diversities. On this basis, we elucidated whether the successful invasion and competitive displacement of B. tabaci MED and MEAM1 in China has been facilitated by disparities in their population genetics. This work provides crucial information for developing informed strategies to manage these destructive pests and to advance our understanding of genetic influences on species invasiveness and competitiveness more broadly.
3. Discussion
B. tabaci has increasingly been shown to exhibit complex genetic diversity, mostly as a unique cryptic species complex [
33]. However, few studies have focused on the population genetics of the two invasive cryptic species within this complex, and even less is known about the role of genetic factors in their invasion and population displacement [
34]. In this study, our genetic analysis revealed that the MED population presented greater genetic diversity than did the MEAM1 population in China, which is consistent with their population dominance and substitution process. Therefore, we hypothesize that disparities in genetic diversity are likely to have driven population shifts between these two invasive whiteflies.
The results of our analysis on the basis of the
mtCOI gene indicate that both the MED and MEAM1 populations present limited genetic variation at this mitochondrial locus, despite their wide geographic distribution. This low genetic diversity could be due to various factors, such as recent population expansions following bottleneck events, selective sweeps, or a combination thereof [
31,
35]. The significant negative values obtained from
Tajima’s D and
Fu’s F statistics are consistent with a demographic expansion scenario. Nuclear genetic diversity, however, as indicated by microsatellite data, is more substantial within these populations. The discrepancy between mitochondrial and nuclear genetic diversity may stem from the different modes of inheritance and evolutionary dynamics between these two types of markers. Mitochondrial DNA is inherited exclusively through the maternal line, whereas microsatellites reflect biparental inheritance and may be assessed to capture a more complete history of genetic exchange within and among populations [
36].
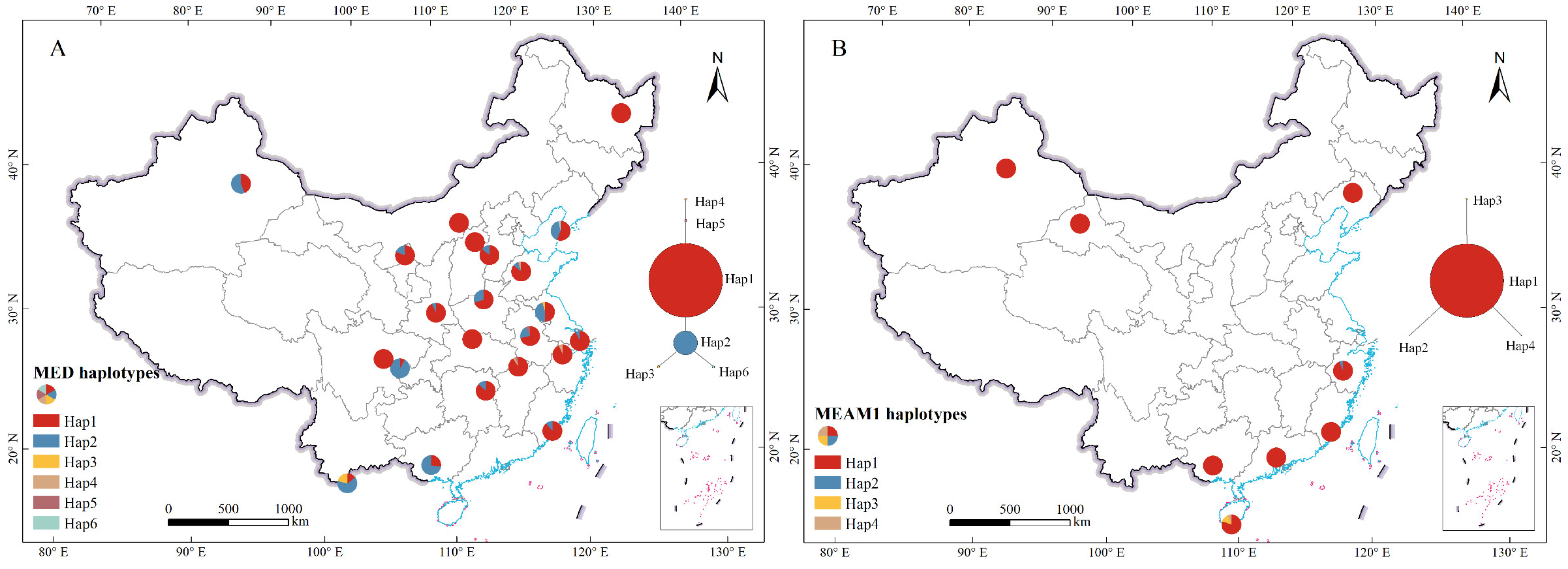
Previous studies on the basis of the
mtCOI gene have shown that the MED population in China has low haplotype diversity and does not form significant geographic clusters, although global populations can be distinguished into two subclades [
32,
37,
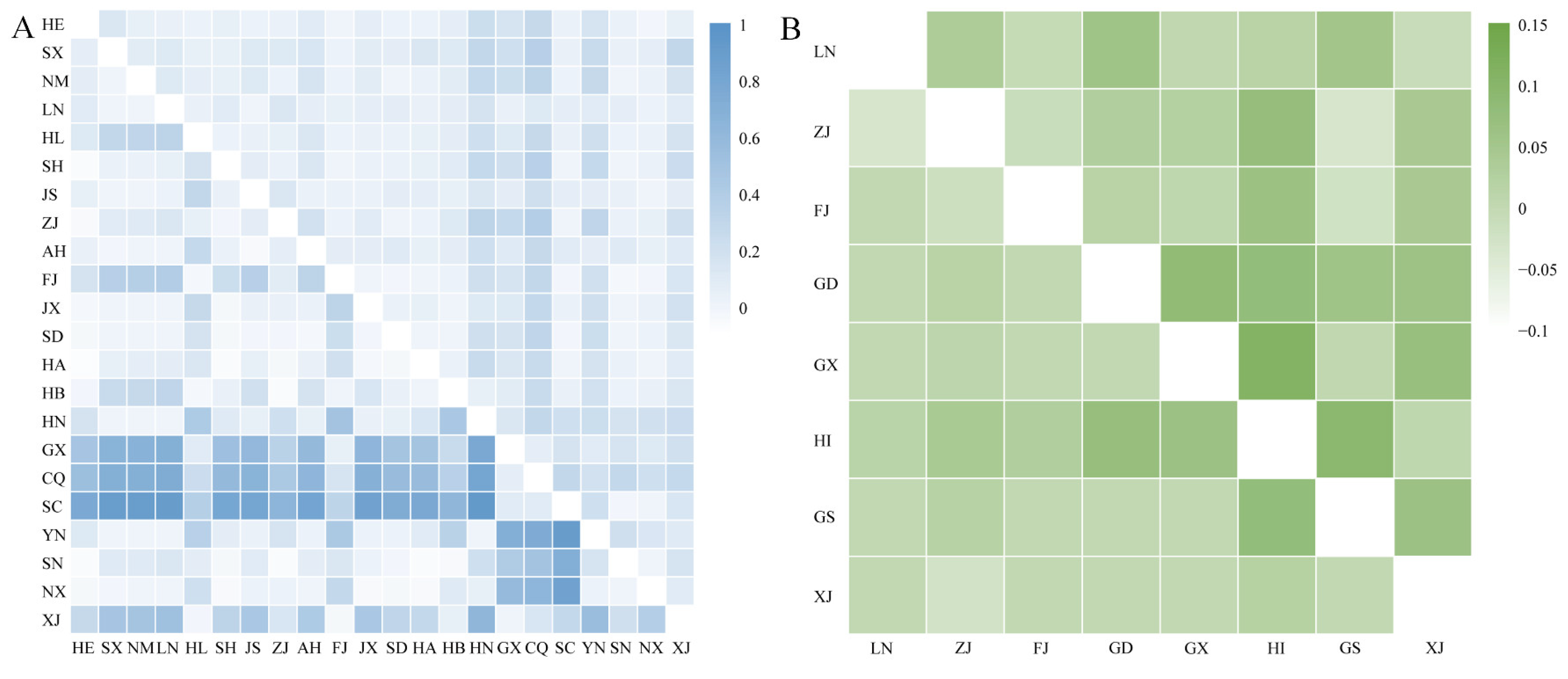
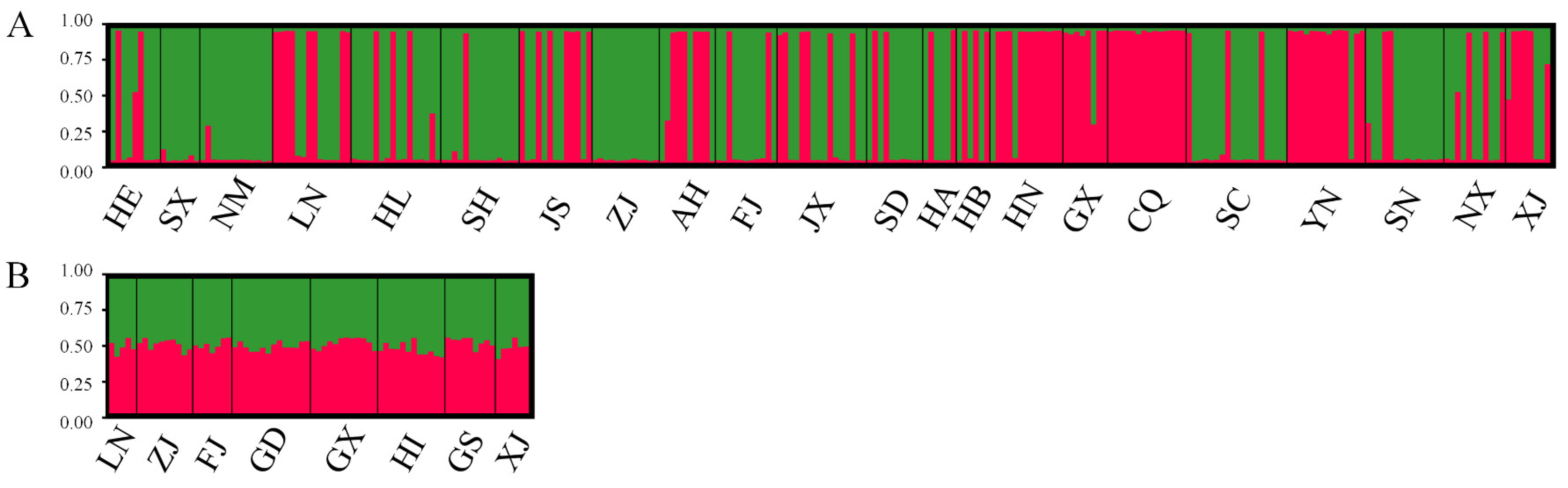
38]. The present study also confirmed those previous observations. The star-shaped radiation distribution in the haplotype network implies a common ancestor for the haplotypes, with subsequent mutations leading to the current diversity. The dominance of a single haplotype in MED populations may suggest a founder effect, where a small number of individuals colonized new areas and gave rise to the current populations. Nevertheless, the microsatellite marker analysis results in this study suggested that there may be two subdivisions of MED populations in China, while the population structure was not distinguished by geographic barriers. Similarly, Simón et al. [
39] reported substantial population structure differences among MED populations on the basis of microsatellite marker analysis. In contrast, there may be a more homogenous population with less geographic influence on the genetic structure of MEAM1. Such homogeneity among different populations could result from extensive gene flow. Indeed, pairwise fixation index values revealed that there was no significant correlation between the evolutionary relationships of the haplotypes and the distributions of geographic populations of both MED and MEAM1. In addition, the results of AMOVA and structural analysis also provided no evidence for the clustering of geographic population structure. The low genetic diversity observed in invasive species could be influenced by genetic bottlenecks, founder effects, and insecticide use [
40,
41,
42]. The persistence of a single population may also be due to its dispersal following annual population explosions [
35], as our results show that the northern populations of MEAM1 have only one haplotype.
The present study, which was based on analysis of
mtCOI gene sequences and microsatellite markers, revealed that the genetic diversity of MED was greater than that of MEAM1, which is consistent with the results of other reports based on analysis of AFLP or RAPD markers in China [
43,
44] as well as in the United States [
29]. Although the results derived from these different molecular tools were not perfectly consistent regarding genetic diversity and gene flow between different geographic populations, the results consistently confirmed the objective disparities in genetic diversity between the two invasive cryptic species. This difference has been suggested to affect the invasiveness of these whitefly populations [
43]. As corroborated by a field survey, MED has replaced MEAM1 as the dominant whitefly throughout China [
20], as MED was the most prevalent cryptic species among the sampled sites in that study.
Interestingly, there are important biological differences between the two cryptic species. MEAM1 outperforms MED in terms of fecundity, nymphal survival, and population increase, aided by reproductive disturbance [
45]. Thus, MEAM1 tended to outcompete and displace MED in the absence of anthropogenic disturbance. However, the relatively high insecticide resistance of MED gives it a competitive advantage over MEAM1 in the field [
13,
42]. As discussed above, insecticide use can significantly influence the genetic diversity of
B. tabaci populations, potentially explaining the lower genetic variation observed. Nevertheless, MED exhibits relatively high genetic diversity, suggesting a greater capacity to adapt to changes in the external environment, including exposure to pesticides. Therefore, under the selective pressure of extensive and frequent insecticide applications, MED can prevail in competition with MEAM1, leading to significant shifts in dominance in field populations.
While previous research has revealed the global dispersal capability of MED and MEAM1 populations, this study presents a new distribution map and directly compares the genetic diversity of these two invasive species across China, providing new perspectives on the genetic basis of their invasion success and competitive displacement. These results highlight the importance of considering genetic diversity when formulating management strategies, as it may influence the adaptability and dispersal potential of populations. Further research should explore the role of host plants and environmental factors in shaping the genetic structure of these populations to provide insights into the ecology and evolution of these invasive species. Because the biotypes and genetic structure of B. tabaci can change rapidly over time, future research should also include long-term monitoring to track changes in population genetic structure.

 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}