

Identification and Validation of New DNA-PKcs Inhibitors through High-Throughput Virtual Screening and Experimental Verification
 , , and
, , and
Abstract
:1. Introduction
2. Results
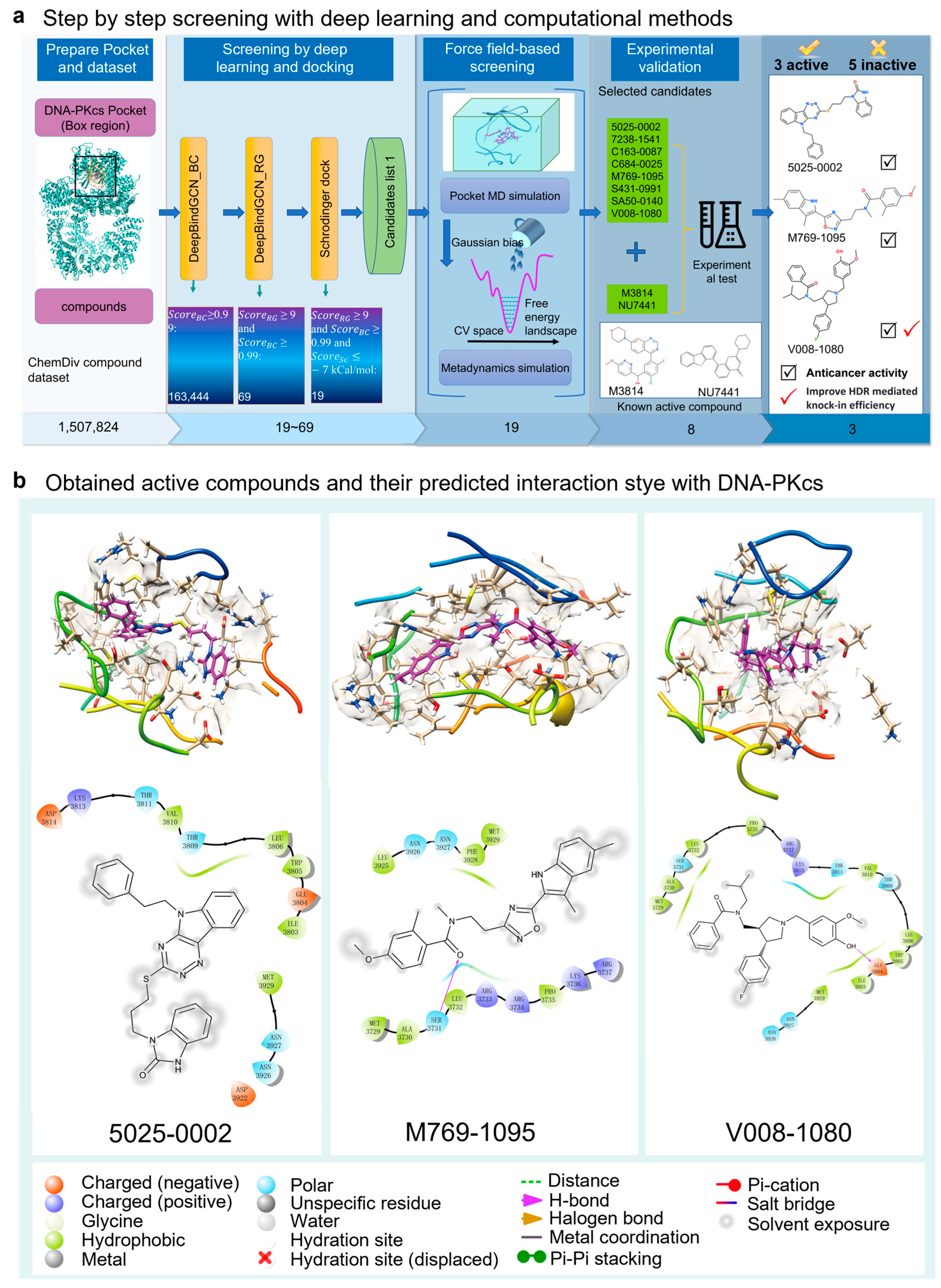
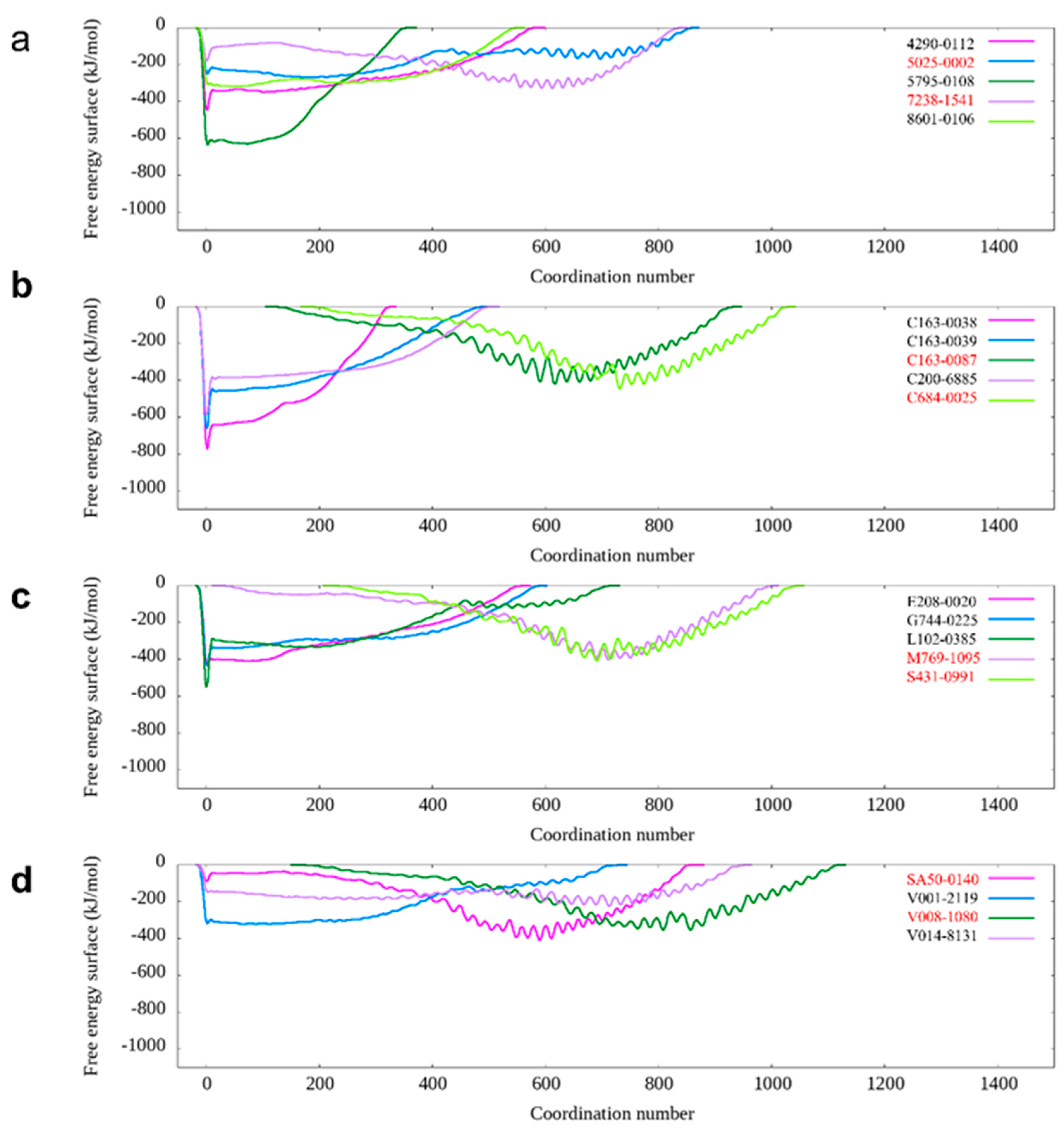
2.1. Computational Methods Aid DNA-PKcs Inhibitor Screening
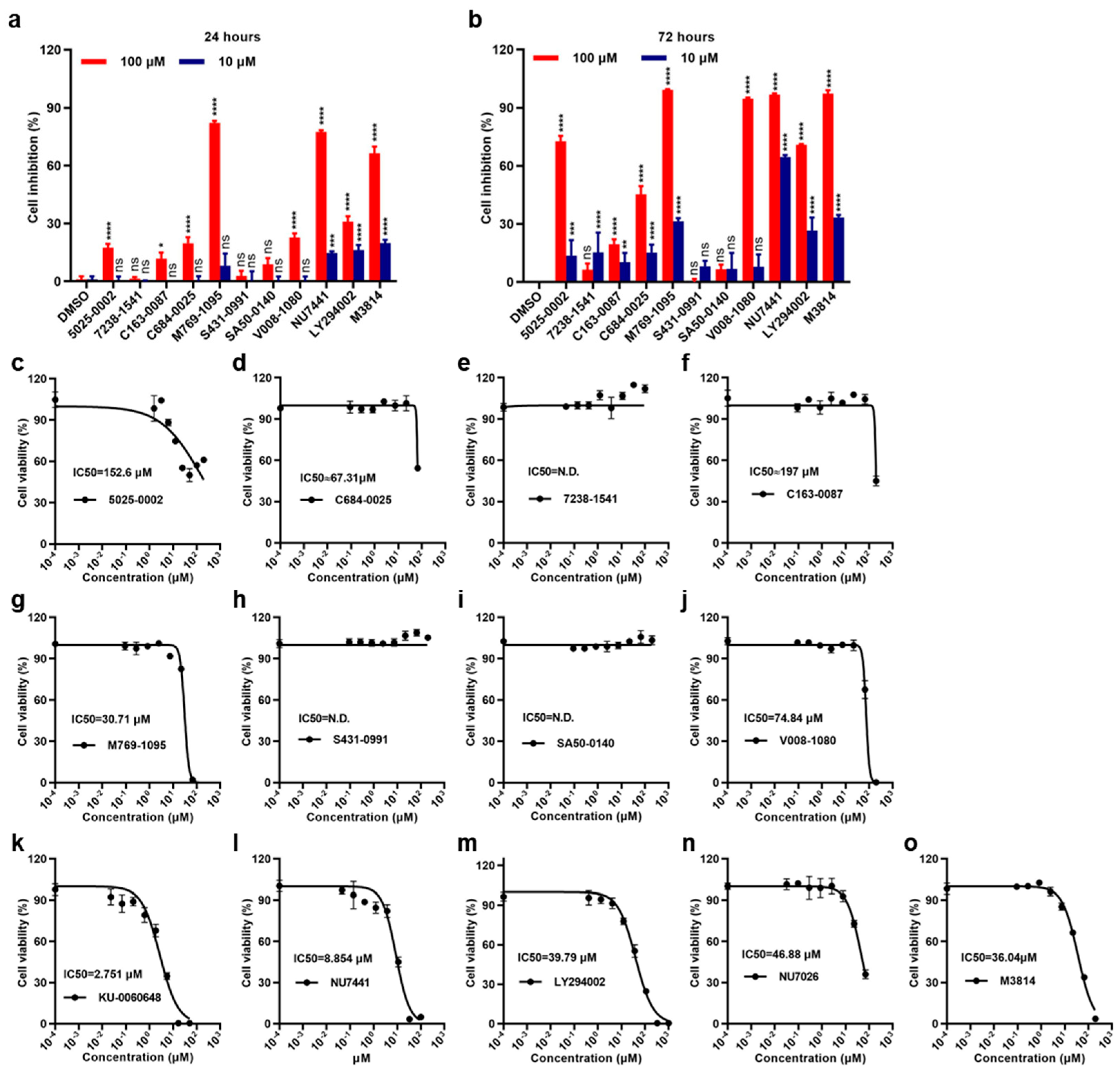
2.2. Effects of Small Molecules on 786-O Cell Viability and Proliferation Inhibition
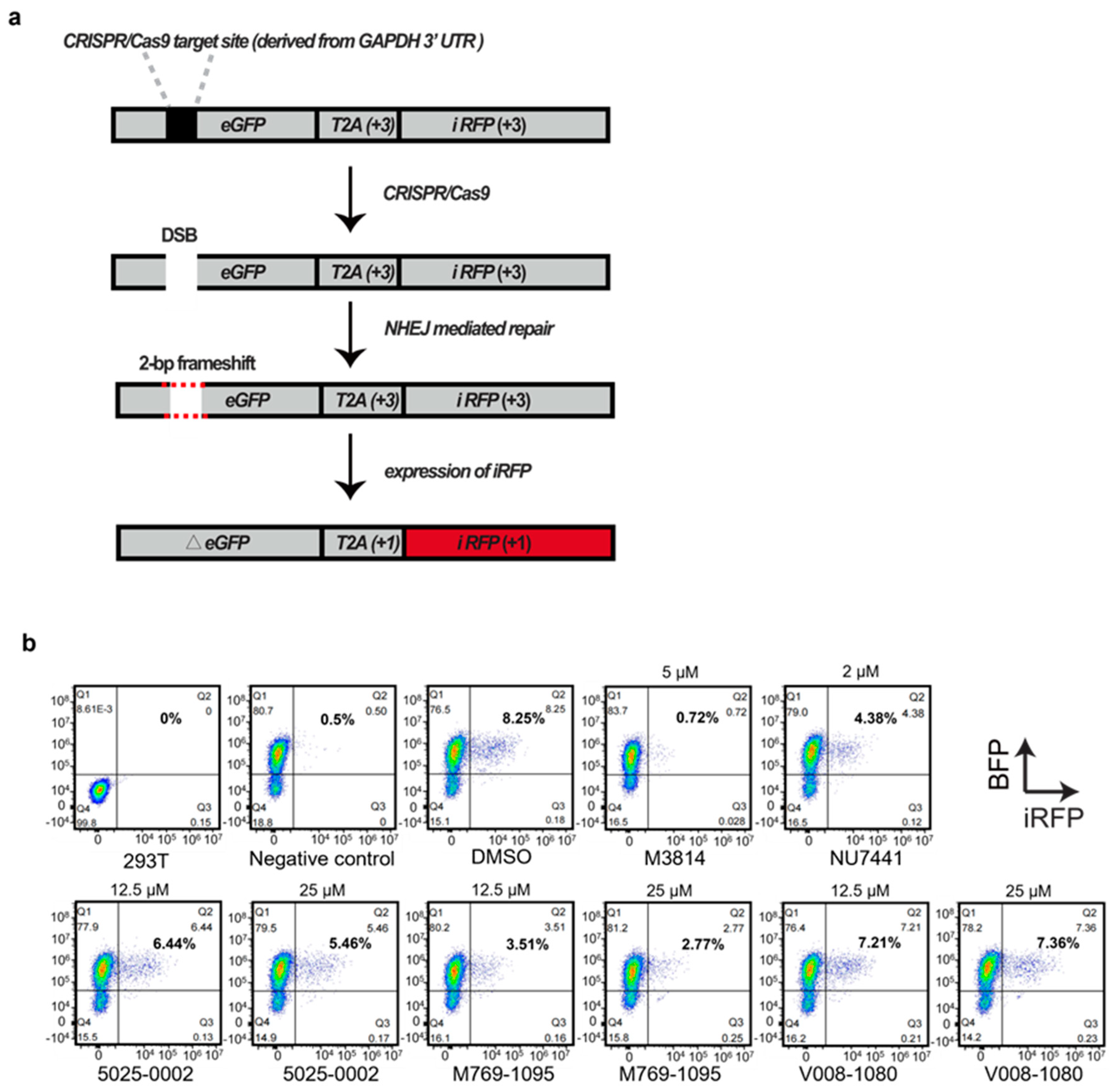
2.3. Increase in HDR-Mediated Knock-In by CRISPR/Cas9
2.4. Molecular Docking for Inhibitor and DNA-PKcs
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Protein Acquisition and Pocket Identification
5.2. Screening by DeepBindGCN
5.3. Schrödinger Docking Process
5.4. Force Field-Based Screening
5.5. Cell Culture
5.6. Nucleofection
5.7. Evaluation of Knock-In Events
5.8. Traffic Light Reporter (TLR) Assay
5.9. Cell Proliferation Assay
5.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carter, T.; Vancurova, I.; Sun, I.; Lou, W.; DeLeon, S. A DNA-activated protein kinase from hela cell nuclei. Mol. Cell Biol. 1990, 10, 6460–6471. [Google Scholar] [PubMed]
- Jackson, S.P.; MacDonald, J.J.; Lees-Miller, S.; Tjian, R. Gc box binding induces phosphorylation of sp1 by a DNA-dependent protein kinase. Cell 1990, 63, 155–165. [Google Scholar] [CrossRef]
- Lees-Miller, S.P.; Chen, Y.R.; Anderson, C.W. Human cells contain a DNA-activated protein kinase that phosphorylates simian virus 40 t antigen, mouse p53, and the human ku autoantigen. Mol. Cell Biol. 1990, 10, 6472–6481. [Google Scholar]
- Aparicio, T.; Baer, R.; Gautier, J. DNA double-strand break repair pathway choice and cancer. DNA Repair 2014, 19, 169–175. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Yue, X.; Bai, C.; Xie, D.; Ma, T.; Zhou, P.K. DNA-pkcs: A multi-faceted player in DNA damage response. Front. Genet. 2020, 11, 607428. [Google Scholar] [CrossRef]
- Jackson, S.P. DNA-dependent protein kinase. Int. J. Biochem. Cell Biol. 1997, 29, 935–938. [Google Scholar] [CrossRef]
- Goodwin, J.F.; Knudsen, K.E. Beyond DNA repair: DNA-pk function in cancer. Cancer Discov. 2014, 4, 1126–1139. [Google Scholar] [CrossRef]
- Ciszewski, W.M.; Tavecchio, M.; Dastych, J.; Curtin, N.J. DNA-pk inhibition by nu7441 sensitizes breast cancer cells to ionizing radiation and doxorubicin. Breast Cancer Res. Treat. 2014, 143, 47–55. [Google Scholar] [CrossRef]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. Azd7648 is a potent and selective DNA-pk inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef] [PubMed]
- Zenke, F.T.; Zimmermann, A.; Sirrenberg, C.; Dahmen, H.; Kirkin, V.; Pehl, U.; Grombacher, T.; Wilm, C.; Fuchss, T.; Amendt, C.; et al. Pharmacologic inhibitor of DNA-pk, m3814, potentiates radiotherapy and regresses human tumors in mouse models. Mol. Cancer Ther. 2020, 19, 1091–1101. [Google Scholar] [CrossRef]
- Smithson, M.; Irwin, R.K.; Williams, G.; McLeod, M.C.; Choi, E.K.; Ganguly, A.; Pepple, A.; Cho, C.S.; Willey, C.D.; Leopold, J.; et al. Inhibition of DNA-pk may improve response to neoadjuvant chemoradiotherapy in rectal cancer. Neoplasia 2022, 25, 53–61. [Google Scholar] [CrossRef]
- Bibikova, M.; Carroll, D.; Segal, D.J.; Trautman, J.K.; Smith, J.; Kim, Y.G.; Chandrasegaran, S. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol. Cell Biol. 2001, 21, 289–297. [Google Scholar] [CrossRef]
- Choulika, A.; Perrin, A.; Dujon, B.; Nicolas, J.F. Induction of homologous recombination in mammalian chromosomes by using the i-scei system of saccharomyces cerevisiae. Mol. Cell Biol. 1995, 15, 1968–1973. [Google Scholar] [CrossRef]
- Sander, J.D.; Cade, L.; Khayter, C.; Reyon, D.; Peterson, R.T.; Joung, J.K.; Yeh, J.R. Targeted gene disruption in somatic zebrafish cells using engineered talens. Nat. Biotechnol. 2011, 29, 697–698. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using crispr/cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. Rna-guided human genome engineering via cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Robert, F.; Barbeau, M.; Ethier, S.; Dostie, J.; Pelletier, J. Pharmacological inhibition of DNA-pk stimulates cas9-mediated genome editing. Genome Med. 2015, 7, 93. [Google Scholar] [CrossRef]
- Wimberger, S.; Akrap, N.; Firth, M.; Brengdahl, J.; Engberg, S.; Schwinn, M.K.; Slater, M.R.; Lundin, A.; Hsieh, P.P.; Li, S.; et al. Simultaneous inhibition of DNA-pk and polϴ improves integration efficiency and precision of genome editing. Nat. Commun. 2023, 14, 4761. [Google Scholar] [CrossRef]
- Riesenberg, S.; Chintalapati, M.; Macak, D.; Kanis, P.; Maricic, T.; Paabo, S. Simultaneous precise editing of multiple genes in human cells. Nucleic Acids Res. 2019, 47, e116. [Google Scholar] [CrossRef]
- Jekimovs, C.; Bolderson, E.; Suraweera, A.; Adams, M.; O’Byrne, K.J.; Richard, D.J. Chemotherapeutic compounds targeting the DNA double-strand break repair pathways: The good, the bad, and the promising. Front. Oncol. 2014, 4, 86. [Google Scholar] [CrossRef]
- Gavande, N.S.; VanderVere-Carozza, P.S.; Hinshaw, H.D.; Jalal, S.I.; Sears, C.R.; Pawelczak, K.S.; Turchi, J.J. DNA repair targeted therapy: The past or future of cancer treatment? Pharmacol. Ther. 2016, 160, 65–83. [Google Scholar] [CrossRef]
- Hu, S.; Hui, Z.; Lirussi, F.; Garrido, C.; Ye, X.Y.; Xie, T. Small molecule DNA-pk inhibitors as potential cancer therapy: A patent review (2010-present). Expert. Opin. Ther. Pat. 2021, 31, 435–452. [Google Scholar] [CrossRef]
- Zhang, H.; Saravanan, K.M.; Zhang, J.Z.H. Deepbindgcn: Integrating molecular vector representation with graph convolutional neural networks for protein-ligand interaction prediction. Molecules 2023, 28, 4691. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, Y.; Li, J.; Wang, M.; Saravanan, K.M.; Wei, J.; Tze-Yang Ng, J.; Tofazzal Hossain, M.; Liu, M.; Zhang, H.; et al. A novel virtual screening procedure identifies pralatrexate as inhibitor of SARS-CoV-2 rdrp and it reduces viral replication in vitro. PLoS Comput. Biol. 2020, 16, e1008489. [Google Scholar] [CrossRef]
- Zhang, H.; Fan, H.; Wang, J.; Hou, T.; Saravanan, K.M.; Xia, W.; Kan, H.W.; Li, J.; Zhang, J.Z.H.; Liang, X.; et al. Revolutionizing gpcr-ligand predictions: Deepgpcr with experimental validation for high-precision drug discovery. Brief. Bioinform. 2024, 25, bbae281. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, L.; Graf, L.; Yu, B.; Liu, Y.; Kochs, G.; Zhao, Y.; Gao, S. Conformational dynamics of dynamin-like mxa revealed by single-molecule fret. Nat. Commun. 2017, 8, 15744. [Google Scholar] [CrossRef]
- Hu, J.L.; Liang, H.; Zhang, H.; Yang, M.Z.; Sun, W.; Zhang, P.; Luo, L.; Feng, J.X.; Bai, H.; Liu, F.; et al. Fam46b is a prokaryotic-like cytoplasmic poly(a) polymerase essential in human embryonic stem cells. Nucleic Acids Res. 2020, 48, 2733–2748. [Google Scholar] [CrossRef]
- Zheng, B.; Mao, J.H.; Li, X.Q.; Qian, L.; Zhu, H.; Gu, D.H.; Pan, X.D. Over-expression of DNA-pkcs in renal cell carcinoma regulates mtorc2 activation, hif-2alpha expression and cell proliferation. Sci. Rep. 2016, 6, 29415. [Google Scholar] [CrossRef]
- He, X.; Tan, C.; Wang, F.; Wang, Y.; Zhou, R.; Cui, D.; You, W.; Zhao, H.; Ren, J.; Feng, B. Knock-in of large reporter genes in human cells via crispr/cas9-induced homology-dependent and independent DNA repair. Nucleic Acids Res. 2016, 44, e85. [Google Scholar] [CrossRef] [PubMed]
- Certo, M.T.; Ryu, B.Y.; Annis, J.E.; Garibov, M.; Jarjour, J.; Rawlings, D.J.; Scharenberg, A.M. Tracking genome engineering outcome at individual DNA breakpoints. Nat. Methods 2011, 8, 671–676. [Google Scholar] [CrossRef]
- Leahy, J.J.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; Richardson, C.; Rigoreau, L.; Smith, G.C. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-pk) inhibitor (nu7441) by screening of chromenone libraries. Bioorg Med. Chem. Lett. 2004, 14, 6083–6087. [Google Scholar] [CrossRef]
- Liang, S.; Thomas, S.E.; Chaplin, A.K.; Hardwick, S.W.; Chirgadze, D.Y.; Blundell, T.L. Structural insights into inhibitor regulation of the DNA repair protein DNA-pkcs. Nature 2022, 601, 643–648. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, S.H.; Hu, J.L.; Wu, Y.T.; Ma, X.Y.; Chen, Y.; Yu, B.; Liao, S.; Huang, H.; Gao, S. Structural and functional characterization of multiple myeloma associated cytoplasmic poly(a) polymerase fam46c. Cancer Commun. 2021, 41, 615–630. [Google Scholar] [CrossRef]
- He, Z.; Tian, T.; Guo, D.; Wu, H.; Chen, Y.; Zhang, Y.; Wan, Q.; Zhao, H.; Wang, C.; Shen, H.; et al. Cytoplasmic retention of a nucleocytoplasmic protein tbc1d3 by microtubule network is required for enhanced egfr signaling. PLoS ONE 2014, 9, e94134. [Google Scholar] [CrossRef]
- Chen, D.Q.; Xie, Y.; Cao, L.Q.; Fleishman, J.S.; Chen, Y.; Wu, T.; Yang, D.H. The role of abcc10/mrp7 in anti-cancer drug resistance and beyond. Drug Resist. Updat. 2024, 73, 101062. [Google Scholar] [CrossRef]
- Liu, J.; Fan, H.; Liang, X.; Chen, Y. Polycomb repressor complex: Its function in human cancer and therapeutic target strategy. Biomed. Pharmacother. 2023, 169, 115897. [Google Scholar] [CrossRef]
- Moyret-Lalle, C.; Prodhomme, M.K.; Burlet, D.; Kashiwagi, A.; Petrilli, V.; Puisieux, A.; Seimiya, H.; Tissier, A. Role of emt in the DNA damage response, double-strand break repair pathway choice and its implications in cancer treatment. Cancer Sci. 2022, 113, 2214–2223. [Google Scholar] [CrossRef]
- Tarazi, H.; Saleh, E.; El-Awady, R. In-silico screening for DNA-dependent protein kinase (DNA-pk) inhibitors: Combined homology modeling, docking, molecular dynamic study followed by biological investigation. Biomed. Pharmacother. 2016, 83, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Gavande, N.S.; VanderVere-Carozza, P.S.; Pawelczak, K.S.; Mendoza-Munoz, P.; Vernon, T.L.; Hanakahi, L.A.; Summerlin, M.; Dynlacht, J.R.; Farmer, A.H.; Sears, C.R.; et al. Discovery and development of novel DNA-pk inhibitors by targeting the unique ku-DNA interaction. Nucleic Acids Res. 2020, 48, 11536–11550. [Google Scholar] [CrossRef] [PubMed]
- Pawelczak, K.S.; Gavande, N.S.; VanderVere-Carozza, P.S.; Turchi, J.J. Modulating DNA repair pathways to improve precision genome engineering. ACS Chem. Biol. 2018, 13, 389–396. [Google Scholar] [CrossRef]
- Rdkit: Open-Source Cheminformatics Software. Available online: https://rdkit.org/ (accessed on 2 March 2023).
- Jaeger, S.; Fulle, S.; Turk, S. Mol2vec: Unsupervised machine learning approach with chemical intuition. J. Chem. Inf. Model. 2018, 58, 27–35. [Google Scholar] [CrossRef]
- Nguyen, T.; Le, H.; Quinn, T.P.; Nguyen, T.; Le, T.D.; Venkatesh, S. Graphdta: Predicting drug-target binding affinity with graph neural networks. Bioinformatics 2021, 37, 1140–1147. [Google Scholar] [CrossRef]
- Laio, A.; Gervasio, F.L. Metadynamics: A method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 2008, 71, 126601. [Google Scholar] [CrossRef]
- Schrödinger, L.; DeLano, W. PyMOL. 2020. Available online: http://www.pymol.org/pymol (accessed on 24 September 2023).
- Saleh, N.; Ibrahim, P.; Saladino, G.; Gervasio, F.L.; Clark, T. An Efficient Metadynamics-Based Protocol To Model the Binding Affinity and the Transition State Ensemble of G-Protein-Coupled Receptor Ligands. J. Chem. Inf. Model. 2017, 57, 1210–1217. [Google Scholar] [CrossRef]
- Ruiz-Carmona, S.; Schmidtke, P.; Luque, F.J.; Baker, L.; Matassova, N.; Davis, B.; Roughley, S.; Murray, J.; Hubbard, R.; Barril, X. Dynamic undocking and the quasi-bound state as tools for drug discovery. Nat. Chem. 2017, 9, 201–206. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 3, 435–447. [Google Scholar] [CrossRef]
- Hornak, V.; Simmerling, C. Generation of accurate protein loop conformations through low-barrier molecular dynamics. Proteins Struct. Funct. Genet. 2003, 51, 577–590. [Google Scholar] [CrossRef]
- Sousa Da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log( N ) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef]
- Williams, T.; Kelley, C.; Bröker, H.-B.; Campbell, J.; Cunningham, R.; Denholm, D.; Elber, G.; Fearick, R.; Grammes, C.; Hart, L. gnuplot 4.6. An Interactive Plotting Program. 2012. Available online: http://gnuplot.sourceforge.net/ (accessed on 2 November 2023).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemdiv ID | DeepBindGCN_BC | DeepBindGCN_RG | Schrödinger Docking (Kcal/mol) |
|---|---|---|---|
| L102-0385 | 0.9988 | 9.1991 | −8.4419 |
| 8601-0106 | 1.0000 | 9.1074 | −8.0588 |
| 5795-0108 | 1.0000 | 9.3339 | −7.9719 |
| C684-0025 | 0.9985 | 9.0321 | −7.9543 |
| 4290-0112 | 0.9959 | 9.0631 | −7.8938 |
| V008-1080 | 0.9983 | 9.3616 | −7.7966 |
| C200-6885 | 0.9975 | 9.0352 | −7.5277 |
| C163-0038 | 1.0000 | 9.4493 | −7.4668 |
| V014-8131 | 1.0000 | 9.1249 | −7.4660 |
| E208-0020 | 1.0000 | 9.1201 | −7.4659 |
| S431-0991 | 0.9999 | 9.1192 | −7.4172 |
| C163-0087 | 1.0000 | 9.0191 | −7.3098 |
| V001-2119 | 0.9947 | 9.0161 | −7.2953 |
| G744-0225 | 0.9996 | 9.0588 | −7.2909 |
| SA50-0140 | 1.0000 | 9.1702 | −7.2194 |
| C163-0039 | 1.0000 | 9.4007 | −7.1913 |
| 5025-0002 | 0.9999 | 9.0076 | −7.1717 |
| 7238-1541 | 0.9986 | 9.1096 | −7.1709 |
| M769-1095 | 0.9936 | 9.0299 | −7.1306 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, L.; Yu, P.; Fan, H.; Xia, W.; Zhao, Y.; Zhang, P.; Zhang, J.Z.H.; Zhang, H.; Chen, Y. Identification and Validation of New DNA-PKcs Inhibitors through High-Throughput Virtual Screening and Experimental Verification. Int. J. Mol. Sci. 2024, 25, 7982. https://doi.org/10.3390/ijms25147982
Dai L, Yu P, Fan H, Xia W, Zhao Y, Zhang P, Zhang JZH, Zhang H, Chen Y. Identification and Validation of New DNA-PKcs Inhibitors through High-Throughput Virtual Screening and Experimental Verification. International Journal of Molecular Sciences. 2024; 25(14):7982. https://doi.org/10.3390/ijms25147982
Chicago/Turabian StyleDai, Liujiang, Pengfei Yu, Hongjie Fan, Wei Xia, Yaopeng Zhao, Pengfei Zhang, John Z. H. Zhang, Haiping Zhang, and Yang Chen. 2024. "Identification and Validation of New DNA-PKcs Inhibitors through High-Throughput Virtual Screening and Experimental Verification" International Journal of Molecular Sciences 25, no. 14: 7982. https://doi.org/10.3390/ijms25147982