
Ribosome Pausing Negatively Regulates Protein Translation in Maize Seedlings during Dark-to-Light Transitions
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Translational Regulation Responds Quickly to Early Light Exposure
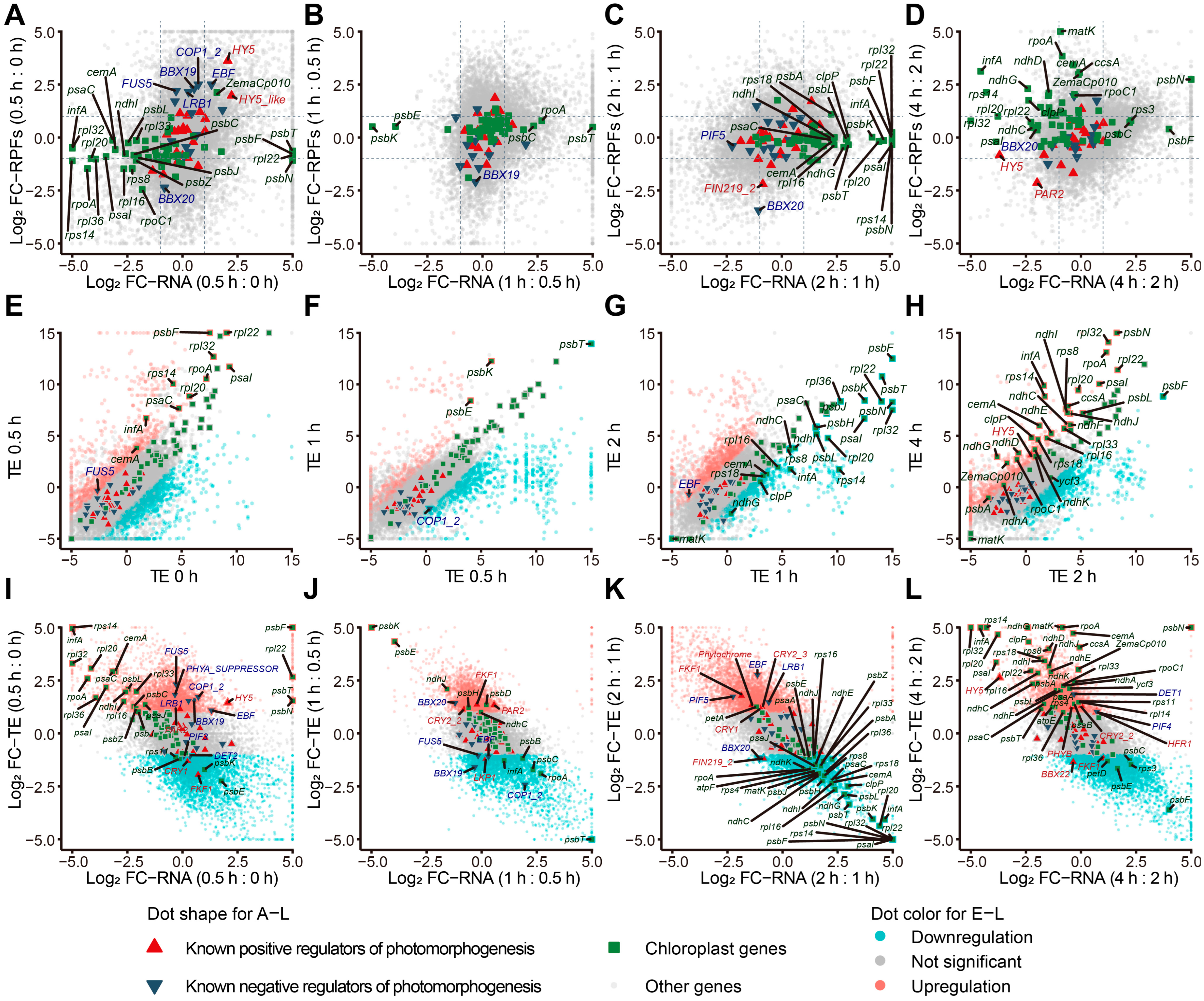
2.2. Genes Are Differentially Regulated at the Transcript and Translation Levels during Photomorphogenesis
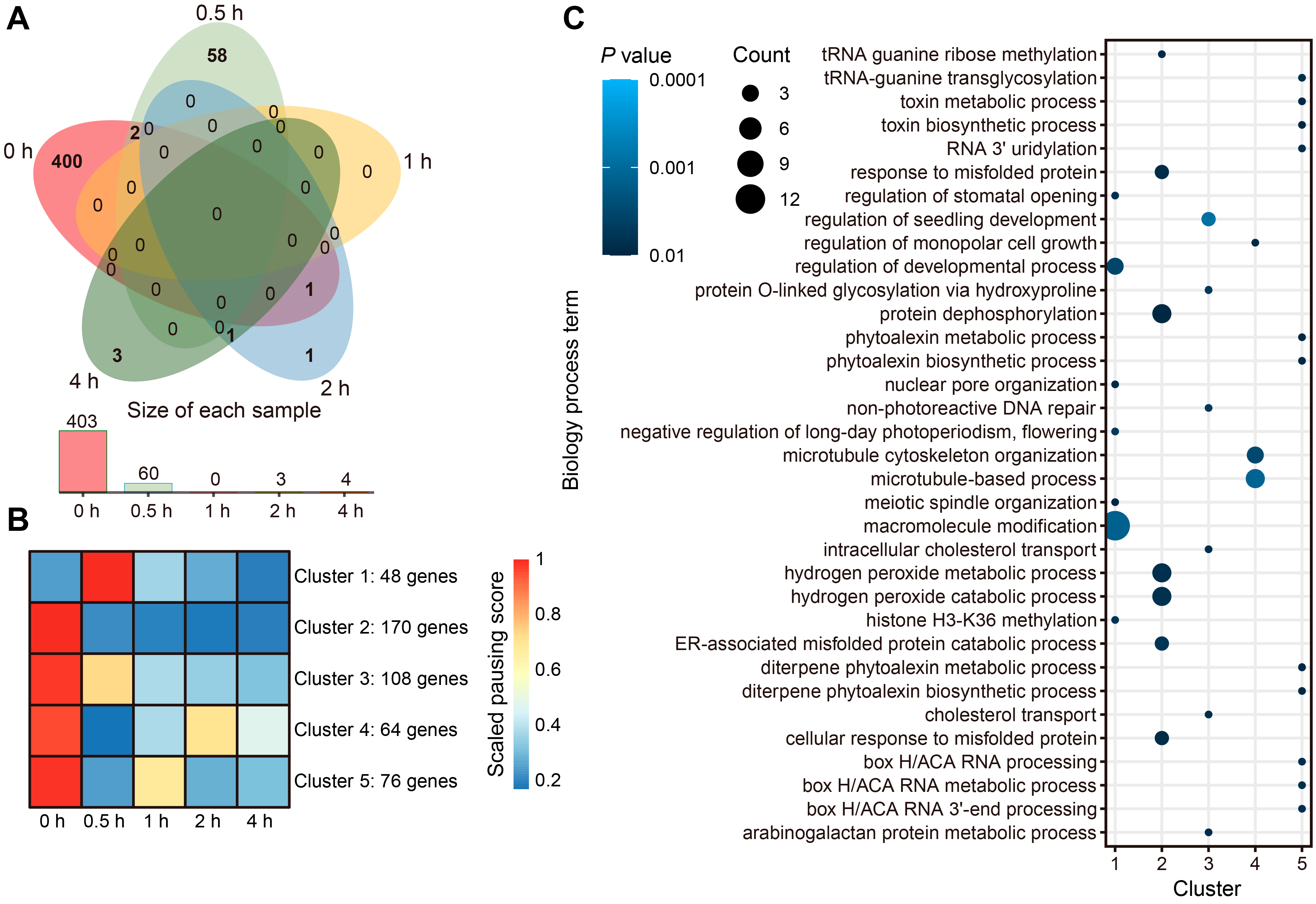
2.3. Light Exposure Widely Alleviates Ribosome Pausing in Maize
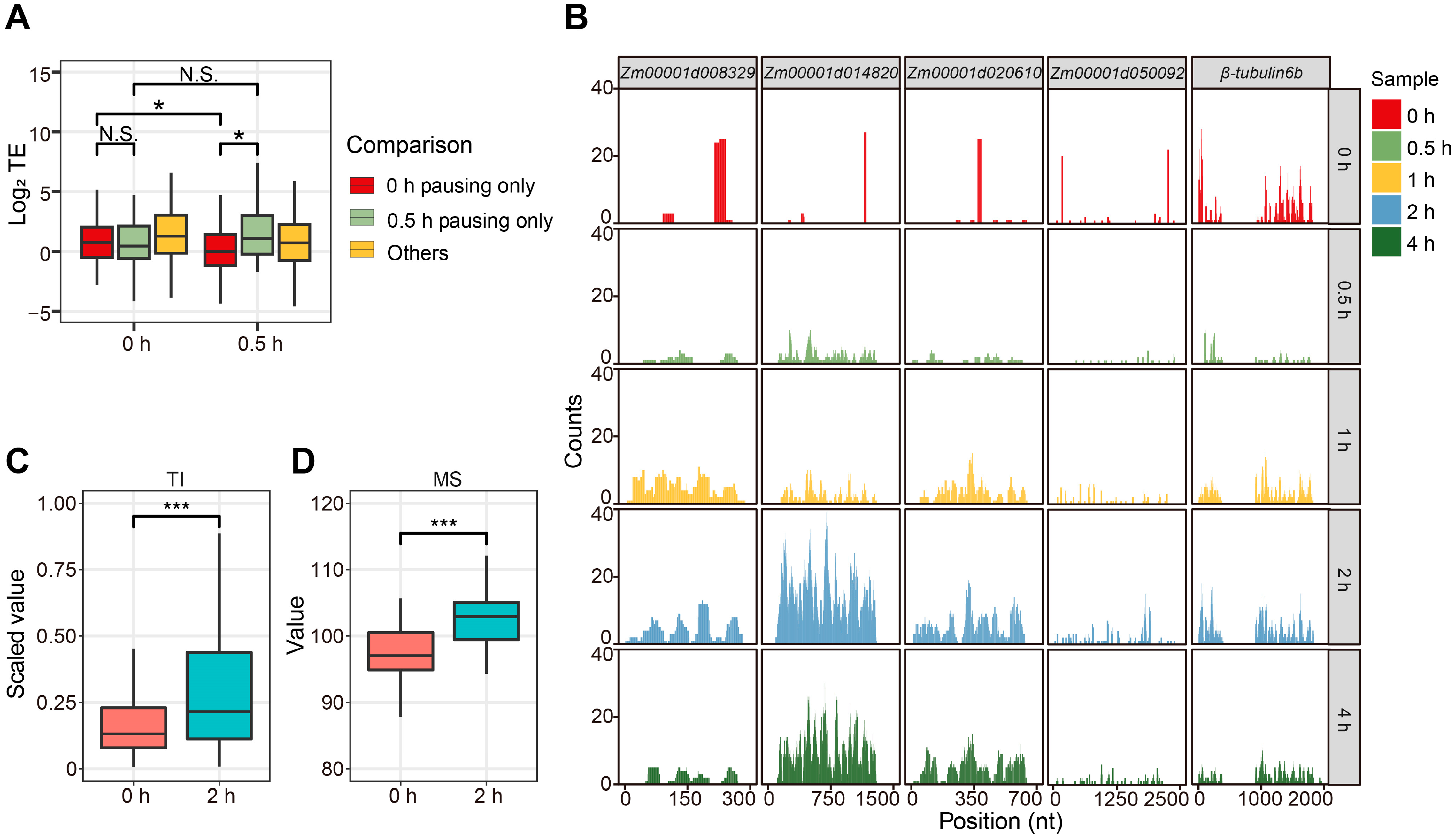
2.4. Ribosome Pausing Negatively Regulates Translation in Maize
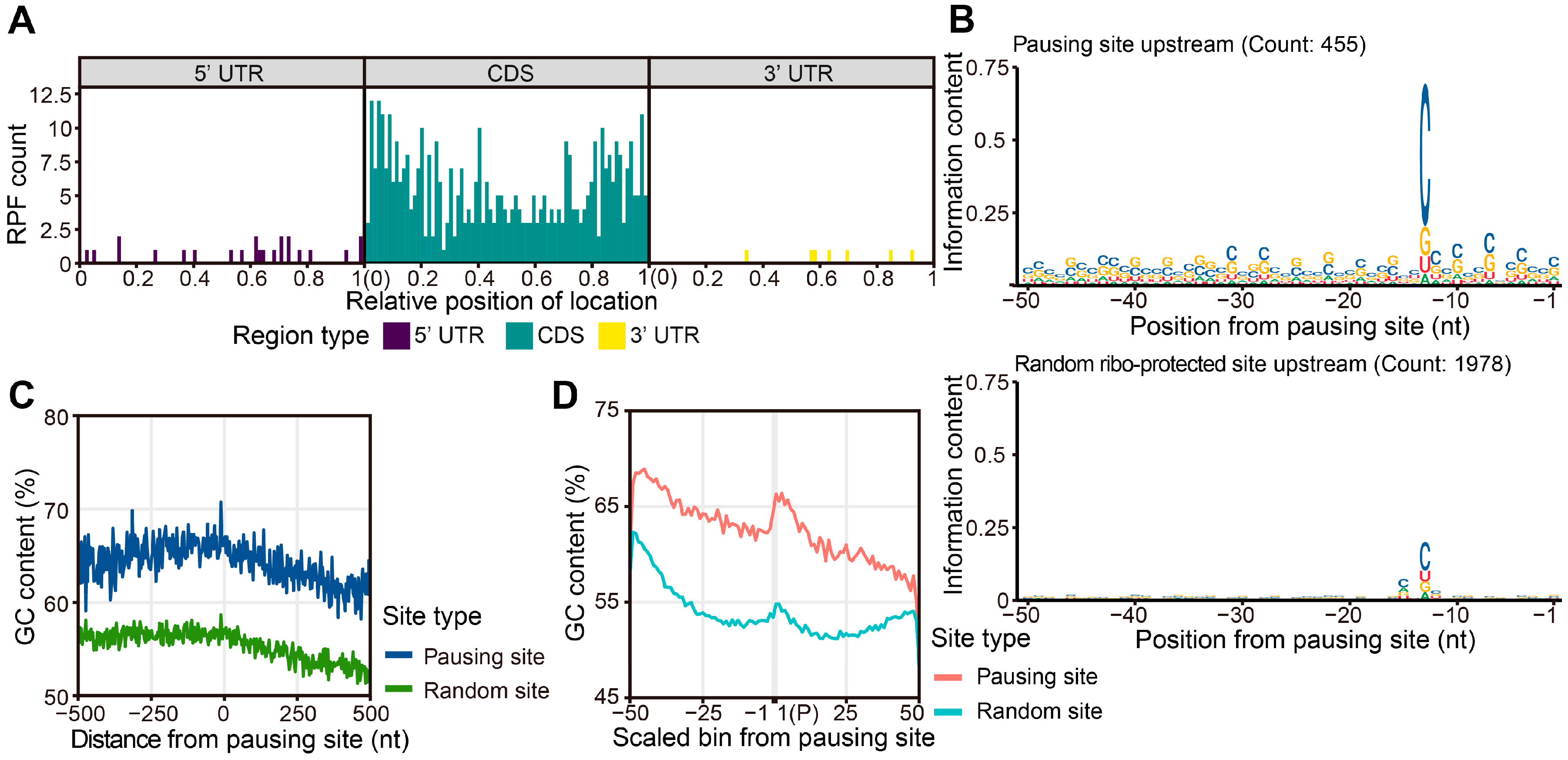
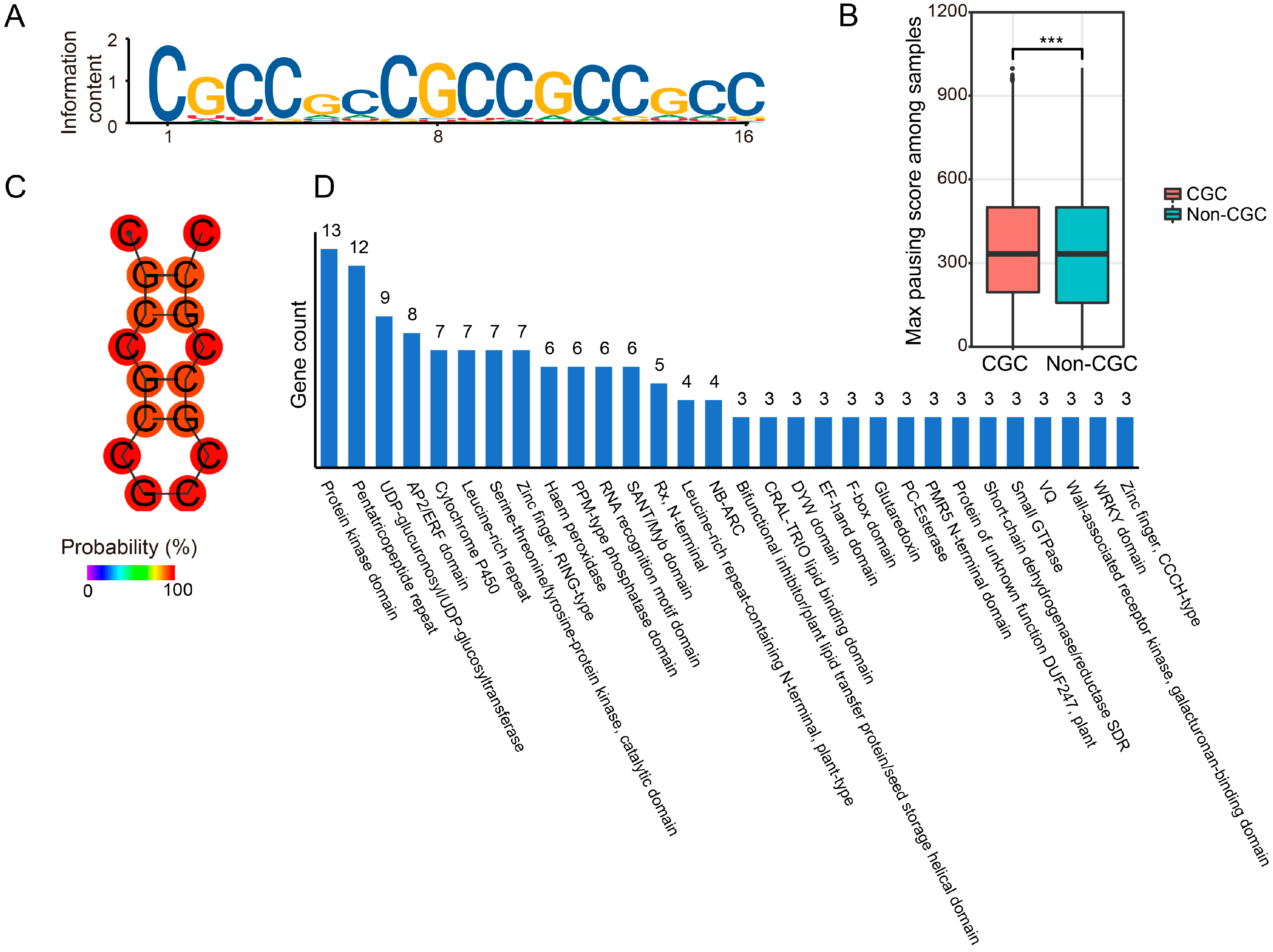
2.5. A High GC Content Leads to Ribosome Pausing
3. Discussion
4. Materials and Methods
4.1. Maize Seedling Growth and Treatments
4.2. Ribo-Seq and RNA-Seq Library Construction
4.3. Quantitative PCR
4.4. TMT-Labeled Mass Spectrometry Analysis
4.5. Sequencing Quality Control
4.6. Sequence Alignment to the Maize Reference Genome
4.7. Feature Read Counts and Differential Expression Analysis for RNA-Seq and Ribo-Seq Reads
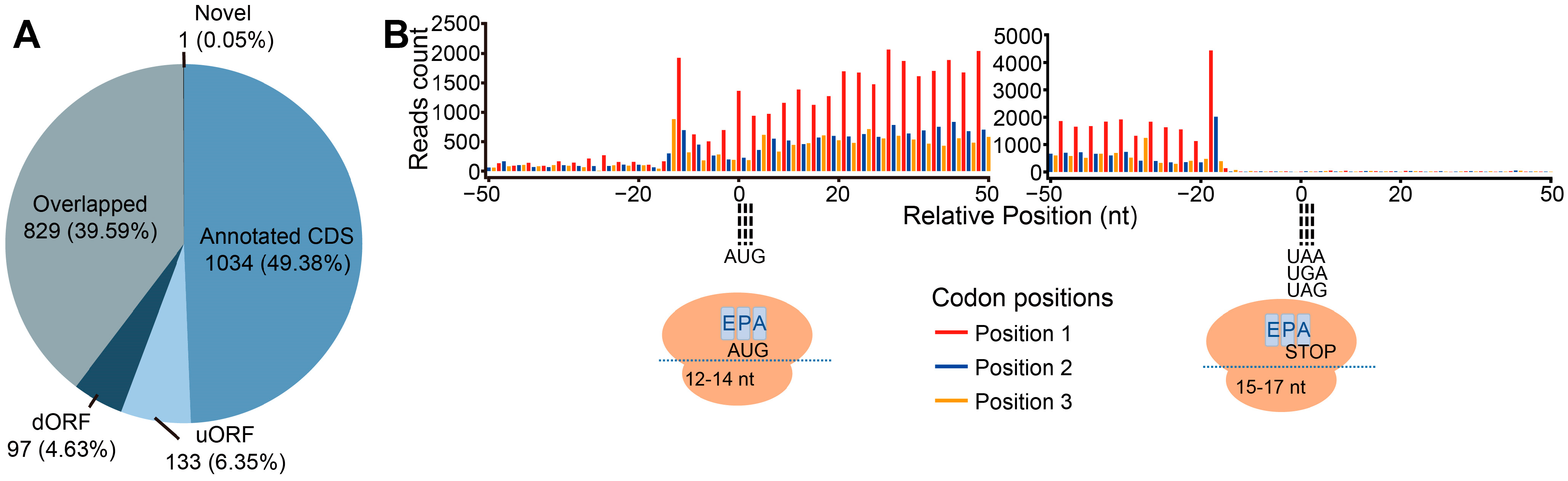
4.8. Trait Detection of Ribosome-Protected Fraction and uORF Prediction
4.9. Ribosome-Pausing Detection and Analysis
4.10. Translation Intensity (TI) Calculation
4.11. Gene Ontology and KEGG Enrichment Analysis
4.12. Graphical Visualization of Results
4.13. Secondary Structure Prediction and Free Energy Calculation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Z.; Sun, X.; Zhao, Y.; Guo, X.; Jiang, H.; Li, H.; Gu, Z. Evolution of gene regulation during transcription and translation. Genome Biol. Evol. 2015, 7, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.D.; Coller, J. Pausing on Polyribosomes: Make Way for Elongation in Translational Control. Cell 2015, 163, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, F.; Hentze, M.W. Molecular mechanisms of translational control. Nat. Rev. Mol. Cell Biol. 2004, 5, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Son, S.; Park, S.R. Plant translational reprogramming for stress resilience. Front. Plant Sci. 2023, 14, 1151587. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.; Weissman, J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Lareau, L.F.; Weissman, J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 2011, 147, 789–802. [Google Scholar] [CrossRef] [PubMed]
- McGlincy, N.J.; Ingolia, N.T. Transcriptome-wide measurement of translation by ribosome profiling. Methods 2017, 126, 112–129. [Google Scholar] [CrossRef]
- Hsu, P.Y.; Calviello, L.; Wu, H.L.; Li, F.W.; Rothfels, C.J.; Ohler, U.; Benfey, P.N. Super-resolution ribosome profiling reveals unannotated translation events in Arabidopsis. Proc. Natl. Acad. Sci. USA 2016, 113, E7126–E7135. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Brar, G.A.; Stern-Ginossar, N.; Harris, M.S.; Talhouarne, G.J.; Jackson, S.E.; Wills, M.R.; Weissman, J.S. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 2014, 8, 1365–1379. [Google Scholar] [CrossRef]
- Guerrero-Gonzalez, M.L.; Rodriguez-Kessler, M.; Jimenez-Bremont, J.F. uORF, a regulatory mechanism of the Arabidopsis polyamine oxidase 2. Mol. Biol. Rep. 2014, 41, 2427–2443. [Google Scholar] [CrossRef]
- Manske, F.; Ogoniak, L.; Jurgens, L.; Grundmann, N.; Makalowski, W.; Wethmar, K. The new uORFdb: Integrating literature, sequence, and variation data in a central hub for uORF research. Nucleic Acids Res. 2022, 51, D328–D336. [Google Scholar] [CrossRef] [PubMed]
- Russell, P.J.; Slivka, J.A.; Boyle, E.P.; Burghes, A.H.M.; Kearse, M.G. Translation reinitiation after uORFs does not fully protect mRNAs from nonsense-mediated decay. RNA 2023, 29, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Dever, T.E.; Ivanov, I.P.; Sachs, M.S. Conserved Upstream Open Reading Frame Nascent Peptides That Control Translation. Annu. Rev. Genet. 2020, 54, 237–264. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, A.G.; Ivanov, I.P.; Sonenberg, N. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science 2016, 352, 1413–1416. [Google Scholar] [CrossRef] [PubMed]
- Chew, G.L.; Pauli, A.; Schier, A.F. Conservation of uORF repressiveness and sequence features in mouse, human and zebrafish. Nat. Commun. 2016, 7, 11663. [Google Scholar] [CrossRef] [PubMed]
- Darnell, A.M.; Subramaniam, A.R.; O’Shea, E.K. Translational Control through Differential Ribosome Pausing during Amino Acid Limitation in Mammalian Cells. Mol. Cell 2018, 71, 229–243.e11. [Google Scholar] [CrossRef]
- Stein, K.C.; Morales-Polanco, F.; van der Lienden, J.; Rainbolt, T.K.; Frydman, J. Ageing exacerbates ribosome pausing to disrupt cotranslational proteostasis. Nature 2022, 601, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Gawronski, P.; Jensen, P.E.; Karpinski, S.; Leister, D.; Scharff, L.B. Pausing of Chloroplast Ribosomes Is Induced by Multiple Features and Is Linked to the Assembly of Photosynthetic Complexes. Plant Physiol. 2018, 176, 2557–2569. [Google Scholar] [CrossRef]
- Manjunath, H.; Zhang, H.; Rehfeld, F.; Han, J.; Chang, T.C.; Mendell, J.T. Suppression of Ribosomal Pausing by eIF5A Is Necessary to Maintain the Fidelity of Start Codon Selection. Cell Rep. 2019, 29, 3134–3146.e6. [Google Scholar] [CrossRef]
- Dong, L.; Mao, Y.; Zhou, A.; Liu, X.M.; Zhou, J.; Wan, J.; Qian, S.B. Relaxed initiation pausing of ribosomes drives oncogenic translation. Sci. Adv. 2021, 7, eabd6927. [Google Scholar] [CrossRef]
- Chotewutmontri, P.; Barkan, A. Dynamics of Chloroplast Translation during Chloroplast Differentiation in Maize. PLoS Genet. 2016, 12, e1006106. [Google Scholar] [CrossRef] [PubMed]
- Chotewutmontri, P.; Barkan, A. Multilevel effects of light on ribosome dynamics in chloroplasts program genome-wide and psbA-specific changes in translation. PLoS Genet. 2018, 14, e1007555. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.J.; Wu, S.H.; Wu, J.F.; Lin, W.D.; Wu, Y.C.; Tsai, T.Y.; Tsai, H.L.; Wu, S.H. Translational landscape of photomorphogenic Arabidopsis. Plant Cell 2013, 25, 3699–3710. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Shi, J.; Chen, J.; Zhang, M.; Sun, S.; Xie, S.; Li, X.; Zeng, B.; Peng, L.; Hauck, A.; et al. Ribosome profiling reveals dynamic translational landscape in maize seedlings under drought stress. Plant J. 2015, 84, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Juntawong, P.; Girke, T.; Bazin, J.; Bailey-Serres, J. Translational dynamics revealed by genome-wide profiling of ribosome footprints in Arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, E203–E212. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Song, B.; Cui, J.; Wang, L.; Wang, S.; Luo, L.; Gao, L.; Mo, B.; Yu, Y.; Liu, L. Comparative ribosome profiling reveals distinct translational landscapes of salt-sensitive and -tolerant rice. BMC Genom. 2021, 22, 612. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Zhong, L.; Du, J.; Zhu, C.; Peng, X.; He, X.; Fu, J.; Ouyang, L.; Bian, J.; Hu, L.; et al. Ribosome profiling reveals the effects of nitrogen application translational regulation of yield recovery after abrupt drought-flood alternation in rice. Plant Physiol. Biochem. 2020, 155, 42–58. [Google Scholar] [CrossRef]
- Xu, G.; Greene, G.H.; Yoo, H.; Liu, L.; Marques, J.; Motley, J.; Dong, X. Global translational reprogramming is a fundamental layer of immune regulation in plants. Nature 2017, 545, 487–490. [Google Scholar] [CrossRef]
- Xu, G.; Yuan, M.; Ai, C.; Liu, L.; Zhuang, E.; Karapetyan, S.; Wang, S.; Dong, X. uORF-mediated translation allows engineered plant disease resistance without fitness costs. Nature 2017, 545, 491–494. [Google Scholar] [CrossRef]
- Xiang, Y.; Huang, W.; Tan, L.; Chen, T.; He, Y.; Irving, P.S.; Weeks, K.M.; Zhang, Q.C.; Dong, X. Pervasive downstream RNA hairpins dynamically dictate start-codon selection. Nature 2023, 621, 423–430. [Google Scholar] [CrossRef]
- Han, N.C.; Kelly, P.; Ibba, M. Translational quality control and reprogramming during stress adaptation. Exp. Cell Res. 2020, 394, 112161. [Google Scholar] [CrossRef]
- Xiao, Z.; Zou, Q.; Liu, Y.; Yang, X. Genome-wide assessment of differential translations with ribosome profiling data. Nat. Commun. 2016, 7, 11194. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Shen, L.; Chen, Y.; Bao, S.; Thong, Z.; Yu, H. A MYB-domain protein EFM mediates flowering responses to environmental cues in Arabidopsis. Dev. Cell 2014, 30, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Murakami, N.; Fuji, S.; Yamauchi, S.; Hosotani, S.; Mano, J.; Takemiya, A. Reactive Carbonyl Species Inhibit Blue-Light-Dependent Activation of the Plasma Membrane H+-ATPase and Stomatal Opening. Plant Cell Physiol. 2022, 63, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Zuber, H.; Scheer, H.; Ferrier, E.; Sement, F.M.; Mercier, P.; Stupfler, B.; Gagliardi, D. Uridylation and PABP Cooperate to Repair mRNA Deadenylated Ends in Arabidopsis. Cell Rep. 2016, 14, 2707–2717. [Google Scholar] [CrossRef] [PubMed]
- Gamble, C.E.; Brule, C.E.; Dean, K.M.; Fields, S.; Grayhack, E.J. Adjacent Codons Act in Concert to Modulate Translation Efficiency in Yeast. Cell 2016, 166, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Li, G.W.; Oh, E.; Weissman, J.S. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature 2012, 484, 538–541. [Google Scholar] [CrossRef] [PubMed]
- Merret, R.; Nagarajan, V.K.; Carpentier, M.C.; Park, S.; Favory, J.J.; Descombin, J.; Picart, C.; Charng, Y.Y.; Green, P.J.; Deragon, J.M.; et al. Heat-induced ribosome pausing triggers mRNA co-translational decay in Arabidopsis thaliana. Nucleic Acids Res. 2015, 43, 4121–4132. [Google Scholar] [CrossRef] [PubMed]
- Tesina, P.; Lessen, L.N.; Buschauer, R.; Cheng, J.; Wu, C.C.; Berninghausen, O.; Buskirk, A.R.; Becker, T.; Beckmann, R.; Green, R. Molecular mechanism of translational stalling by inhibitory codon combinations and poly(A) tracts. EMBO J. 2020, 39, e103365. [Google Scholar] [CrossRef]
- Woolstenhulme, C.J.; Guydosh, N.R.; Green, R.; Buskirk, A.R. High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep. 2015, 11, 13–21. [Google Scholar] [CrossRef]
- Tsai, C.J.; Sauna, Z.E.; Kimchi-Sarfaty, C.; Ambudkar, S.V.; Gottesman, M.M.; Nussinov, R. Synonymous mutations and ribosome stalling can lead to altered folding pathways and distinct minima. J. Mol. Biol. 2008, 383, 281–291. [Google Scholar] [CrossRef]
- Wruck, F.; Tian, P.; Kudva, R.; Best, R.B.; von Heijne, G.; Tans, S.J.; Katranidis, A. The ribosome modulates folding inside the ribosomal exit tunnel. Commun. Biol. 2021, 4, 523. [Google Scholar] [CrossRef]
- Spahr, P.F.; Hollingworth, B.R. Purification and Mechanism of Action of Ribonuclease from Escherichia coli Ribosomes. J. Biol. Chem. 1961, 236, 823–831. [Google Scholar] [CrossRef]
- Wong, G.K.; Wang, J.; Tao, L.; Tan, J.; Zhang, J.; Passey, D.A.; Yu, J. Compositional gradients in Gramineae genes. Genome Res. 2002, 12, 851–856. [Google Scholar] [CrossRef]
- Osterlund, M.T.; Hardtke, C.S.; Wei, N.; Deng, X.W. Targeted destabilization of HY5 during light-regulated development of Arabidopsis. Nature 2000, 405, 462–466. [Google Scholar] [CrossRef]
- Han, X.; Huang, X.; Deng, X.W. The Photomorphogenic Central Repressor COP1: Conservation and Functional Diversification during Evolution. Plant Commun. 2020, 1, 100044. [Google Scholar] [CrossRef]
- Collart, M.A.; Weiss, B. Ribosome pausing, a dangerous necessity for co-translational events. Nucleic Acids Res. 2020, 48, 1043–1055. [Google Scholar] [CrossRef]
- Ikeuchi, K.; Izawa, T.; Inada, T. Recent Progress on the Molecular Mechanism of Quality Controls Induced by Ribosome Stalling. Front. Genet. 2018, 9, 743. [Google Scholar] [CrossRef]
- Joazeiro, C.A.P. Ribosomal Stalling During Translation: Providing Substrates for Ribosome-Associated Protein Quality Control. Annu. Rev. Cell Dev. Biol. 2017, 33, 343–368. [Google Scholar] [CrossRef]
- Buskirk, A.R.; Green, R. Ribosome pausing, arrest and rescue in bacteria and eukaryotes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160183. [Google Scholar] [CrossRef]
- Wen, J.D.; Lancaster, L.; Hodges, C.; Zeri, A.C.; Yoshimura, S.H.; Noller, H.F.; Bustamante, C.; Tinoco, I. Following translation by single ribosomes one codon at a time. Nature 2008, 452, 598–603. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, H.; Broitman, S.L.; Reiche, M.; Farrell, I.; Cooperman, B.S.; Goldman, Y.E. Dynamics of translation by single ribosomes through mRNA secondary structures. Nat. Struct. Mol. Biol. 2013, 20, 582–588. [Google Scholar] [CrossRef]
- Desai, V.P.; Frank, F.; Lee, A.; Righini, M.; Lancaster, L.; Noller, H.F.; Tinoco, I., Jr.; Bustamante, C. Co-temporal Force and Fluorescence Measurements Reveal a Ribosomal Gear Shift Mechanism of Translation Regulation by Structured mRNAs. Mol. Cell 2019, 75, 1007–1019.e5. [Google Scholar] [CrossRef]
- Caliskan, N.; Katunin, V.I.; Belardinelli, R.; Peske, F.; Rodnina, M.V. Programmed -1 frameshifting by kinetic partitioning during impeded translocation. Cell 2014, 157, 1619–1631. [Google Scholar] [CrossRef]
- Bao, C.; Zhu, M.; Nykonchuk, I.; Wakabayashi, H.; Mathews, D.H.; Ermolenko, D.N. Specific length and structure rather than high thermodynamic stability enable regulatory mRNA stem-loops to pause translation. Nat. Commun. 2022, 13, 988. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Brar, G.A.; Rouskin, S.; McGeachy, A.M.; Weissman, J.S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012, 7, 1534–1550. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Gaspar, J.M. NGmerge: Merging paired-end reads via novel empirically-derived models of sequencing errors. BMC Bioinform. 2018, 19, 536. [Google Scholar] [CrossRef]
- Andrews, S.; Krueger, F.; Segonds-Pichon, A.; Biggins, L.; Krueger, C.; Wingett, S. FastQC; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Portwood, J.L.; Woodhouse, M.R.; Cannon, E.K.; Gardiner, J.M.; Harper, L.C.; Schaeffer, M.L.; Walsh, J.R.; Sen, T.Z.; Cho, K.T.; Schott, D.A. MaizeGDB 2018: The maize multi-genome genetics and genomics database. Nucleic Acids Res. 2019, 47, D1146–D1154. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.; Anders, S.; Huber, W. Differential analysis of count data–the DESeq2 package. Genome Biol. 2014, 15, 10–1186. [Google Scholar]
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Xiao, Z.; Huang, R.; Xing, X.; Chen, Y.; Deng, H.; Yang, X. De novo annotation and characterization of the translatome with ribosome profiling data. Nucleic Acids Res. 2018, 46, e61. [Google Scholar] [CrossRef]
- Kumari, R.; Michel, A.M.; Baranov, P.V. PausePred and Rfeet: Webtools for inferring ribosome pauses and visualizing footprint density from ribosome profiling data. RNA 2018, 24, 1297–1304. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Santana-Garcia, W.; Castro-Mondragon, J.A.; Padilla-Gálvez, M.; Nguyen, N.T.T.; Elizondo-Salas, A.; Ksouri, N.; Gerbes, F.; Thieffry, D.; Vincens, P.; Contreras-Moreira, B. RSAT 2022: Regulatory sequence analysis tools. Nucleic Acids Res. 2022, 50, W670–W676. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R.; Kolde, M.R. Package ‘pheatmap’. R Package 2015, 1, 790. [Google Scholar]
- Consortium, G.O. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Slowikowski, K. ggrepel: Automatically Position Non-Overlapping Text Labels with ‘ggplot2’. Available online: https://ggrepel.slowkow.com/ (accessed on 13 June 2024).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, M.; Fan, W.; Zhong, D.; Dai, X.; Wang, Q.; Liu, W.; Li, S. Ribosome Pausing Negatively Regulates Protein Translation in Maize Seedlings during Dark-to-Light Transitions. Int. J. Mol. Sci. 2024, 25, 7985. https://doi.org/10.3390/ijms25147985
Hou M, Fan W, Zhong D, Dai X, Wang Q, Liu W, Li S. Ribosome Pausing Negatively Regulates Protein Translation in Maize Seedlings during Dark-to-Light Transitions. International Journal of Molecular Sciences. 2024; 25(14):7985. https://doi.org/10.3390/ijms25147985
Chicago/Turabian StyleHou, Mingming, Wei Fan, Deyi Zhong, Xing Dai, Quan Wang, Wanfei Liu, and Shengben Li. 2024. "Ribosome Pausing Negatively Regulates Protein Translation in Maize Seedlings during Dark-to-Light Transitions" International Journal of Molecular Sciences 25, no. 14: 7985. https://doi.org/10.3390/ijms25147985