
Microbial and Metabolic Gut Profiling across Seven Malignancies Identifies Fecal Faecalibacillus intestinalis and Formic Acid as Commonly Altered in Cancer Patients
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
2.1. Patients Overview
2.2. Metagenomic and Metabolomic Analyses of Pretreatment Fecal Samples
2.2.1. Bacterial Diversity
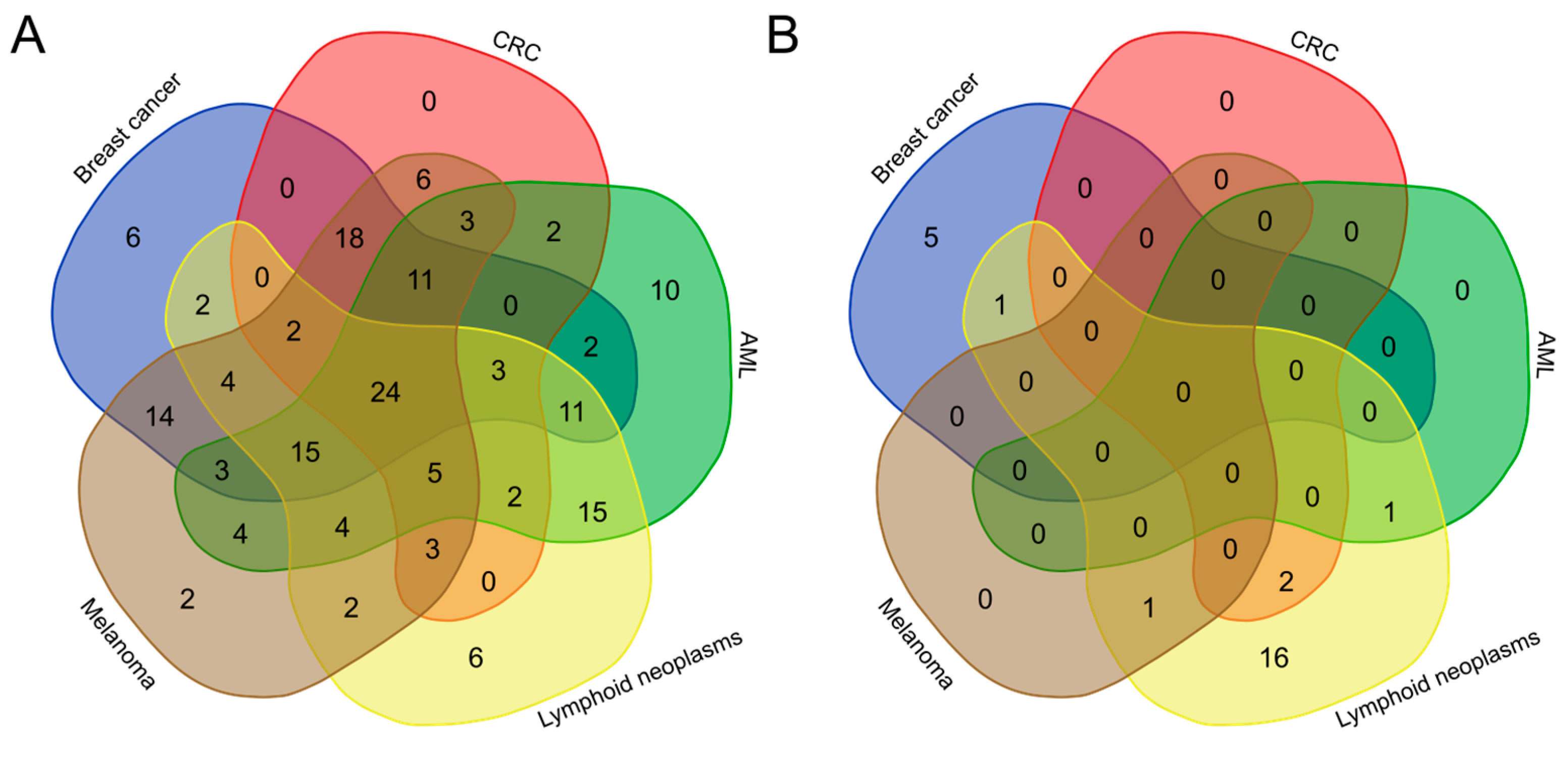
2.2.2. Taxonomic Profiling
2.2.3. Correlation between Bacteria Populations and Metabolites
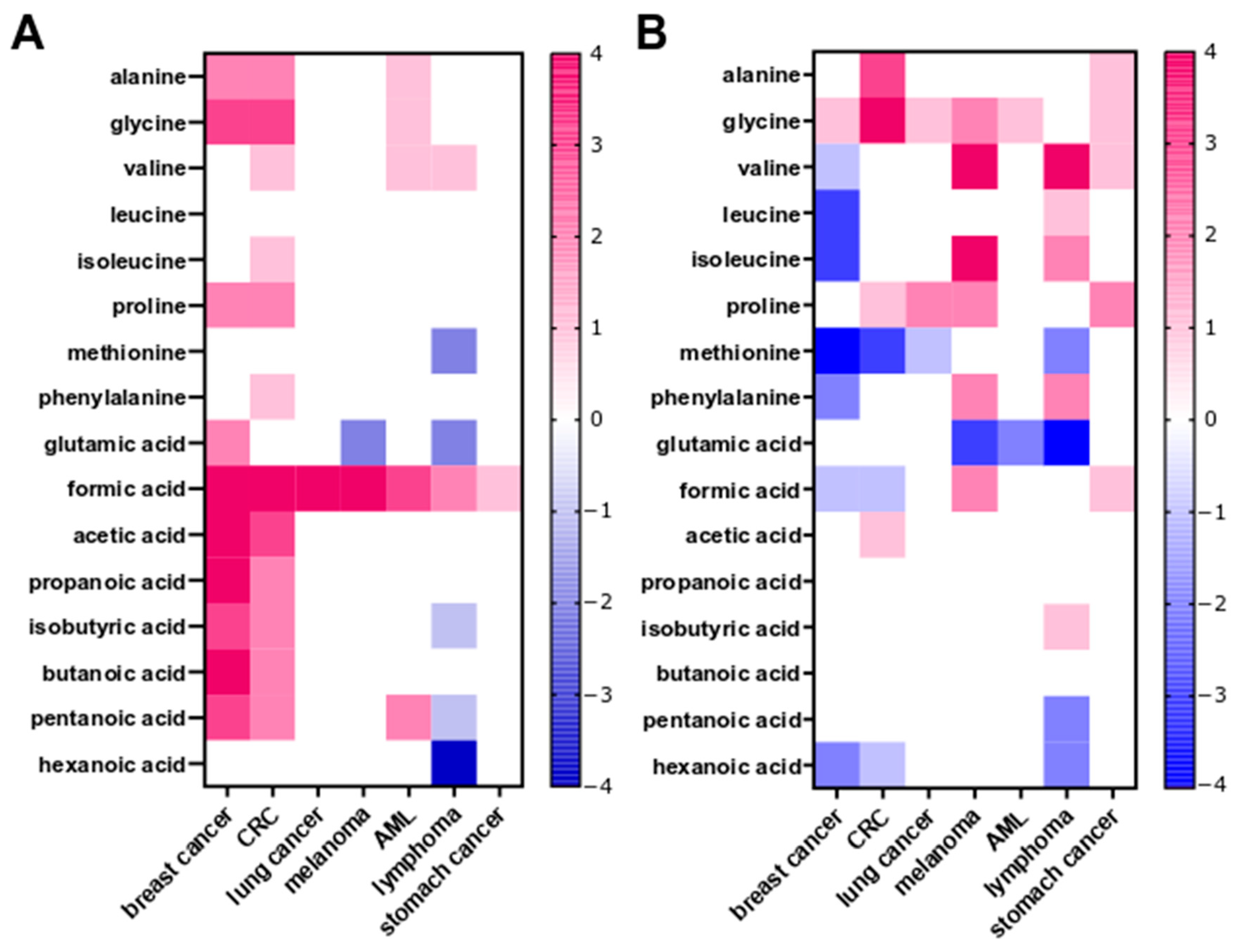
2.2.4. Fecal SCFA and Amino Acid Profiling
2.3. Metagenomic and Metabolomic Analyses to Compare Pretreatment and Post-Treatment Fecal Samples
2.4. Functional Analyses
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Metagenomics Analysis
4.3. SCFA and Amino Acid Profiling
4.4. Statistical Analysis
4.4.1. Bacteria and Metabolites
4.4.2. Associations between Bacteria and Metabolites
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Escalona, M.A.R.; Poloni, J.d.F.; Krause, M.J.; Dorn, M. Meta-Analyses of Host Metagenomes from Colorectal Cancer Patients Reveal Strong Relationship between Colorectal Cancer-Associated Species. Mol. Omics 2023, 19, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Dai, X.; Zhou, C.-C.; Li, K.; Zhang, Y.; Lou, X.-Y.; Zhu, Y.-M.; Sun, Y.-L.; Peng, B.-X.; Cui, W. Integrated Analysis of the Faecal Metagenome and Serum Metabolome Reveals the Role of Gut Microbiome-Associated Metabolites in the Detection of Colorectal Cancer and Adenoma. Gut 2022, 71, 1315–1325. [Google Scholar] [CrossRef]
- Wu, S.; Powers, S.; Zhu, W.; Hannun, Y.A. Substantial Contribution of Extrinsic Risk Factors to Cancer Development. Nature 2016, 529, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; An, R.; Wang, L.; Shan, J.; Wang, X. Specific Gut Microbiome and Serum Metabolome Changes in Lung Cancer Patients. Front. Cell. Infect. Microbiol. 2021, 11, 725284. [Google Scholar] [CrossRef]
- Chen, J.; Domingue, J.C.; Sears, C.L. Microbiota Dysbiosis in Select Human Cancers: Evidence of Association and Causality. Semin. Immunol. 2017, 32, 25–34. [Google Scholar] [CrossRef]
- Dzutsev, A.; Goldszmid, R.S.; Viaud, S.; Zitvogel, L.; Trinchieri, G. The Role of the Microbiota in Inflammation, Carcinogenesis, and Cancer Therapy. Eur. J. Immunol. 2015, 45, 17–31. [Google Scholar] [CrossRef]
- Mao, Q.; Jiang, F.; Yin, R.; Wang, J.; Xia, W.; Dong, G.; Ma, W.; Yang, Y.; Xu, L.; Hu, J. Interplay between the Lung Microbiome and Lung Cancer. Cancer Lett. 2018, 415, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Abu-Freha, N.; Cohen, B.; Gordon, M.; Weissmann, S.; Kestenbaum, E.H.; Vosko, S.; Abu-Tailakh, M.; Ben-Shoshan, L.; Cohen, D.L.; Shirin, H. Colorectal Cancer among Inflammatory Bowel Disease Patients: Risk Factors and Prevalence Compared to the General Population. Front. Med. 2023, 10, 1225616. [Google Scholar] [CrossRef]
- Liu, D.; Cao, M.; Wang, H.; Cao, W.; Zheng, C.; Li, Y.; Wang, Y. Association between Inflammatory Bowel Disease and Cancer Risk: Evidence Triangulation from Genetic Correlation, Mendelian Randomization, and Colocalization Analyses across East Asian and European Populations. BMC Med. 2024, 22, 137. [Google Scholar] [CrossRef]
- Marzullo, P.; Di Renzo, L.; Pugliese, G.; De Siena, M.; Barrea, L.; Muscogiuri, G.; Colao, A.; Savastano, S. From Obesity through Gut Microbiota to Cardiovascular Diseases: A Dangerous Journey. Int. J. Obes. Suppl. 2020, 10, 35–49. [Google Scholar] [CrossRef]
- Zhuang, Z.; Zhou, P.; Wang, J.; Lu, X.; Chen, Y. The Characteristics, Mechanisms and Therapeutics: Exploring the Role of Gut Microbiota in Obesity. Diabetes Metab. Syndr. Obes. 2023, 16, 3691–3705. [Google Scholar] [CrossRef] [PubMed]
- Crudele, L.; Gadaleta, R.M.; Cariello, M.; Moschetta, A. Gut Microbiota in the Pathogenesis and Therapeutic Approaches of Diabetes. eBioMedicine 2023, 97, 104821. [Google Scholar] [CrossRef]
- Qiu, P.; Ishimoto, T.; Fu, L.; Zhang, J.; Zhang, Z.; Liu, Y. The Gut Microbiota in Inflammatory Bowel Disease. Front. Cell. Infect. Microbiol. 2022, 12, 733992. [Google Scholar] [CrossRef] [PubMed]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, V.F.; Elias-Oliveira, J.; Pereira, Í.S.; Pereira, J.A.; Barbosa, S.C.; Machado, M.S.G.; Carlos, D. Akkermansia Muciniphila and Gut Immune System: A Good Friendship That Attenuates Inflammatory Bowel Disease, Obesity, and Diabetes. Front. Immunol. 2022, 13, 934695. [Google Scholar] [CrossRef] [PubMed]
- Noor, J.; Chaudhry, A.; Batool, S.; Noor, R.; Fatima, G. Exploring the Impact of the Gut Microbiome on Obesity and Weight Loss: A Review Article. Cureus 2023, 15, e40948. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Deepinder, F.; Morales, W.; Hwang, L.; Weitsman, S.; Chang, C.; Gunsalus, R.; Pimentel, M. Methanobrevibacter Smithii Is the Predominant Methanogen in Patients with Constipation-Predominant IBS and Methane on Breath. Dig. Dis. Sci. 2012, 57, 3213–3218. [Google Scholar] [CrossRef] [PubMed]
- Ullah, H.; Arbab, S.; Tian, Y.; Liu, C.; Chen, Y.; Qijie, L.; Khan, M.I.U.; Hassan, I.U.; Li, K. The Gut Microbiota–Brain Axis in Neurological Disorder. Front. Neurosci. 2023, 17, 1225875. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Du, L.; Shi, D.; Kong, C.; Liu, J.; Liu, G.; Li, X.; Ma, Y. Dysbiosis of Human Gut Microbiome in Young-Onset Colorectal Cancer. Nat. Commun. 2021, 12, 6757. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Jobin, C. The Microbiome and Cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef]
- Garrett, W.S. Cancer and the Microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Pascal, V.; Pozuelo, M.; Borruel, N.; Casellas, F.; Campos, D.; Santiago, A.; Martinez, X.; Varela, E.; Sarrabayrouse, G.; Machiels, K.; et al. A Microbial Signature for Crohn’s Disease. Gut 2017, 66, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Mekadim, C.; Skalnikova, H.K.; Cizkova, J.; Cizkova, V.; Palanova, A.; Horak, V.; Mrazek, J. Dysbiosis of Skin Microbiome and Gut Microbiome in Melanoma Progression. BMC Microbiol. 2022, 22, 63. [Google Scholar] [CrossRef] [PubMed]
- Gagnière, J. Gut Microbiota Imbalance and Colorectal Cancer. World J. Gastroenterol. 2016, 22, 501–518. [Google Scholar] [CrossRef]
- Xiong, H.; Wang, J.; Chang, Z.; Hu, H.; Yuan, Z.; Zhu, Y.; Hu, Z.; Wang, C.; Liu, Y.; Wang, Y.; et al. Gut Microbiota Display Alternative Profiles in Patients with Early-Onset Colorectal Cancer. Front. Cell. Infect. Microbiol. 2022, 12, 1036946. [Google Scholar] [CrossRef] [PubMed]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Bingula, R.; Filaire, M.; Radosevic-Robin, N.; Berthon, J.-Y.; Bernalier-Donadille, A.; Vasson, M.-P.; Thivat, E.; Kwiatkowski, F.; Filaire, E. Characterisation of Gut, Lung, and Upper Airways Microbiota in Patients with Non-Small Cell Lung Carcinoma. Medicine 2018, 97, e13676. [Google Scholar] [CrossRef]
- Rashidi, A.; Koyama, M.; Dey, N.; McLean, J.S.; Hill, G.R. Colonization Resistance Is Dispensable for Segregation of Oral and Gut Microbiota. BMC Med. Genom. 2023, 16, 31. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, S.; Yamada, T.; Yachida, S. Significance of the Gut Microbiome in Multistep Colorectal Carcinogenesis. Cancer Sci. 2020, 111, 766–773. [Google Scholar] [CrossRef]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.-J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Böhm, J.; Brunetti, F.; Habermann, N.; et al. Potential of Fecal Microbiota for Early-Stage Detection of Colorectal Cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhong, H.; Li, Y.; Shi, Z.; Ren, H.; Zhang, Z.; Zhou, X.; Tang, S.; Han, X.; Lin, Y.; et al. Sex- and Age-Related Trajectories of the Adult Human Gut Microbiota Shared across Populations of Different Ethnicities. Nat. Aging 2021, 1, 87–100. [Google Scholar] [CrossRef]
- Siegwald, L.; Caboche, S.; Even, G.; Viscogliosi, E.; Audebert, C.; Chabé, M. The Impact of Bioinformatics Pipelines on Microbiota Studies: Does the Analytical “Microscope” Affect the Biological Interpretation? Microorganisms 2019, 7, 393. [Google Scholar] [CrossRef]
- Samsom, K.G. Bridging the Gap: Implementation of Whole Genome Sequencing in Routine Clinical Care. Ph.D. Thesis, Utrecht University, Utrecht, The Netherlands, 2023; p. E17745820230503. [Google Scholar]
- Seo, B.; Jeon, K.; Baek, I.; Lee, Y.M.; Baek, K.; Ko, G. Faecalibacillus Intestinalis Gen. Nov., Sp. Nov. and Faecalibacillus Faecis Sp. Nov., Isolated from Human Faeces. Int. J. Syst. Evol. Microbiol. 2019, 69, 2120–2128. [Google Scholar] [CrossRef] [PubMed]
- Maturana, J.L.; Cárdenas, J.P. Insights on the Evolutionary Genomics of the Blautia Genus: Potential New Species and Genetic Content Among Lineages. Front. Microbiol. 2021, 12, 660920. [Google Scholar] [CrossRef] [PubMed]
- Koike, K.; Tourlousse, D.M.; Hamajima, M.; Sekiguchi, Y. Complete Genome Sequence of Coprobacter Fastidiosus JCM 33896 T. Microbiol. Resour. Announc. 2023, 12, e00326-23. [Google Scholar] [CrossRef]
- Fraccascia, D.; Chanyi, R.M.; Altermann, E.; Roy, N.C.; Flint, S.H.; McNabb, W.C. Complete Genome Sequences of Eight Faecalibacterium Sp. Strains Isolated from Healthy Human Stool. Microbiol. Resour. Announc. 2023, 12, e0082422. [Google Scholar] [CrossRef] [PubMed]
- Kant, R.; Rasinkangas, P.; Satokari, R.; Pietilä, T.E.; Palva, A. Genome Sequence of the Butyrate-Producing Anaerobic Bacterium Anaerostipes Hadrus PEL 85. Genome Announc. 2015, 3, e00224-15. [Google Scholar] [CrossRef]
- Shetty, S.A.; Zuffa, S.; Bui, T.P.N.; Aalvink, S.; Smidt, H.; De Vos, W.M. Reclassification of Eubacterium Hallii as Anaerobutyricum Hallii Gen. Nov., Comb. Nov., and Description of Anaerobutyricum Soehngenii Sp. Nov., a Butyrate and Propionate-Producing Bacterium from Infant Faeces. Int. J. Syst. Evol. Microbiol. 2018, 68, 3741–3746. [Google Scholar] [CrossRef]
- Marquez-Ortiz, R.A.; Leon, M.; Abril, D.; Escobar-Perez, J.; Florez-Sarmiento, C.; Parra-Izquierdo, V.; Chalem, P.; Romero-Sanchez, C. Colonoscopy Aspiration Lavages for Mucosal Metataxonomic Profiling of Spondylarthritis-Associated Gastrointestinal Tract Alterations. Sci. Rep. 2023, 13, 7015. [Google Scholar] [CrossRef]
- Renson, A.; Mullan Harris, K.; Dowd, J.B.; Gaydosh, L.; McQueen, M.B.; Krauter, K.S.; Shannahan, M.; Aiello, A.E. Early Signs of Gut Microbiome Aging: Biomarkers of Inflammation, Metabolism, and Macromolecular Damage in Young Adulthood. J. Gerontol. Ser. A 2020, 75, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sheng, D.; Jin, C.; Zhao, G.; Zhang, L. Identifying and Ranking Causal Microbial Biomarkers for Colorectal Cancer at Different Cancer Subsites and Stages: A Mendelian Randomization Study. Front. Oncol. 2023, 13, 1224705. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, Y.-J.; Trapecar, M.; Wright, C.; Schneider, K.; Kemmit, J.; Hernandez-Gordillo, V.; Yoon, J.Y.; Alm, E.J.; Breault, D.T.; et al. An Immune-Competent Human Gut Microphysiological System Enables Inflammation-Modulation of Faecalibacterium Prausnitzii. Res. Sq. 2023; rs.3.rs-3373576. [Google Scholar] [CrossRef]
- Yeh, T.-K.; Lin, H.-J.; Liu, P.-Y.; Wang, J.-H.; Hsueh, P.-R. Antibiotic Resistance in Enterobacter Hormaechei. Int. J. Antimicrob. Agents 2022, 60, 106650. [Google Scholar] [CrossRef] [PubMed]
- Mezzatesta, M.L.; Gona, F.; Stefani, S. Enterobacter Cloacae Complex: Clinical Impact and Emerging Antibiotic Resistance. Future Microbiol. 2012, 7, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Fukuzawa, S.; Sato, T.; Aoki, K.; Yamamoto, S.; Ogasawara, N.; Nakajima, C.; Suzuki, Y.; Horiuchi, M.; Takahashi, S.; Yokota, S.-I. High Prevalence of Colistin Heteroresistance in Specific Species and Lineages of Enterobacter Cloacae Complex Derived from Human Clinical Specimens. Ann. Clin. Microbiol. Antimicrob. 2023, 22, 60. [Google Scholar] [CrossRef] [PubMed]
- Muchaamba, F.; Barmettler, K.; Treier, A.; Houf, K.; Stephan, R. Microbiology and Epidemiology of Escherichia Albertii-An Emerging Elusive Foodborne Pathogen. Microorganisms 2022, 10, 875. [Google Scholar] [CrossRef]
- Khan, R.A.; Devi, K.R.; Pratim Barman, M.; Bhagawati, M.; Sarmah, R. Bacteria in the Oral Cavity of Individuals Consuming Intoxicating Substances. PLoS ONE 2023, 18, e0285753. [Google Scholar] [CrossRef]
- Rodríguez-Medina, N.; Barrios-Camacho, H.; Duran-Bedolla, J.; Garza-Ramos, U. Klebsiella Variicola: An Emerging Pathogen in Humans. Emerg. Microbes Infect. 2019, 8, 973–988. [Google Scholar] [CrossRef]
- Mielko, K.A.; Jabłoński, S.J.; Milczewska, J.; Sands, D.; Łukaszewicz, M.; Młynarz, P. Metabolomic Studies of Pseudomonas Aeruginosa. World J. Microbiol. Biotechnol. 2019, 35, 178. [Google Scholar] [CrossRef]
- Niyogi, S.K. Shigellosis. J. Microbiol. 2005, 43, 133–143. [Google Scholar]
- Duceppe, M.-O.; Phipps-Todd, B.; Carrillo, C.; Huang, H. Draft Genome Sequences of Eight Canadian Citrobacter Braakii and Citrobacter Freundii Strains. Microbiol. Resour. Announc. 2019, 8, e00273-19. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, Z.; Xiao, N.; Tang, J.; He, Y.; Guo, J.; Zhao, X. Genetic Characterization of Bla NDM-1-Carrying Citrobacter Portucalensis Sequence Type 328 and Citrobacter Freundii Sequence Type 98. Infect. Drug Resist. 2022, 15, 2235–2242. [Google Scholar] [CrossRef] [PubMed]
- Strakova, N.; Korena, K.; Karpiskova, R. Klebsiella Pneumoniae Producing Bacterial Toxin Colibactin as a Risk of Colorectal Cancer Development—A Systematic Review. Toxicon 2021, 197, 126–135. [Google Scholar] [CrossRef]
- Martin, R.M.; Bachman, M.A. Colonization, Infection, and the Accessory Genome of Klebsiella Pneumoniae. Front. Cell. Infect. Microbiol. 2018, 8, 4. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc Database of Metabolic Pathways and Enzymes—A 2019 Update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.; Wang, Y.; Sui, X.; Fan, J.; Li, S.; Lei, X.; Shi, C.; Sun, W.; Song, M.; Wang, H.; et al. Gut Microbiota-Mediated Nucleotide Synthesis Attenuates the Response to Neoadjuvant Chemoradiotherapy in Rectal Cancer. Cancer Cell 2023, 41, 124–138.e6. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, P.S.; O’Prey, J.; Cardaci, S.; Barthet, V.J.A.; Sakamaki, J.; Beaumatin, F.; Roseweir, A.; Gay, D.M.; Mackay, G.; Malviya, G.; et al. Mannose Impairs Tumour Growth and Enhances Chemotherapy. Nature 2018, 563, 719–723. [Google Scholar] [CrossRef]
- Siddiqui, A.; Ceppi, P. A Non-Proliferative Role of Pyrimidine Metabolism in Cancer. Mol. Metab. 2020, 35, 100962. [Google Scholar] [CrossRef]
- Wang, X.; Yang, K.; Xie, Q.; Wu, Q.; Mack, S.C.; Shi, Y.; Kim, L.J.Y.; Prager, B.C.; Flavahan, W.A.; Liu, X.; et al. Purine Synthesis Promotes Maintenance of Brain Tumor Initiating Cells in Glioma. Nat. Neurosci. 2017, 20, 661–673. [Google Scholar] [CrossRef]
- Yang, Y.; Long, J.; Wang, C.; Blot, W.J.; Pei, Z.; Shu, X.; Wu, F.; Rothman, N.; Wu, J.; Lan, Q.; et al. Prospective Study of Oral Microbiome and Gastric Cancer Risk among Asian, African American and European American Populations. Int. J. Cancer 2022, 150, 916–927. [Google Scholar] [CrossRef]
- Davrandi, M.; Harris, S.; Smith, P.J.; Murray, C.D.; Lowe, D.M. The Relationship Between Mucosal Microbiota, Colitis, and Systemic Inflammation in Chronic Granulomatous Disorder. J. Clin. Immunol. 2022, 42, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Shaw, J.L.; Haigis, M.C.; Greka, A. Lipid Metabolism in Sickness and in Health: Emerging Regulators of Lipotoxicity. Mol. Cell 2021, 81, 3708–3730. [Google Scholar] [CrossRef]
- Chattopadhyay, I.; Gundamaraju, R.; Jha, N.K.; Gupta, P.K.; Dey, A.; Mandal, C.C.; Ford, B.M. Interplay between Dysbiosis of Gut Microbiome, Lipid Metabolism, and Tumorigenesis: Can Gut Dysbiosis Stand as a Prognostic Marker in Cancer? Dis. Markers 2022, 2022, 2941248. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid Metabolic Reprogramming in Cancer Cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.-Y.; Fang, X. Fatty Acid Oxidation: An Emerging Facet of Metabolic Transformation in Cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Hadjiagapiou, C.; Schmidt, L.; Dudeja, P.K.; Layden, T.J.; Ramaswamy, K. Mechanism(s) of Butyrate Transport in Caco-2 Cells: Role of Monocarboxylate Transporter 1. Am. J. Physiol.-Gastrointest. Liver Physiol. 2000, 279, G775–G780. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Ogawa, S.A.; Chau, L.; Whelan, K.A.; Hamilton, K.E.; Chen, J.; Tan, L.; Chen, E.Z.; Keilbaugh, S.; Fogt, F.; et al. Mitochondrial Dysfunction in Inflammatory Bowel Disease Alters Intestinal Epithelial Metabolism of Hepatic Acylcarnitines. J. Clin. Investig. 2021, 131, e133371. [Google Scholar] [CrossRef] [PubMed]
- Ivleva, E.A.; Grivennikov, S.I. Microbiota-Driven Mechanisms at Different Stages of Cancer Development. Neoplasia 2022, 32, 100829. [Google Scholar] [CrossRef]
- Fernandes, M.R.; Aggarwal, P.; Costa, R.G.F.; Cole, A.M.; Trinchieri, G. Targeting the Gut Microbiota for Cancer Therapy. Nat. Rev. Cancer 2022, 22, 703–722. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut Microbiota Functions: Metabolism of Nutrients and Other Food Components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Pramanik, S. Structural Diversity, Functional Aspects and Future Therapeutic Applications of Human Gut Microbiome. Arch. Microbiol. 2021, 203, 5281–5308. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.; Murphy, K.G.; Frost, G.; Chambers, E.S. Short-Chain Fatty Acids as Potential Regulators of Skeletal Muscle Metabolism and Function. Nat. Metab. 2020, 2, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Oliphant, K.; Allen-Vercoe, E. Macronutrient Metabolism by the Human Gut Microbiome: Major Fermentation by-Products and Their Impact on Host Health. Microbiome 2019, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, S.; Quigley, M.E.; Hopkins, M.J.; Newton, D.F.; Macfarlane, G.T. Polysaccharide Degradation by Human Intestinal Bacteria during Growth under Multi-Substrate Limiting Conditions in a Three-Stage Continuous Culture System. FEMS Microbiol. Ecol. 1998, 26, 231–243. [Google Scholar] [CrossRef]
- Pietzke, M.; Meiser, J.; Vazquez, A. Formate Metabolism in Health and Disease. Mol. Metab. 2020, 33, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Pham, V.T.; Lacroix, C.; Braegger, C.P.; Chassard, C. Lactate-Utilizing Community Is Associated with Gut Microbiota Dysbiosis in Colicky Infants. Sci. Rep. 2017, 7, 11176. [Google Scholar] [CrossRef]
- Hughes, E.R.; Winter, M.G.; Duerkop, B.A.; Spiga, L.; Furtado de Carvalho, T.; Zhu, W.; Gillis, C.C.; Büttner, L.; Smoot, M.P.; Behrendt, C.L.; et al. Microbial Respiration and Formate Oxidation as Metabolic Signatures of Inflammation-Associated Dysbiosis. Cell Host Microbe 2017, 21, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Kulecka, M.; Zeber-Lubecka, N.; Bałabas, A.; Czarnowski, P.; Bagińska, K.; Głowienka, M.; Kluska, A.; Piątkowska, M.; Dąbrowska, M.; Waker, E.; et al. Diarrheal-Associated Gut Dysbiosis in Cancer and Inflammatory Bowel Disease Patients Is Exacerbated by Clostridioides Difficile Infection. Front. Cell. Infect. Microbiol. 2023, 13, 1190910. [Google Scholar] [CrossRef]
- Mikó, E.; Kovács, T.; Sebő, É.; Tóth, J.; Csonka, T.; Ujlaki, G.; Sipos, A.; Szabó, J.; Méhes, G.; Bai, P. Microbiome—Microbial Metabolome—Cancer Cell Interactions in Breast Cancer—Familiar, but Unexplored. Cells 2019, 8, 293. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Beaugerie, L.; Petit, J.-C. Antibiotic-Associated Diarrhoea. Best Pract. Res. Clin. Gastroenterol. 2004, 18, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Wang, X.-W.; Wu, A.-K.; Fan, Y.; Friedman, J.; Dahlin, A.; Waldor, M.K.; Weinstock, G.M.; Weiss, S.T.; Liu, Y.-Y. Deciphering Functional Redundancy in the Human Microbiome. Nat. Commun. 2020, 11, 6217. [Google Scholar] [CrossRef] [PubMed]
- Moya, A.; Ferrer, M. Functional Redundancy-Induced Stability of Gut Microbiota Subjected to Disturbance. Trends Microbiol. 2016, 24, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Does Functional Redundancy Affect Ecological Stability and Resilience? A Review and Meta-Analysis—NASA/ADS. Available online: https://ui.adsabs.harvard.edu/abs/2020Ecosp..11E3184B/abstract (accessed on 17 May 2024).
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef]
- Wilmanski, T.; Rappaport, N.; Diener, C.; Gibbons, S.M.; Price, N.D. From Taxonomy to Metabolic Output: What Factors Define Gut Microbiome Health? Gut Microbes 2021, 13, 1907270. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and Metabolomic Analyses Reveal Distinct Stage-Specific Phenotypes of the Gut Microbiota in Colorectal Cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.M.; Manghi, P.; Asnicar, F.; Pasolli, E.; Armanini, F.; Zolfo, M.; Beghini, F.; Manara, S.; Karcher, N.; Pozzi, C.; et al. Metagenomic Analysis of Colorectal Cancer Datasets Identifies Cross-Cohort Microbial Diagnostic Signatures and a Link with Choline Degradation. Nat. Med. 2019, 25, 667–678. [Google Scholar] [CrossRef]
- Pérez-Cobas, A.E.; Gomez-Valero, L.; Buchrieser, C. Metagenomic Approaches in Microbial Ecology: An Update on Whole-Genome and Marker Gene Sequencing Analyses. Microb. Genom. 2020, 6, e000409. [Google Scholar] [CrossRef]
- Durazzi, F.; Sala, C.; Castellani, G.; Manfreda, G.; Remondini, D.; De Cesare, A. Comparison between 16S rRNA and Shotgun Sequencing Data for the Taxonomic Characterization of the Gut Microbiota. Sci. Rep. 2021, 11, 3030. [Google Scholar] [CrossRef]
- Langille, M.G.I. Exploring Linkages between Taxonomic and Functional Profiles of the Human Microbiome. mSystems 2018, 3, e00163-17. [Google Scholar] [CrossRef]
- Gupta, V.K.; Kim, M.; Bakshi, U.; Cunningham, K.Y.; Davis, J.M.; Lazaridis, K.N.; Nelson, H.; Chia, N.; Sung, J. A Predictive Index for Health Status Using Species-Level Gut Microbiome Profiling. Nat. Commun. 2020, 11, 4635. [Google Scholar] [CrossRef] [PubMed]
- Navgire, G.S.; Goel, N.; Sawhney, G.; Sharma, M.; Kaushik, P.; Mohanta, Y.K.; Mohanta, T.K.; Al-Harrasi, A. Analysis and Interpretation of Metagenomics Data: An Approach. Biol. Proced. Online 2022, 24, 18. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, F.; Zeng, Z.; Xu, M.; Sun, F.; Yang, L.; Bi, X.; Lin, Y.; Gao, Y.; Hao, H.; et al. Advances in Metagenomics and Its Application in Environmental Microorganisms. Front. Microbiol. 2021, 12, 766364. [Google Scholar] [CrossRef] [PubMed]
- Kulecka, M.; Fraczek, B.; Balabas, A.; Czarnowski, P.; Zeber-Lubecka, N.; Zapala, B.; Baginska, K.; Glowienka, M.; Szot, M.; Skorko, M.; et al. Characteristics of the Gut Microbiome in Esports Players Compared with Those in Physical Education Students and Professional Athletes. Front. Nutr. 2023, 9, 1092846. [Google Scholar] [CrossRef]
- Zeber-Lubecka, N.; Kulecka, M.; Jagiełło-Gruszfeld, A.; Dąbrowska, M.; Kluska, A.; Piątkowska, M.; Bagińska, K.; Głowienka, M.; Surynt, P.; Tenderenda, M.; et al. Breast Cancer but Not the Menopausal Status Is Associated with Small Changes of the Gut Microbiota. Front. Oncol. 2024, 14, 1279132. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; He, K.; Chen, J.; Zhang, X. LinDA: Linear Models for Differential Abundance Analysis of Microbiome Compositional Data. Genome Biol. 2022, 23, 95. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- rCCA Nutrimouse Case Study|mixOmics. Available online: http://mixomics.org/case-studies/rcca-nutrimouse-case-study/ (accessed on 27 January 2024).
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating Taxonomic, Functional, and Strain-Level Profiling of Diverse Microbial Communities with bioBakery 3. eLife 2021, 10, e65088. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Cases | Controls | ||
|---|---|---|---|---|
| Women | Men | Women | Men | |
| n/Median; Range (Years) | n/Median; Range (Years) | n/Median; Range (Years) | n/Median; Range (Years) | |
| Colorectal cancer | 20/66; 36–82 | 20/67; 35–82 | 20/68; 49–79 | 20/61; 50–81 |
| Stomach cancer | 15/68; 37–78 | 30/68; 40–87 | 15/70; 37–82 | 30/62; 41–81 |
| Breast cancer | 71/50; 30–79 | 71/54; 30–82 | ||
| Lung cancer | 17/64; 54–81 | 17/61; 35–85 | 17/64; 52–82 | 17/61; 40–81 |
| Melanoma | 23/65; 48–84 | 27/66; 34–88 | 23/65; 47–82 | 27/60; 42–81 |
| Lymphoid neoplasms | 35/58; 22–78 | 25/57; 31–74 | 35/59; 22–82 | 25/58; 30–81 |
| Acute myeloid leukemia | 26/60; 20–68 | 14/51; 23–74 | 26/60; 22–73 | 14/50; 23–75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulecka, M.; Czarnowski, P.; Bałabas, A.; Turkot, M.; Kruczkowska-Tarantowicz, K.; Żeber-Lubecka, N.; Dąbrowska, M.; Paszkiewicz-Kozik, E.; Walewski, J.; Ługowska, I.; et al. Microbial and Metabolic Gut Profiling across Seven Malignancies Identifies Fecal Faecalibacillus intestinalis and Formic Acid as Commonly Altered in Cancer Patients. Int. J. Mol. Sci. 2024, 25, 8026. https://doi.org/10.3390/ijms25158026
Kulecka M, Czarnowski P, Bałabas A, Turkot M, Kruczkowska-Tarantowicz K, Żeber-Lubecka N, Dąbrowska M, Paszkiewicz-Kozik E, Walewski J, Ługowska I, et al. Microbial and Metabolic Gut Profiling across Seven Malignancies Identifies Fecal Faecalibacillus intestinalis and Formic Acid as Commonly Altered in Cancer Patients. International Journal of Molecular Sciences. 2024; 25(15):8026. https://doi.org/10.3390/ijms25158026
Chicago/Turabian StyleKulecka, Maria, Paweł Czarnowski, Aneta Bałabas, Maryla Turkot, Kamila Kruczkowska-Tarantowicz, Natalia Żeber-Lubecka, Michalina Dąbrowska, Ewa Paszkiewicz-Kozik, Jan Walewski, Iwona Ługowska, and et al. 2024. "Microbial and Metabolic Gut Profiling across Seven Malignancies Identifies Fecal Faecalibacillus intestinalis and Formic Acid as Commonly Altered in Cancer Patients" International Journal of Molecular Sciences 25, no. 15: 8026. https://doi.org/10.3390/ijms25158026
APA StyleKulecka, M., Czarnowski, P., Bałabas, A., Turkot, M., Kruczkowska-Tarantowicz, K., Żeber-Lubecka, N., Dąbrowska, M., Paszkiewicz-Kozik, E., Walewski, J., Ługowska, I., Koseła-Paterczyk, H., Rutkowski, P., Kluska, A., Piątkowska, M., Jagiełło-Gruszfeld, A., Tenderenda, M., Gawiński, C., Wyrwicz, L., Borucka, M., ... Ostrowski, J. (2024). Microbial and Metabolic Gut Profiling across Seven Malignancies Identifies Fecal Faecalibacillus intestinalis and Formic Acid as Commonly Altered in Cancer Patients. International Journal of Molecular Sciences, 25(15), 8026. https://doi.org/10.3390/ijms25158026