
A Novel Affordable and Reliable Framework for Accurate Detection and Comprehensive Analysis of Somatic Mutations in Cancer

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Evaluating Musta in a Hepatocellular Carcinoma (HCC) Dataset
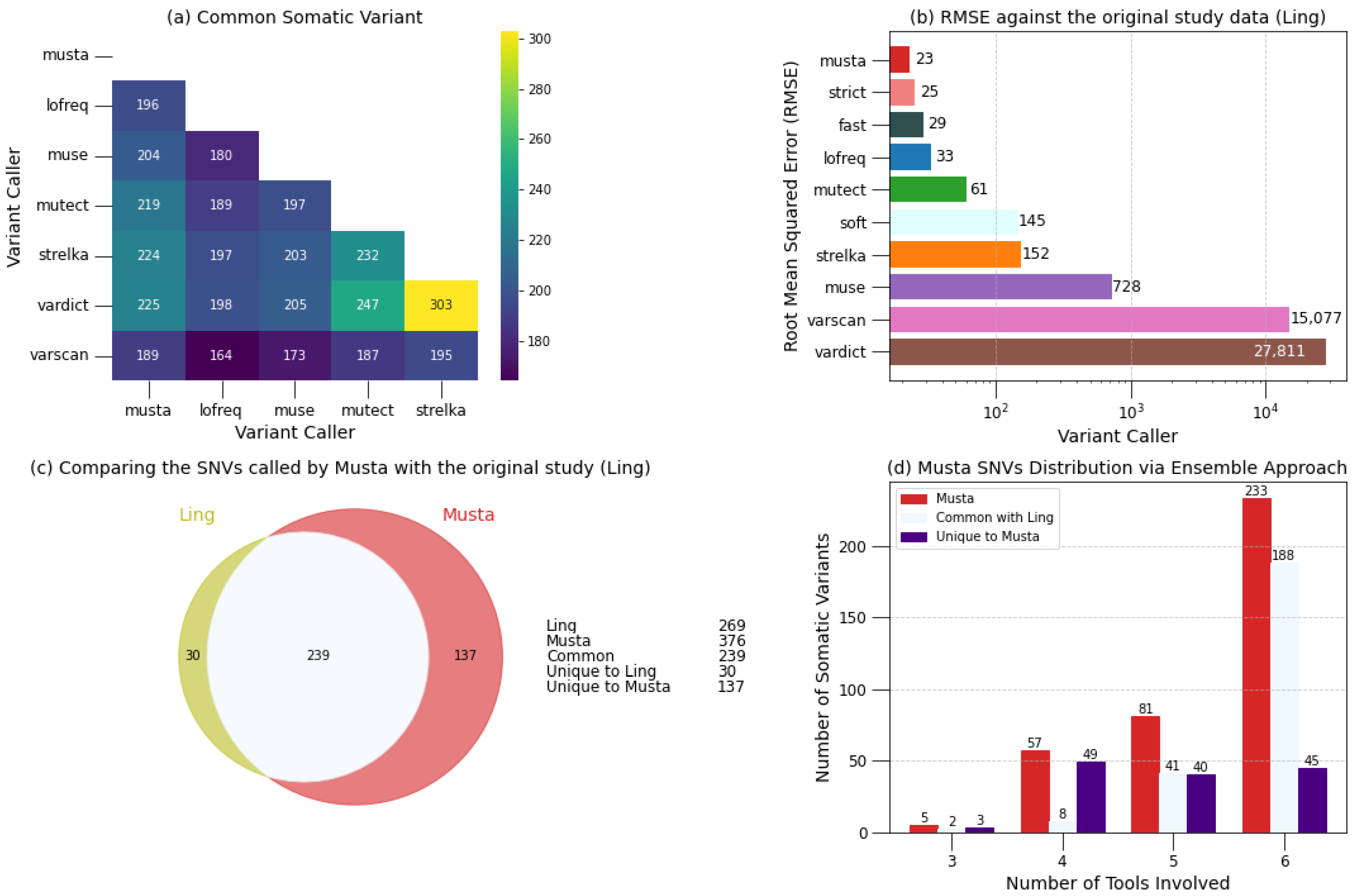
2.1.1. Detection
2.1.2. Classification
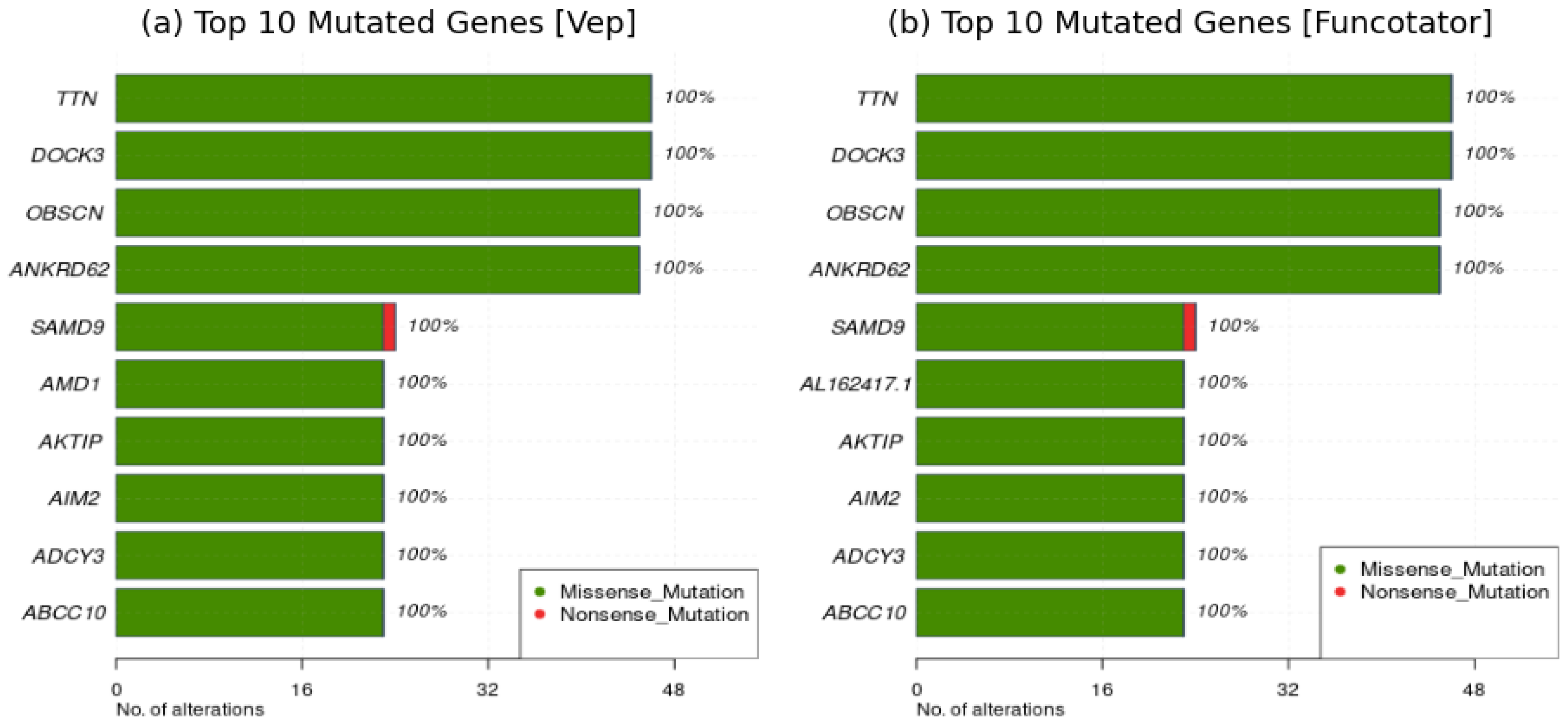
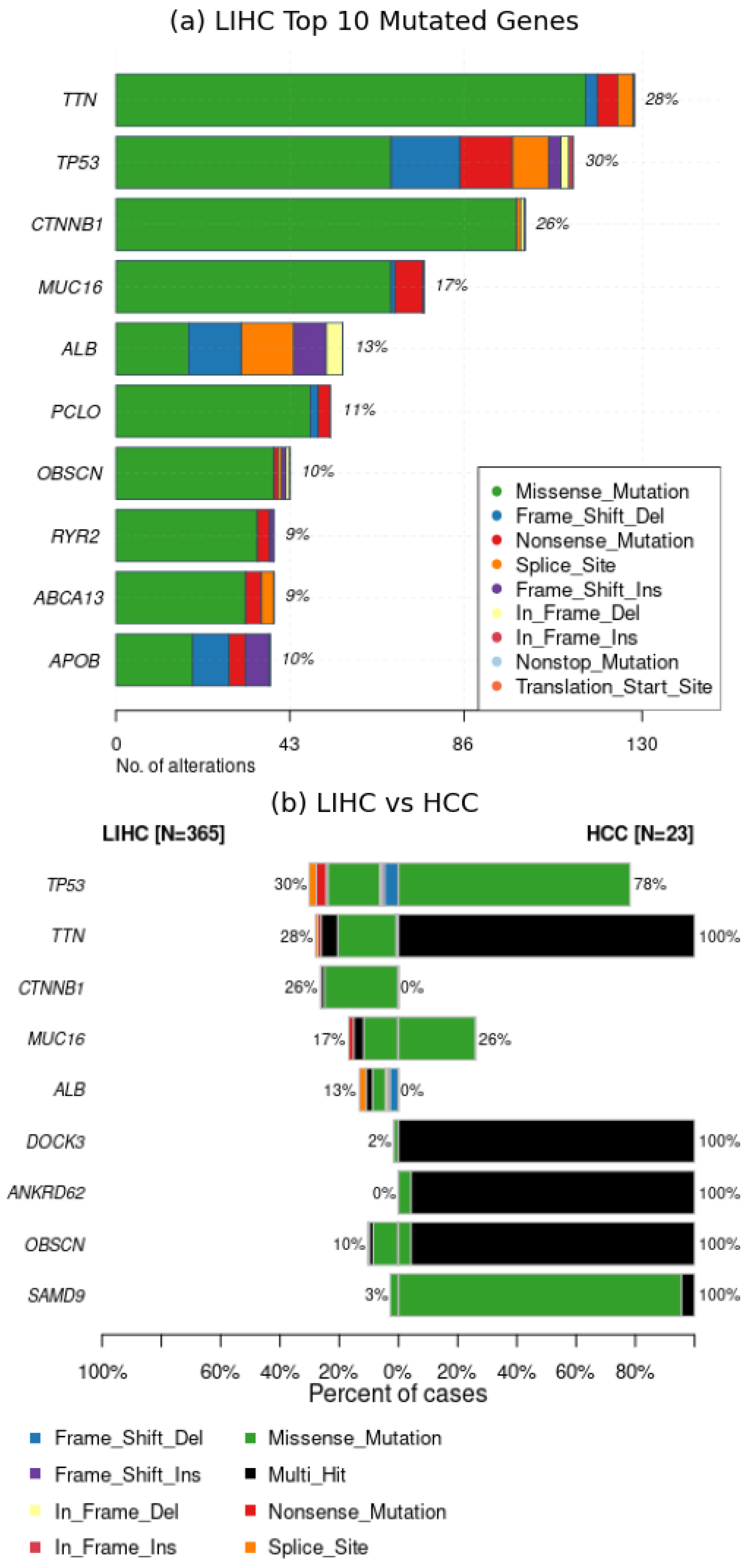
2.1.3. Interpretation
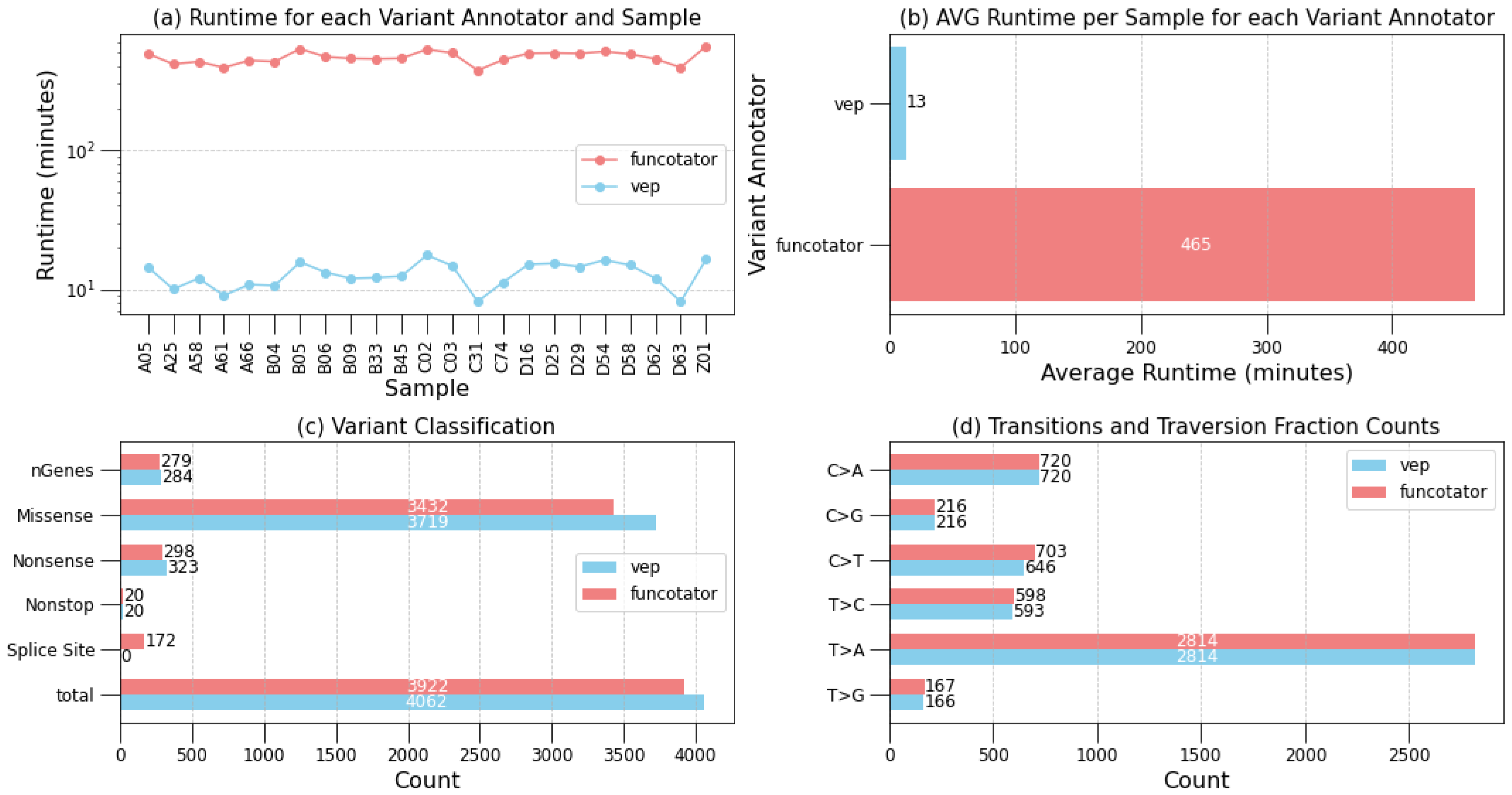
2.2. Evaluating the Scalability and Portability of Musta
3. Discussion
4. Materials and Methods
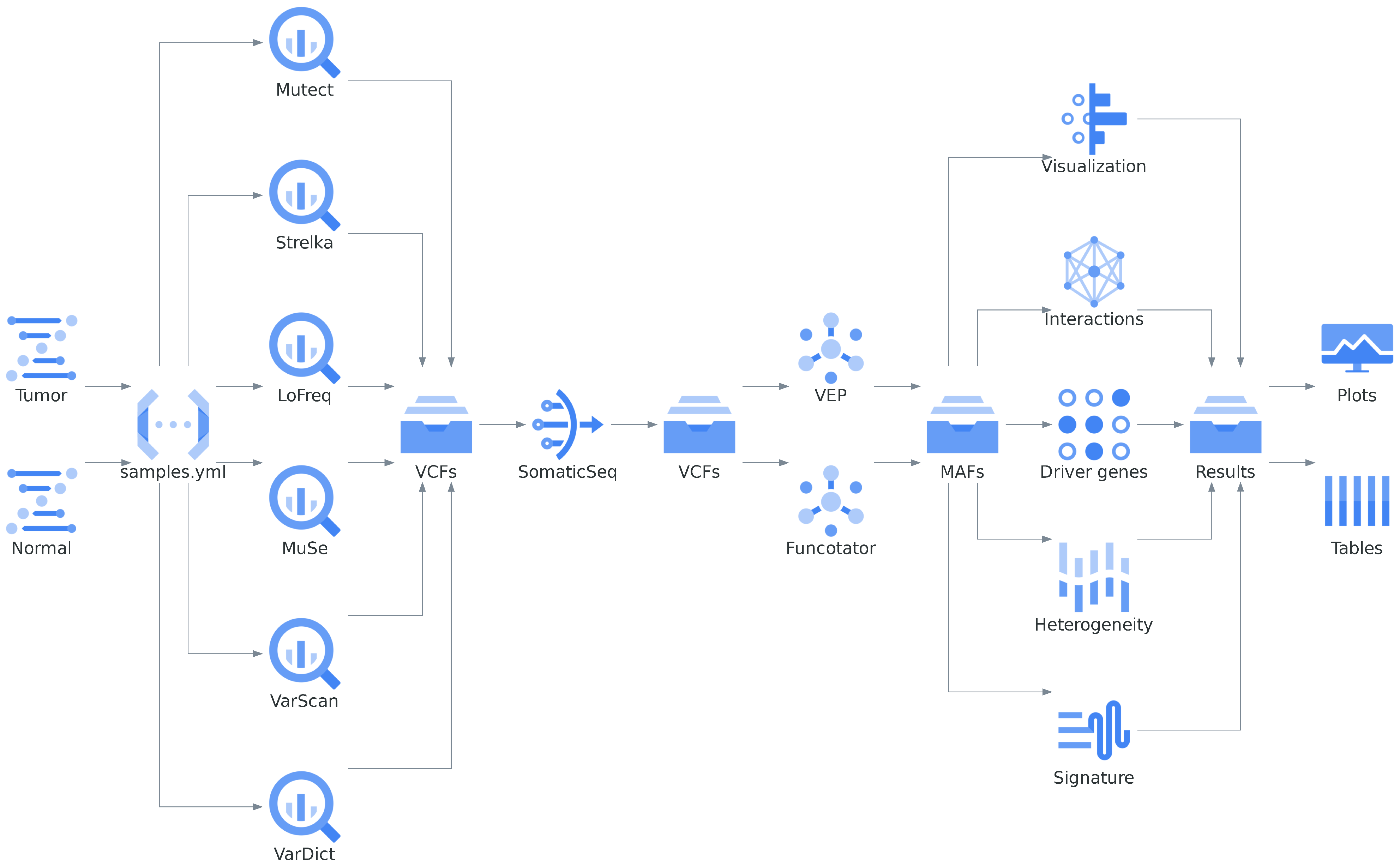
4.1. Overview
- COMMAND: This can be one of the following: detect, classify, or interpret. It selects the specific module the user wants to run. The command musta COMMAND --help will show a brief usage help for each command.
- --workdir WORKING-DIR: This parameter designates the working directory, which is the destination folder for analysis. This is the folder where (i) the Snakemake pipeline will be deployed, (ii) analysis logs will be stored, and (iii) all analysis outputs will be generated.
- --samples-file SAMPLES-FILE: this parameter points to a YAML file that lists the datasets the user wants to analyze.
4.1.1. Snakemake Core
- Snakefile: This file represents the workflow’s core. It contains all the rules and commands that define the sequence of tasks and dependencies within the pipeline. Each rule specifies how to create output files from input files or other rules, and may invoke commands, scripts, or generate output directly. The Snakefile essentially orchestrates the entire analysis process.
- config.yaml: The config.yaml file complements the Snakefile by providing essential input files and parameters for the workflow. It specifies the data sources, settings, and configurations needed for Musta to execute the analysis correctly. This file helps to configure the workflow according to the user’s specific requirements and data. Furthermore, through boolean flags in the config.yaml, allow users to specify which module to execute and which tools to enable.
4.1.2. Python framework
- Pipeline download. Initially, it downloads the pipeline into the working directory (WORKING-DIR).
- Environment configuration. The tool sets up internal paths within the Docker container environment.
- Edit samples.yaml. It edits the samples.yaml file, replacing user-local paths with Docker volume paths.
- Edit config.yaml. It manipulates the config.yaml file to control execution by toggling module and tool flags and constructing the workflow of rules.
- Workflow execution. Finally, this tool initiates the Snakemake workflow, ensuring the execution of the defined tasks.
4.1.3. Docker Container
4.1.4. Command Line Interface
- Input file and dataset check. The CLI performs an initial check on input files and datasets. It ensures that they exist, are in the correct format, and that all required accessory files are present. For example, it verifies that BAM files are indexed (with corresponding BAI files for each BAM), checks that VCF files are in compressed (GZ) format and indexed, and confirms the completeness of reference files.
- Constructing the Docker command. After verifying input files, the CLI constructs the Docker command. For each input file, volumes are mounted in Docker, and the list of arguments for the Musta Python tool is assembled. Finally, the Docker RUN command is prepared.
4.2. Workflow Modules
4.2.1. Detection
- 1.
- Selecting diverse and accurate component callers. Choosing an optimal number of component callers while ensuring diversity is crucial. Base learners must balance high accuracy with diversity to build a robust ensemble [40,41]. Diversity is essential because the benefit of an ensemble diminishes if all callers perform similarly. Conversely, too much diversity can lead to contradictory results, so a balanced selection of diverse component callers is essential.
- 2.
- Combining individual caller results:
- (a)
- (b)
- Complex machine learning-based methods such as stacking, Bayesian approaches, decision trees, and deep learning [44,45,46,47,48,49] leverage prediction results or metrics from individual callers as input features. Machine learning algorithms are used to combine these features, offering increased robustness against noise and errors. However, these methods often demand more computational resources and may be less interpretable.
- Excluding specific variant callers. If users wish, Musta allows specific variant callers to be excluded from the analysis. This customization ensures that only the desired variant callers are effectively applied to the data.
- Selecting preset combinations. Alternatively, users can choose from preset combinations of variant callers. Musta offers predefined sets of variant callers that have been optimized for specific analysis scenarios:--strict run only restrictive variant callers: Mutect2, LoFreq, Strelka2.--soft run only permissive variant callers: VarScan2, VarDict, MuSe.--fast run only fast variant callers: LoFreq, VarScan2, Strelka2, MuSe.
4.2.2. Classification
4.2.3. Interpretation
- 1.
- Variant visualization. Within the Musta pipeline framework, Maftools presents a range of graphical representations that assist in detecting mutation patterns and recurring characteristics within the dataset.
- Summary plots provide an at-a-glance summary by showcasing variant counts per sample and their distribution based on classification. They offer a high-level overview of the mutation landscape within the dataset.
- Onco plots illustrate mutations across samples, revealing distribution patterns. They are definitely valuable in cancer patient studies, providing a comprehensive mutational view.
- Lollipop plots take the interpretation a step further by depicting mutations on protein structures. This visual representation helps researchers to gain a better understanding of the precise locations of mutations and their potential impact on protein structure and thus function.
- Transitions and transversions plots categorize mutations into transitions and transversions, providing insights into the mutational spectrum within the dataset. Understanding these mutation types is crucial for unraveling mutational patterns.
- Oncostrip allows researchers to zoom in on specific genes, simplifying the exploration of features like mutual exclusivity. This focused view aids in uncovering relationships and interactions among genes of interest.
- 2.
- Somatic interactions. Recent advances in cancer genomics research have shown that many disease-causing genes in cancer are often mutated in a mutually exclusive manner [73,74]. Identification of such gene sets can reveal de novo pathways and underlying mechanisms of tumorigenesis. For this, Musta performs a Fisher’s exact test on all combinations of genes, to detect such mutually exclusive or co-occurring sets of genes.
- 3.
- Detecting cancer driver genes. Cancer driver genes provide selective growth advantage to cancer cells when mutated [3]. Several mathematical approaches have been developed to identify such driver genes [5,75,76,77]. In the context of the Musta framework, detection of such associated genes is based on the oncodriveCLUST algorithm [78]: the concept is that a majority of the mutations in oncogenes are clustered around mutational hotspots, whereas mutations on passenger genes are randomly distributed.
- 4.
- Pfam domains. In each type of cancer, specific protein domains are notably enriched with mutations [79,80]. The process of identifying and categorizing protein domains based on their mutation frequency serves the dual purpose of discerning the predominant domain affected within a particular cancer cohort. This approach further aids in pinpointing highly disrupted pathways and protein families that share similar functions, offering insights into the intricacies of deregulated mechanisms.
- 5.
- Tumor heterogeneity. Tumors are generally heterogeneous, composed of multiple clones and undergoing continuous evolution [81]. Heterogeneity can be inferred by clustering and classifying variants into sub clones, according to their allele frequencies [82,83]. Although clustering of variant allele frequencies gives us a fair idea on heterogeneity, it is also possible to measure the extent of heterogeneity in terms of a numerical value. MATH score is a simple quantitative measure of intra-tumor heterogeneity, which expresses the width of the variant allele frequency (VAF) distribution [84]. High MATH scores are found to be associated with poor prognosis and survival [85].
- 6.
- Mutational signature analysis. As cancer progresses, it develops a characteristic mutational pattern that unveils the underlying mutagenic processes at play. This revealing pattern can be deciphered through dimensional reduction techniques like non-negative matrix factorization (NMF) [23]. By decomposing a matrix containing nucleotide substitutions categorized into 96 substitution classes, based on their surrounding bases, specific mutational signatures, unique to each cancer type, emerge. These mutational signatures offer a blurred picture of the intricate mutational landscape of cancers. However, by cross-referencing them with validated signatures, we can achieve a clearer and more focused picture. This further enriches our understanding of the distinct processes that drive cancer progression.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mardis, E.R. A decade’s perspective on DNA sequencing technology. Nature 2011, 470, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Garraway, L.A. Genomics-driven oncology: Framework for an emerging paradigm. J. Clin. Oncol. 2013, 31, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Getz, G. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Swanton, C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Jäger, N. Bioinformatics workflows for clinical applications in precision oncology. Nat. Semin. Cancer Biol. 2022, 84, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Hu, Z.; Yang, Z.; Yang, F.; Li, Y.; Lin, P.; Chen, K.; Dong, L.; Cao, L.; Tao, Y.; et al. Extremely high genetic diversity in a single tumor points to prevalence of non-darwinian cell evolution. Proc. Natl Acad. Sci. USA 2015, 112, E6496–E6505. [Google Scholar] [CrossRef]
- Li, Z.; Gao, H.; Zhang, X.; Liu, Q.; Chen, G. Mutational and transcriptional alterations and clinicopathological factors predict the prognosis of stage I hepatocellular carcinoma. BMC Gastroenterol. 2022, 22, 427. [Google Scholar] [CrossRef]
- Shen, J.; Qi, L.; Zou, Z.; Du, J.; Kong, W.; Zhao, L.; Wei, J.; Lin, L.; Ren, M.; Liu, B. Identification of a novel gene signature for the prediction of recurrence in HCC patients by machine learning of genome-wide databases. Sci. Rep. 2020, 10, 4435. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, L.; Sun, Z.; Jiang, H.; Zhang, J. A radiomics signature associated with underlying gene expression pattern for the prediction of prognosis and treatment response in hepatocellular carcinoma. Eur. J. Radiol. 2023, 167, 111086. [Google Scholar] [CrossRef]
- Wang, Q.; Zhai, Y.Y.; Dai, J.H.; Li, K.Y.; Deng, Q.; Han, Z.G. SAMD9L inactivation promotes cell proliferation via facilitating G1-S transition in hepatitis B virus-associated hepatocellular carcinoma. Int. J. Biol. Sci. 2014, 10, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Shi, D.; Xing, K.; Zhou, H.; Lu, L.; Yu, D.; Wu, W. AMD1 upregulates hepatocellular carcinoma cells stemness by FTO mediated mRNA demethylation. Clin. Transl. Med. 2021, 11, e352. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Huang, J.M.; Li, T.M.; Liu, J.Q.; Wei, Z.L.; Lan, C.L.; Zhu, G.Z.; Liao, X.W.; Ye, X.P.; Peng, T. Clinical Significance and Potential Mechanisms of ATP Binding Cassette Subfamily C Genes in Hepatocellular Carcinoma. Front. Genet. 2022, 13, 805961. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qiu, Z.; Wei, L.; Tang, R.; Lian, B.; Zhao, Y.; He, X.; Xie, L. Integrated analysis of mutation data from various sources identifies key genes and signaling pathways in hepatocellular carcinoma. PLoS ONE 2014, 9, e100854. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Xiao, W.; Zhang, J.; Zheng, X.; Jiang, J. The role of AIM2 in human hepatocellular carcinoma and its clinical significance. Pathol. Res. Pract. 2023, 245, 154454. [Google Scholar] [CrossRef] [PubMed]
- Mroweh, M.; Roth, G.; Decaens, T.; Marche, P.N.; Lerat, H.; Macek Jílková, Z. Targeting Akt in Hepatocellular Carcinoma and Its Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 1794. [Google Scholar] [CrossRef] [PubMed]
- Ellrott, K.; Bailey, M.H.; Saksena, G.; Covington, K.R.; Kandoth, C.; Stewart, C.; Hess, J.; Ma, S.; Chiotti, K.E.; McLellan, M.; et al. MC3 Working Group; Cancer Genome Atlas Research Network. Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst. 2018, 28, 271–281. [Google Scholar] [CrossRef]
- Mölder, F.; Jablonski, K.P.; Letcher, B.; Hall, M.B.; Tomkins-Tinch, C.H.; Sochat, V.; Forster, J.; Lee, S.; Twardziok, S.O.; Kanitz, A.; et al. Holtgrewe M, Rahmann S, Nahnsen S, Köster J. Sustainable data analysis with Snakemake. F1000Research 2021, 10, 33. [Google Scholar] [CrossRef]
- Docker: Lightweight Linux Containers for Consistent Development and Deployment. Available online: https://www.docker.com (accessed on 21 July 2024).
- The Snakemake API Reference. Available online: https://snakemake.readthedocs.io/en/v7.0.0/api_reference/snakemake.html (accessed on 22 July 2024).
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 22 July 2024).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F. Trim Galore. A Wrapper Tool around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ Files. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 22 July 2024).
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; DePristo, M.A. From FastQ data to high-confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Kim, E.; Lee, I.; Marcotte, E.M. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci. Rep. 2015, 5, 17875. [Google Scholar] [CrossRef] [PubMed]
- Xu, C. A review of somatic single nucleotide variant calling algorithms for next-generation sequencing data. Comput. Struct. Biotechnol. 2018, 16, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Kroigard, A.B.; Thomassen, M.; Laenkholm, A.V.; Kruse, T.A.; Larsen, M.J. Evaluation of nine somatic variant callers for detection of somatic mutations in exome and targeted deep sequencing data. PLoS ONE 2016, 11, e0151664. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.D.; Kortschak, R.D.; Parker, W.T.; Schreiber, A.W.; Branford, S.; Scott, H.S.; Glonek, G.; Adelson, D.L. A comparative analysis of algorithms for somatic SNV detection in cancer. Bioinformatics 2013, 29, 2223–2230. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Jia, P.; Li, F.; Chen, H.; Ji, H.; Hucks, D.; Dahlman, K.b.; Pao, W.; Zhao, Z. Detecting somatic point mutations in cancer genome sequencing data: A comparison of mutation callers. Genome Med. 2013, 5, 91. [Google Scholar] [CrossRef]
- Kim, S.Y.; Speed, T.P. Comparing somatic mutation-callers: Beyond Venn diagrams. BMC Bioinform. 2013, 14, 189. [Google Scholar] [CrossRef]
- O’Rawe, J.; Jiang, T.; Sun, G.; Wu, Y.; Wang, W.; Hu, J.; Bodily, P.; Tian, L.; Hakonarson, H.; Johnson, W.E.; et al. Low concordance of multiple variant-calling pipelines: Practical implications for exome and genome sequencing. Genome Med. 2013, 5, 28. [Google Scholar] [CrossRef]
- Goode, D.L.; Hunter, S.M.; Doyle, M.A.; Ma, T.; Rowley, S.M.; Choong, D.; Ryland, G.L.; Campbell, I.G. A simple consensus approach improves somatic mutation prediction accuracy. Genome Med. 2013, 5, 90. [Google Scholar] [CrossRef] [PubMed]
- Chiara, M.; Gioiosa, S.; Chillemi, G.; D’Antonio, M.; Flati, T.; Picardi, E.; Zambelli, F.; Horner, D.S.; Pesole, G.; Castrignanó, T. CoVaCS: A consensus variant calling system. BMC Genom. 2018, 19, 120. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.K.; Shang, Y.K.; Chen, Z.N.; Bian, H. A three-caller pipeline for variant analysis of cancer whole-exome sequencing data. Mol. Med. Rep. 2017, 15, 2489–2494. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.; Robles-Espinoza, C.D.; Rust, A.G.; Adams, D.J. Cake: A bioinformatics pipeline for the integrated analysis of somatic variants in cancer genomes. Bioinformatics 2013, 29, 2208–2210. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-H. Ensemble Methods: Foundations and Algorithms; Chapman and Hall/CRC: Boca Raton, FL, USA, 2012. [Google Scholar]
- Hansen, L.K.; Salamon, P. Neural network ensembles. IEEE Trans. Pattern Anal. 1990, 12, 993–1001. [Google Scholar] [CrossRef]
- Brown, G.; Wyatt, J.; Harris, R.; Yao, X. Diversity creation methods: A survey and categorisation. Inf. Fusion 2005, 6, 5–20. [Google Scholar] [CrossRef]
- Ewing, A.D.; Houlahan, K.E.; Hu, Y.; Ellrott, K.; Caloian, C.; Yamaguchi, T.N.; Bare, J.C.; P’ng, C.; Waggott, D.; Sabelnykova, V.Y.; et al. Combining tumor genome simulation with crowdsourcing to benchmark somatic single-nucleotide-variant detection. Nat. Methods 2015, 12, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Callari, M.; Sammut, S.J.; De Mattos-Arruda, L.; Bruna, A.; Rueda, O.M.; Chin, S.F.; Caldas, C. Intersect-then-combine approach: Improving the performance of somatic variant calling in whole exome sequencing data using multiple aligners and callers. Genome Med. 2017, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Jacob, L.; Speed, T.P. Combining calls from multiple somatic mutation-callers. BMC Bioinf. 2014, 15, 154. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Weaver, D.; McNeill, N.; Zhang, J.; Mackey, A.J.; Reese, J. BAYSIC: A Bayesian method for combining sets of genome variants with improved specificity and sensitivity. BMC Bioinform. 2014, 15, 104. [Google Scholar] [CrossRef]
- Anzar, I.; Sverchkova, A.; Stratford, R.; Clancy, T. NeoMutate: An ensemble machine learning framework for the prediction of somatic mutations in cancer. BMC Med. Genom. 2019, 12, 63. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.T.; Afshar, P.T.; Chhibber, A.; Mohiyuddin, M.; Fan, Y.; Mu, J.C.; Gibeling, G.; Barr, S.; Asadi, N.B.; Gerstein, M.B.; et al. An ensemble approach to accurately detect somatic mutations using SomaticSeq. Genome Biol. 2015, 16, 197. [Google Scholar] [CrossRef]
- Ainscough, B.J.; Barnell, E.K.; Ronning, P.; Campbell, K.M.; Wagner, A.H.; Fehniger, T.A.; Dunn, G.P.; Uppaluri, R.; Govindan, R.; Rohan, T.E.; et al. A deep learning approach to automate refinement of somatic variant calling from cancer sequencing data. Nat. Genet. 2018, 50, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Sahraeian, S.M.E.; Liu, R.; Lau, B.; Podesta, K.; Mohiyuddin, M.; Lam, H.Y.K. Deep convolutional neural networks for accurate somatic mutation detection. Nat. Commun. 2019, 10, 1041. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Markovets, A.; Ahdesmaki, M.; Johnson, J. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. AACR Annu. Meeting 2015, 44, e108. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids 2012, 40, 11189–11201. [Google Scholar] [CrossRef]
- Fan, Y.; Xi, L.; Hughes, D.S.T.; Zhang, J.; Zhang, J.; Futreal, P.A.; Wheeler, D.A.; Wang, W. MuSE: Accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol. 2016, 17, 178. [Google Scholar] [CrossRef]
- Wang, M.; Luo, W.; Jones, K.; Bian, X.; Williams, R.; Higson, H.; Wu, D.; Hicks, B.; Yeager, M.; Zhu, B. SomaticCombiner: Improving the performance of somatic variant calling based on evaluation tests and a consensus approach. Sci. Rep. 2020, 10, 12898. [Google Scholar] [CrossRef]
- Zhang, C.; El-Kebir, M.; Ochoa, I. Moss enables high sensitivity single-nucleotide variant calling from multiple bulk DNA tumor samples. Nat. Commun. 2021, 12, 2204. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Ding, J.; Morin, R.; Crisan, A.; Ha, G.; Giuliany, R.; Bashashati, A.; Hirst, M.; Turashvili, G.; Oloumi, A.; et al. JointSNVMix: A probabilistic model for accurate detection of somatic mutations in normal/tumour paired next-generation sequencing data. Bioinformatics 2012, 28, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Larson, D.E.; Harris, C.C.; Chen, K.; Koboldt, D.C.; Abbott, T.E.; Dooling, D.J.; Ley, T.J.; Mardis, E.R.; Wilson, R.K.; Ding, L. SomaticSniper: Identification of somatic point mutations in whole genome sequencing data. Bioinformatics 2012, 28, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Bergmann, E.A.; Arora, K.; Vacic, V.; Zody, M.C.; Iossifov, I.; O’Rawe, J.A.; Wu, Y.; Jimenez Barron, L.T.; Rosenbaum, J.; et al. Indel variant analysis of short-read sequencing data with Scalpel. Nat. Protoc. 2016, 11, 2529–2548. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. DbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 14,1456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019, 531210. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Tuteja, S.; Kadri, S.; Yap, K.L. A performance evaluation study: Variant annotation tools - the enigma of clinical next generation sequencing (NGS) based genetic testing. J. Pathol. Inf. 2022, 13, 2153–3539. [Google Scholar] [CrossRef]
- Yen, J.L.; Garcia, S.; Montana, A.; Harris, J.; Chervitz, S.; Morra, M.; West, J.; Chen, R.; Church, D.M. A variant by any name: Quantifying annotation discordance across tools and clinical databases. Genome Med. 2017, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Mutation Annotation Format. Available online: https://wiki.nci.nih.gov/display/TCGA/Mutation+Annotation+Format (accessed on 21 July 2024).
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Van Loo, P.; Wedge, D.C.; Alexandrov, L.B.; Greenman, C.D.; Lau, K.W.; Raine, K.; Jones, D.; Marshall, J.; Ramakrishna, M.; et al. The life history of 21 breast cancers. Cell 2012, 149, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Leiserson, M.D.; Wu, H.; Vandin, F.; Raphael, B.J. CoMEt: A statistical approach to identify combinations of mutually exclusive alterations in cancer. Genome Biol. 2015, 16, 160. [Google Scholar] [CrossRef] [PubMed]
- Yeang, C.H.; McCormick, F.; Levine, A. Combinatorial patterns of somatic gene mutations in cancer. FASEB J. 2008, 22, 2605–2622. [Google Scholar] [CrossRef] [PubMed]
- Dees, N.D.; Zhang, Q.; Kandoth, C.; Wendl, M.C.; Schierding, W.; Koboldt, D.C.; Mooney, T.B.; Callaway, M.B.; Dooling, D.; Mardis, E.R.; et al. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 2012, 22, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Lopez-Bigas, N. Functional impact bias reveals cancer drivers. Nucleic Acids Res. 2012, 40, e169. [Google Scholar] [CrossRef]
- Zapata, L.; Susak, H.; Drechsel, O.; Friedlander, M.R.; Estivill, X.; Ossowski, S. Signatures of positive selection reveal a universal role of chromatin modifiers as cancer driver genes. Sci. Rep. 2017, 7, 13124. [Google Scholar] [CrossRef] [PubMed]
- Tamborero, D.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveCLUST: Exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 2013, 29, 2238–2244. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Petsalaki, E.; Rolland, T.; Hill, D.E.; Vidal, M.; Roth, F.P. Protein domain-level landscape of cancer-type-specific somatic mutations. PLoS Comput. Biol. 2015, 11, e1004147. [Google Scholar] [CrossRef]
- Nehrt, N.L.; Peterson, T.H.; Park, D.; Kann, M.G. Domain landscapes of somatic mutations in cancer. BMC Genom. 2012, 13 (Suppl. 4), S9. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef]
- Miller, C.A.; White, B.S.; Dees, N.D.; Griffith, M.; Welch, J.S.; Griffith, O.L.; Vij, R.; Tomasson, M.H.; Graubert, T.A.; Walter, M.J.; et al. SciClone: Inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput. Biol. 2014, 10, e1003665. [Google Scholar] [CrossRef] [PubMed]
- Mroz, E.A.; Rocco, J.W. MATH, a novel measure of intratumor genetic heterogeneity, is high in poor-outcome classes of head and neck squamous cell carcinoma. Oral Oncol. 2013, 49, 211–215. [Google Scholar] [CrossRef]
- Mroz, E.A.; Tward, A.D.; Hammon, R.J.; Ren, Y.; Rocco, J.W. Intra-tumor genetic heterogeneity and mortality in head and neck cancer: Analysis of data from the Cancer Genome Atlas. PLoS Med. 2015, 12, e1001786. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atzeni, R.; Massidda, M.; Pieroni, E.; Rallo, V.; Pisu, M.; Angius, A. A Novel Affordable and Reliable Framework for Accurate Detection and Comprehensive Analysis of Somatic Mutations in Cancer. Int. J. Mol. Sci. 2024, 25, 8044. https://doi.org/10.3390/ijms25158044
Atzeni R, Massidda M, Pieroni E, Rallo V, Pisu M, Angius A. A Novel Affordable and Reliable Framework for Accurate Detection and Comprehensive Analysis of Somatic Mutations in Cancer. International Journal of Molecular Sciences. 2024; 25(15):8044. https://doi.org/10.3390/ijms25158044
Chicago/Turabian StyleAtzeni, Rossano, Matteo Massidda, Enrico Pieroni, Vincenzo Rallo, Massimo Pisu, and Andrea Angius. 2024. "A Novel Affordable and Reliable Framework for Accurate Detection and Comprehensive Analysis of Somatic Mutations in Cancer" International Journal of Molecular Sciences 25, no. 15: 8044. https://doi.org/10.3390/ijms25158044
APA StyleAtzeni, R., Massidda, M., Pieroni, E., Rallo, V., Pisu, M., & Angius, A. (2024). A Novel Affordable and Reliable Framework for Accurate Detection and Comprehensive Analysis of Somatic Mutations in Cancer. International Journal of Molecular Sciences, 25(15), 8044. https://doi.org/10.3390/ijms25158044