

Protective Effects of Hepatocyte Stress Defenders, Nrf1 and Nrf2, against MASLD Progression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
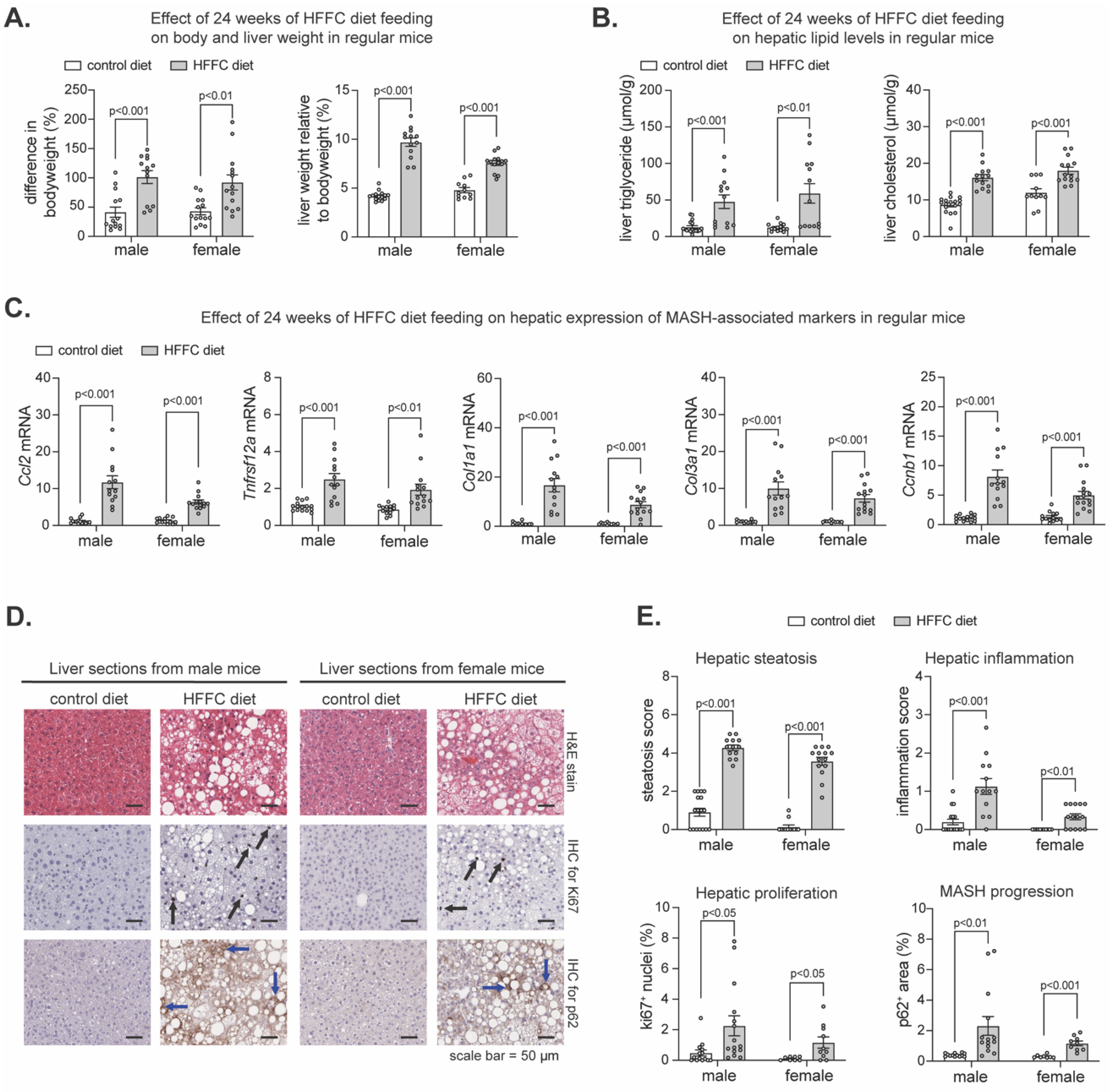
2.1. Effect of HFFC Diet on Liver in Mice with Flox Alleles for Nrf1, Nrf2, or Both
2.2. Effect of Hepatocyte Deficiency for Nrf1, Nrf2, or Both in Mice Chronically Fed HFFC Diet
2.3. Nrf1 Deficiency Enhances Liver Tumorigenesis in Male Mice with MASH
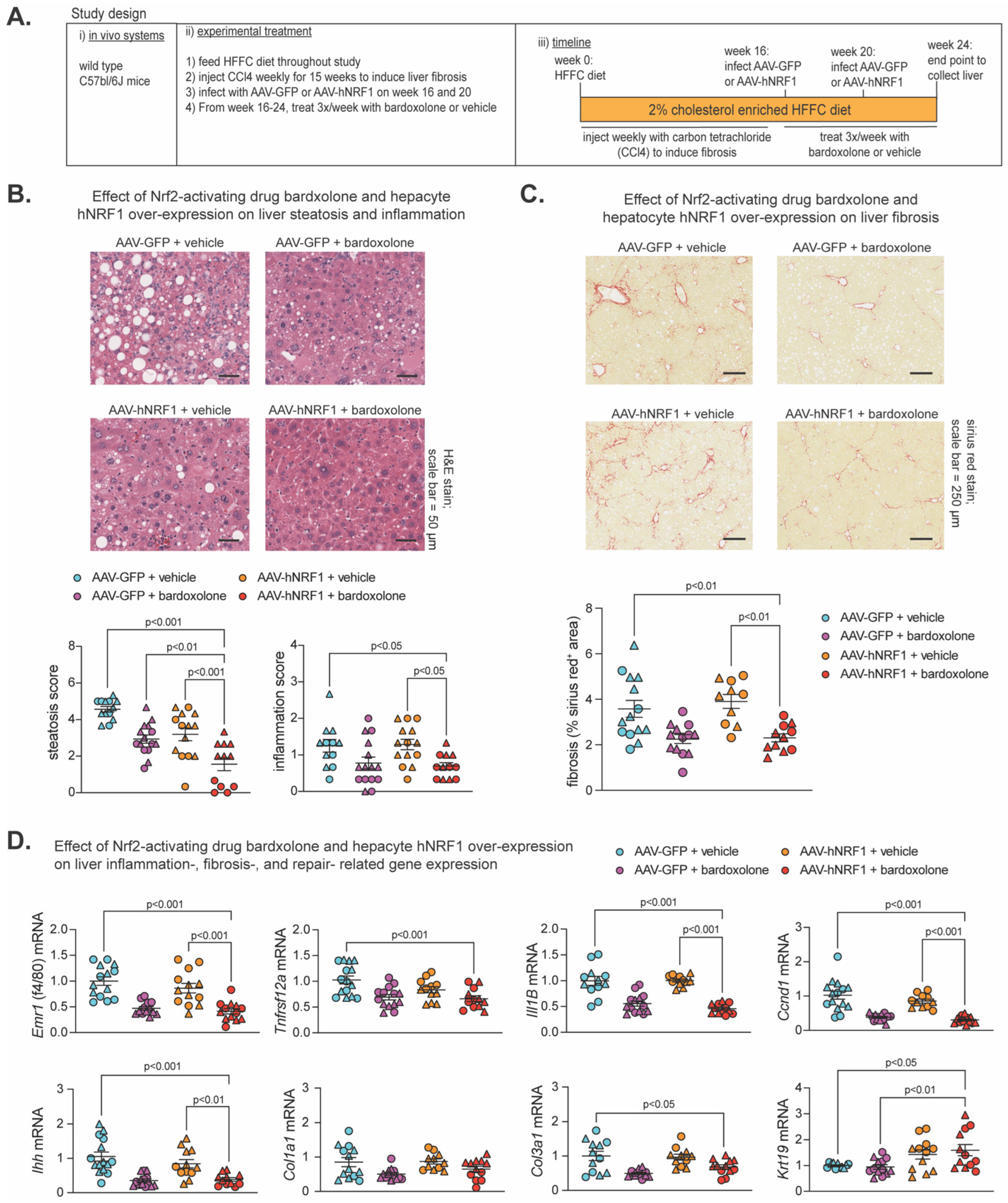
2.4. Effect of Bardoxolone and Nrf1 Overexpression on MASH-Linked Fibrosis
3. Discussion
4. Materials and Methods
4.1. Animals Used in Study
4.2. Tissue Collection and Liver Histological Analysis
4.3. Analysis of Liver Cholesterol and Triglyceride
4.4. Gene Expression Analysis
4.5. Tumor Volume Measurement
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Le, M.H.; Yeo, Y.H.; Li, X.; Li, J.; Zou, B.; Wu, Y.; Ye, Q.; Huang, D.Q.; Zhao, C.; Zhang, J.; et al. 2019 Global NAFLD Prevalence: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2022, 20, 2809–2817.e28. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus, P. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e91. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Hagstrom, H.; Nasr, P.; Fredrikson, M.; Stal, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Ascha, M.S.; Hanouneh, I.A.; Lopez, R.; Tamimi, T.A.; Feldstein, A.F.; Zein, N.N. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010, 51, 1972–1978. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Akl, M.G.; Widenmaier, S.B. Immunometabolic factors contributing to obesity-linked hepatocellular carcinoma. Front. Cell. Dev. Biol. 2022, 10, 1089124. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef]
- Horn, C.L.; Morales, A.L.; Savard, C.; Farrell, G.C.; Ioannou, G.N. Role of Cholesterol-Associated Steatohepatitis in the Development of NASH. Hepatol. Commun. 2022, 6, 12–35. [Google Scholar] [CrossRef]
- Carvalho-Gontijo, R.; Han, C.; Zhang, L.; Zhang, V.; Hosseini, M.; Mekeel, K.; Schnabl, B.; Loomba, R.; Karin, M.; Brenner, D.A.; et al. Metabolic Injury of Hepatocytes Promotes Progression of NAFLD and AALD. Semin. Liver Dis. 2022, 42, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Hagstrom, H.; Nasr, P.; Ekstedt, M.; Hammar, U.; Stal, P.; Hultcrantz, R.; Kechagias, S. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J. Hepatol. 2017, 67, 1265–1273. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiang, Y. Molecular and cellular basis for the unique functioning of Nrf1, an indispensable transcription factor for maintaining cell homoeostasis and organ integrity. Biochem. J. 2016, 473, 961–1000. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Hataya, N.; Katsuoka, F.; Yamamoto, M. NF-E2-related factor 1 (Nrf1) serves as a novel regulator of hepatic lipid metabolism through regulation of the Lipin1 and PGC-1beta genes. Mol. Cell. Biol. 2012, 32, 2760–2770. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Ho, D.V.; Chan, J.Y. Nuclear factor-erythroid 2-related factor 1 regulates expression of proteasome genes in hepatocytes and protects against endoplasmic reticulum stress and steatosis in mice. FEBS J. 2013, 280, 3609–3620. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuji, M.; Katsuoka, F.; Kobayashi, A.; Aburatani, H.; Hayes, J.D.; Yamamoto, M. Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J. Biol. Chem. 2008, 283, 33554–33562. [Google Scholar] [CrossRef] [PubMed]
- Widenmaier, S.B.; Snyder, N.A.; Nguyen, T.B.; Arduini, A.; Lee, G.Y.; Arruda, A.P.; Saksi, J.; Bartelt, A.; Hotamisligil, G.S. NRF1 Is an ER Membrane Sensor that Is Central to Cholesterol Homeostasis. Cell 2017, 171, 1094–1109.e1015. [Google Scholar] [CrossRef]
- Tsujita, T.; Peirce, V.; Baird, L.; Matsuyama, Y.; Takaku, M.; Walsh, S.V.; Griffin, J.L.; Uruno, A.; Yamamoto, M.; Hayes, J.D. Transcription factor Nrf1 negatively regulates the cystine/glutamate transporter and lipid-metabolizing enzymes. Mol. Cell Biol. 2014, 34, 3800–3816. [Google Scholar] [CrossRef]
- Akl, M.G.; Li, L.; Baccetto, R.; Phanse, S.; Zhang, Q.; Trites, M.J.; McDonald, S.; Aoki, H.; Babu, M.; Widenmaier, S.B. Complementary gene regulation by NRF1 and NRF2 protects against hepatic cholesterol overload. Cell Rep. 2023, 42, 112399. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, L.; Leung, L.; Yen, T.S.; Lee, C.; Chan, J.Y. Liver-specific inactivation of the Nrf1 gene in adult mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia. Proc. Natl. Acad. Sci. USA 2005, 102, 4120–4125. [Google Scholar] [CrossRef]
- Kamisako, T.; Tanaka, Y.; Kishino, Y.; Ikeda, T.; Yamamoto, K.; Masuda, S.; Ogawa, H. Role of Nrf2 in the alteration of cholesterol and bile acid metabolism-related gene expression by dietary cholesterol in high fat-fed mice. J. Clin. Biochem. Nutr. 2014, 54, 90–94. [Google Scholar] [CrossRef]
- Kitteringham, N.R.; Abdullah, A.; Walsh, J.; Randle, L.; Jenkins, R.E.; Sison, R.; Goldring, C.E.; Powell, H.; Sanderson, C.; Williams, S.; et al. Proteomic analysis of Nrf2 deficient transgenic mice reveals cellular defence and lipid metabolism as primary Nrf2-dependent pathways in the liver. J. Proteomics 2010, 73, 1612–1631. [Google Scholar] [CrossRef] [PubMed]
- Akl, M.G.; Baccetto, R.; Stebbings, B.M.; Li, L.; Widenmaier, S.B. Euglycemia is affected by stress defense factor hepatocyte NRF1, but not NRF2. Biochem. Biophys. Res. Commun. 2023, 668, 96–103. [Google Scholar] [CrossRef]
- Okada, K.; Warabi, E.; Sugimoto, H.; Horie, M.; Gotoh, N.; Tokushige, K.; Hashimoto, E.; Utsunomiya, H.; Takahashi, H.; Ishii, T.; et al. Deletion of Nrf2 leads to rapid progression of steatohepatitis in mice fed atherogenic plus high-fat diet. J. Gastroenterol. 2013, 48, 620–632. [Google Scholar] [CrossRef]
- Sugimoto, H.; Okada, K.; Shoda, J.; Warabi, E.; Ishige, K.; Ueda, T.; Taguchi, K.; Yanagawa, T.; Nakahara, A.; Hyodo, I.; et al. Deletion of nuclear factor-E2-related factor-2 leads to rapid onset and progression of nutritional steatohepatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G283–G294. [Google Scholar] [CrossRef]
- Trites, M.J.; Stebbings, B.M.; Aoki, H.; Phanse, S.; Akl, M.G.; Li, L.; Babu, M.; Widenmaier, S.B. HDL functionality is dependent on hepatocyte stress defense factors Nrf1 and Nrf2. Front. Physiol. 2023, 14, 1212785. [Google Scholar] [CrossRef]
- Wahl, S.; Drong, A.; Lehne, B.; Loh, M.; Scott, W.R.; Kunze, S.; Tsai, P.C.; Ried, J.S.; Zhang, W.; Yang, Y.; et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 2017, 541, 81–86. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; den Besten, W.; Deshaies, R.J. p97-dependent retrotranslocation and proteolytic processing govern formation of active Nrf1 upon proteasome inhibition. eLife 2014, 3, e01856. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Widenmaier, S.B. Proteostasis in thermogenesis and obesity. Biol. Chem. 2020, 401, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Antonucci, L.; Yamachika, S.; Zhang, Z.; Taniguchi, K.; Umemura, A.; Hatzivassiliou, G.; Roose-Girma, M.; Reina-Campos, M.; Duran, A.; et al. NRF2 activates growth factor genes and downstream AKT signaling to induce mouse and human hepatomegaly. J. Hepatol. 2020, 72, 1182–1195. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Taniguchi, K.; Yamachika, S.; He, F.; Karin, M. p62/SQSTM1-Dr. Jekyll and Mr. Hyde that prevents oxidative stress but promotes liver cancer. FEBS Lett. 2016, 590, 2375–2397. [Google Scholar] [CrossRef] [PubMed]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Wufuer, R.; Fan, Z.; Liu, K.; Zhang, Y. Differential Yet Integral Contributions of Nrf1 and Nrf2 in the Human HepG2 Cells on Antioxidant Cytoprotective Response against Tert-Butylhydroquinone as a Pro-Oxidative Stressor. Antioxidants 2021, 10, 1610. [Google Scholar] [CrossRef] [PubMed]
- Wufuer, R.; Fan, Z.; Yuan, J.; Zheng, Z.; Hu, S.; Sun, G.; Zhang, Y. Distinct Roles of Nrf1 and Nrf2 in Monitoring the Reductive Stress Response to Dithiothreitol (DTT). Antioxidants 2022, 11, 1535. [Google Scholar] [CrossRef] [PubMed]
- Wufuer, R.; Liu, K.; Feng, J.; Wang, M.; Hu, S.; Chen, F.; Lin, S.; Zhang, Y. Distinct mechanisms by which Nrf1 and Nrf2 as drug targets contribute to the anticancer efficacy of cisplatin on hepatoma cells. Free Radic. Biol. Med. 2024, 213, 488–511. [Google Scholar] [CrossRef]
- Katsuoka, F.; Otsuki, A.; Hatanaka, N.; Okuyama, H.; Yamamoto, M. Target Gene Diversity of the Nrf1-MafG Transcription Factor Revealed by a Tethered Heterodimer. Mol. Cell. Biol. 2022, 42, e0052021. [Google Scholar] [CrossRef]
- Sekine, H.; Motohashi, H. Unique and overlapping roles of NRF2 and NRF1 in transcriptional regulation. J. Clin. Biochem. Nutr. 2024, 74, 91–96. [Google Scholar] [CrossRef]
- Kawai, Y.; Garduno, L.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation-deacetylation of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) regulates its transcriptional activity and nucleocytoplasmic localization. J. Biol. Chem. 2011, 286, 7629–7640. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef]
- Sekine, H.; Okazaki, K.; Kato, K.; Alam, M.M.; Shima, H.; Katsuoka, F.; Tsujita, T.; Suzuki, N.; Kobayashi, A.; Igarashi, K.; et al. O-GlcNAcylation Signal Mediates Proteasome Inhibitor Resistance in Cancer Cells by Stabilizing NRF1. Mol. Cell. Biol. 2018, 38, e00252-18. [Google Scholar] [CrossRef]
- Sekine, H.; Okazaki, K.; Ota, N.; Shima, H.; Katoh, Y.; Suzuki, N.; Igarashi, K.; Ito, M.; Motohashi, H.; Yamamoto, M. The Mediator Subunit MED16 Transduces NRF2-Activating Signals into Antioxidant Gene Expression. Mol. Cell. Biol. 2016, 36, 407–420. [Google Scholar] [CrossRef]
- Chen, J.; Wang, M.; Xiang, Y.; Ru, X.; Ren, Y.; Liu, X.; Qiu, L.; Zhang, Y. Nrf1 Is Endowed with a Dominant Tumor-Repressing Effect onto the Wnt/beta-Catenin-Dependent and Wnt/beta-Catenin-Independent Signaling Networks in the Human Liver Cancer. Oxid. Med. Cell. Longev. 2020, 2020, 5138539. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Qiu, L.; Lu, F.; Ru, X.; Li, S.; Xiang, Y.; Yu, S.; Zhang, Y. TALENs-directed knockout of the full-length transcription factor Nrf1alpha that represses malignant behaviour of human hepatocellular carcinoma (HepG2) cells. Sci. Rep. 2016, 6, 23775. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Ren, Y.; Hu, S.; Liu, K.; Qiu, L.; Zhang, Y. TCF11 Has a Potent Tumor-Repressing Effect Than Its Prototypic Nrf1alpha by Definition of Both Similar Yet Different Regulatory Profiles, With a Striking Disparity From Nrf2. Front. Oncol. 2021, 11, 707032. [Google Scholar] [CrossRef]
- Sharma, R.S.; Harrison, D.J.; Kisielewski, D.; Cassidy, D.M.; McNeilly, A.D.; Gallagher, J.R.; Walsh, S.V.; Honda, T.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; et al. Experimental Nonalcoholic Steatohepatitis and Liver Fibrosis Are Ameliorated by Pharmacologic Activation of Nrf2 (NF-E2 p45-Related Factor 2). Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 367–398. [Google Scholar] [CrossRef] [PubMed]
- Mohs, A.; Otto, T.; Schneider, K.M.; Peltzer, M.; Boekschoten, M.; Holland, C.H.; Hudert, C.A.; Kalveram, L.; Wiegand, S.; Saez-Rodriguez, J.; et al. Hepatocyte-specific NRF2 activation controls fibrogenesis and carcinogenesis in steatohepatitis. J. Hepatol. 2021, 74, 638–648. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Electronic address, w.b.e.; Cancer Genome Atlas Research, N. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.A.; Reibe, S.; Shalapour, S.; Ooi, G.J.; Watt, M.J.; Karin, M. Preclinical Models for Studying NASH-Driven HCC: How Useful Are They? Cell Metab. 2019, 29, 18–26. [Google Scholar] [CrossRef]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical models of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H.; HM, A.E.; Oro, D.; Evers, S.S.; Heeboll, S.; Eriksen, P.L.; Thomsen, K.L.; Bengtsson, A.; Veidal, S.S.; Feigh, M.; et al. Human translatability of the GAN diet-induced obese mouse model of non-alcoholic steatohepatitis. BMC Gastroenterol. 2020, 20, 210. [Google Scholar] [CrossRef]
- Mells, J.E.; Fu, P.P.; Kumar, P.; Smith, T.; Karpen, S.J.; Anania, F.A. Saturated fat and cholesterol are critical to inducing murine metabolic syndrome with robust nonalcoholic steatohepatitis. J. Nutr. Biochem. 2015, 26, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Savard, C.; Tartaglione, E.V.; Kuver, R.; Haigh, W.G.; Farrell, G.C.; Subramanian, S.; Chait, A.; Yeh, M.M.; Quinn, L.S.; Ioannou, G.N. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology 2013, 57, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Flensted-Jensen, M.; Oro, D.; Rorbeck, E.A.; Zhang, C.; Madsen, M.R.; Madsen, A.N.; Norlin, J.; Feigh, M.; Larsen, S.; Hansen, H.H. Dietary intervention reverses molecular markers of hepatocellular senescence in the GAN diet-induced obese and biopsy-confirmed mouse model of NASH. BMC Gastroenterol. 2024, 24, 59. [Google Scholar] [CrossRef]
- Hansen, H.H.; Pors, S.; Andersen, M.W.; Vyberg, M.; Nohr-Meldgaard, J.; Nielsen, M.H.; Oro, D.; Madsen, M.R.; Lewinska, M.; Mollerhoj, M.B.; et al. Semaglutide reduces tumor burden in the GAN diet-induced obese and biopsy-confirmed mouse model of NASH-HCC with advanced fibrosis. Sci. Rep. 2023, 13, 23056. [Google Scholar] [CrossRef]
- Mollerhoj, M.B.; Veidal, S.S.; Thrane, K.T.; Oro, D.; Overgaard, A.; Salinas, C.G.; Madsen, M.R.; Pfisterer, L.; Vyberg, M.; Simon, E.; et al. Hepatoprotective effects of semaglutide, lanifibranor and dietary intervention in the GAN diet-induced obese and biopsy-confirmed mouse model of NASH. Clin. Transl. Sci. 2022, 15, 1167–1186. [Google Scholar] [CrossRef]
- Nielsen, M.H.; Gillum, M.P.; Vrang, N.; Jelsing, J.; Hansen, H.H.; Feigh, M.; Oro, D. Hepatoprotective effects of the long-acting fibroblast growth factor 21 analog PF-05231023 in the GAN diet-induced obese and biopsy-confirmed mouse model of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2023, 324, G378–G388. [Google Scholar] [CrossRef] [PubMed]
- Assy, N.; Minuk, G.Y. Liver regeneration: Methods for monitoring and their applications. J. Hepatol. 1997, 26, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Turhan, N.; Sonsuz, A.; Basaranoglu, G. Mallory-Denk Bodies in chronic hepatitis. World J. Gastroenterol. 2011, 17, 2172–2177. [Google Scholar] [CrossRef] [PubMed]
- Stumptner, C.; Fuchsbichler, A.; Zatloukal, K.; Denk, H. In vitro production of Mallory bodies and intracellular hyaline bodies: The central role of sequestosome 1/p62. Hepatology 2007, 46, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Lee, Y.A.; Fujiwara, N.; Ybanez, M.; Allen, B.; Martins, S.; Fiel, M.I.; Goossens, N.; Chou, H.I.; Hoshida, Y.; et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol. 2018, 69, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Kubota, N.; Kado, S.; Kano, M.; Masuoka, N.; Nagata, Y.; Kobayashi, T.; Miyazaki, K.; Ishikawa, F. A high-fat diet and multiple administration of carbon tetrachloride induces liver injury and pathological features associated with non-alcoholic steatohepatitis in mice. Clin. Exp. Pharmacol. Physiol. 2013, 40, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Paquet, K.J.; Kamphausen, U. The carbon-tetrachloride-hepatotoxicity as a model of liver damage. First report: Long-time biochemical changes. Acta Hepatogastroenterol. 1975, 22, 84–88. [Google Scholar]
- Scholten, D.; Trebicka, J.; Liedtke, C.; Weiskirchen, R. The carbon tetrachloride model in mice. Lab Anim. 2015, 49, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Espanol-Suner, R.; Carpentier, R.; Van Hul, N.; Legry, V.; Achouri, Y.; Cordi, S.; Jacquemin, P.; Lemaigre, F.; Leclercq, I.A. Liver progenitor cells yield functional hepatocytes in response to chronic liver injury in mice. Gastroenterology 2012, 143, 1564–1575.e1567. [Google Scholar] [CrossRef]
- Van Hul, N.; Lanthier, N.; Espanol Suner, R.; Abarca Quinones, J.; van Rooijen, N.; Leclercq, I. Kupffer cells influence parenchymal invasion and phenotypic orientation, but not the proliferation, of liver progenitor cells in a murine model of liver injury. Am. J. Pathol. 2011, 179, 1839–1850. [Google Scholar] [CrossRef]
- Zhang, L.; Theise, N.; Chua, M.; Reid, L.M. The stem cell niche of human livers: Symmetry between development and regeneration. Hepatology 2008, 48, 1598–1607. [Google Scholar] [CrossRef]
- Shafritz, D.A.; Dabeva, M.D. Liver stem cells and model systems for liver repopulation. J. Hepatol. 2002, 36, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Meda, C.; Barone, M.; Mitro, N.; Lolli, F.; Pedretti, S.; Caruso, D.; Maggi, A.; Della Torre, S. Hepatic ERalpha accounts for sex differences in the ability to cope with an excess of dietary lipids. Mol. Metab. 2020, 32, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Liby, K.T.; Sporn, M.B. Synthetic oleanane triterpenoids: Multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol. Rev. 2012, 64, 972–1003. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Menke, A.L.; Driessen, A.; Koek, G.H.; Lindeman, J.H.; Stoop, R.; Havekes, L.M.; Kleemann, R.; van den Hoek, A.M. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS ONE 2014, 9, e115922. [Google Scholar] [CrossRef]
- Sapi, J.; Kovacs, L.; Drexler, D.A.; Kocsis, P.; Gajari, D.; Sapi, Z. Tumor Volume Estimation and Quasi-Continuous Administration for Most Effective Bevacizumab Therapy. PLoS ONE 2015, 10, e0142190. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akl, M.G.; Li, L.; Widenmaier, S.B. Protective Effects of Hepatocyte Stress Defenders, Nrf1 and Nrf2, against MASLD Progression. Int. J. Mol. Sci. 2024, 25, 8046. https://doi.org/10.3390/ijms25158046
Akl MG, Li L, Widenmaier SB. Protective Effects of Hepatocyte Stress Defenders, Nrf1 and Nrf2, against MASLD Progression. International Journal of Molecular Sciences. 2024; 25(15):8046. https://doi.org/10.3390/ijms25158046
Chicago/Turabian StyleAkl, May G., Lei Li, and Scott B. Widenmaier. 2024. "Protective Effects of Hepatocyte Stress Defenders, Nrf1 and Nrf2, against MASLD Progression" International Journal of Molecular Sciences 25, no. 15: 8046. https://doi.org/10.3390/ijms25158046