
Unraveling the Impact of miR-146a in Pulmonary Arterial Hypertension Pathophysiology and Right Ventricular Function

 , , , , , ,
, , , , , ,  , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
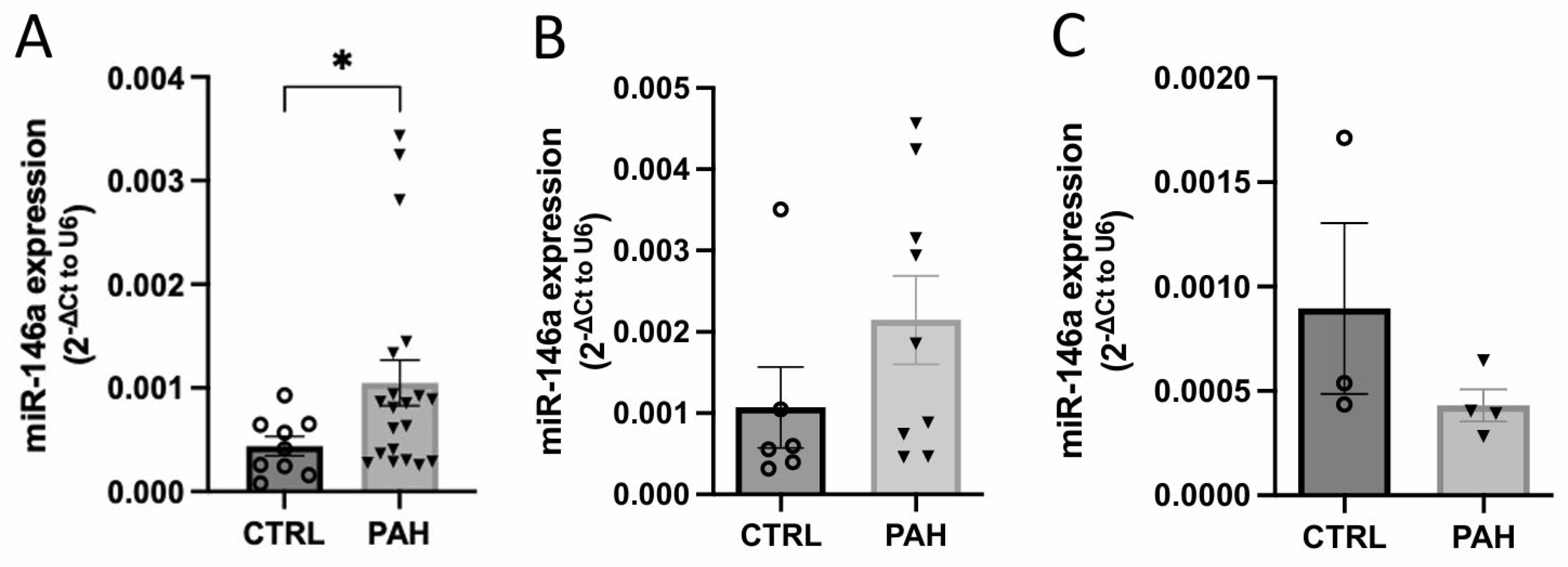
2.1. MiR-146a Expression Is Increased in Human Lung PAH Tissues
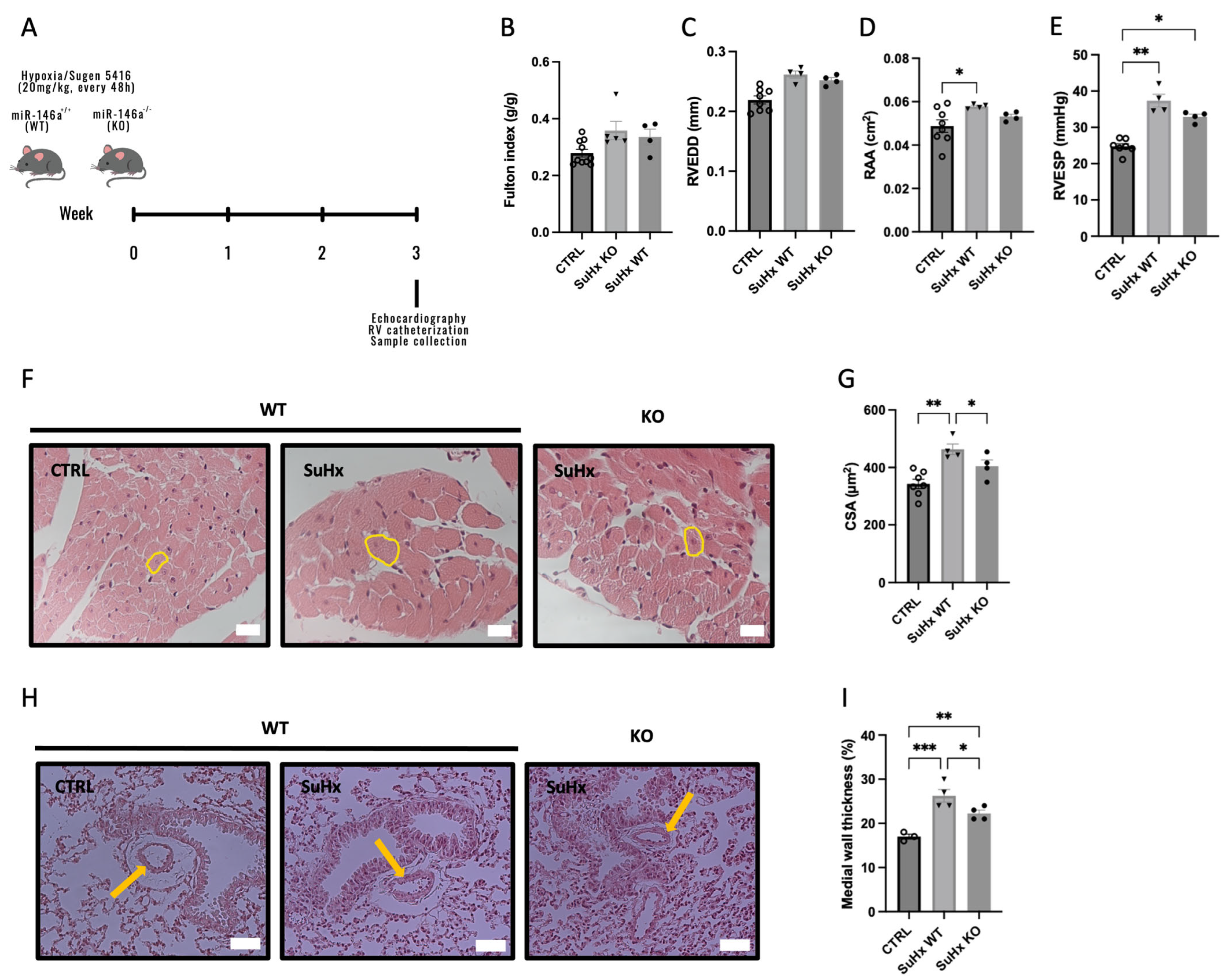
2.2. Knockout (KO) Mice for miR-146a Show Improvements in RV in Response to SuHx
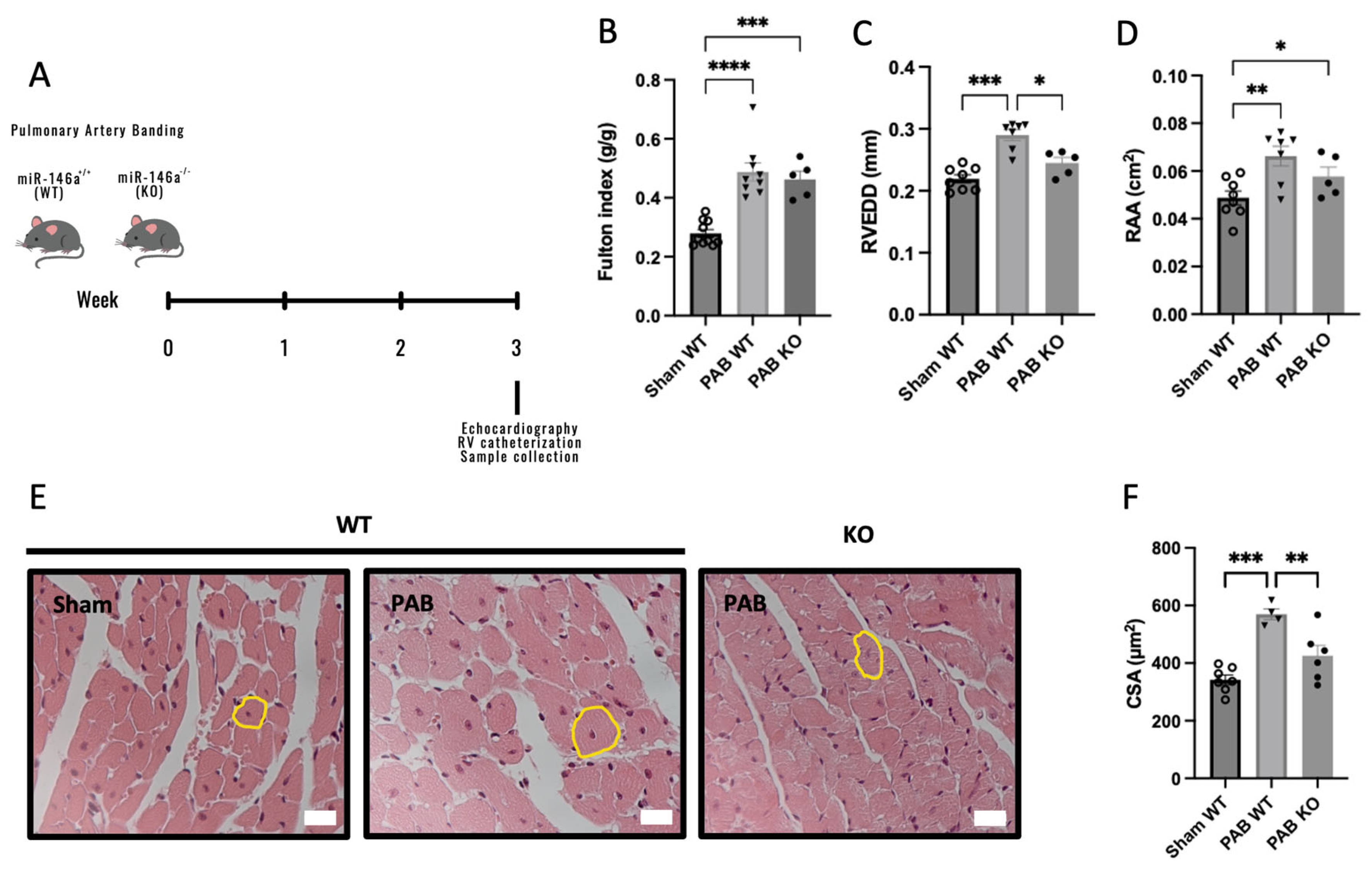
2.3. Knockout (KO) Mice for miR-146a Show Decreased RV Remodeling in Response to Pressure Overload
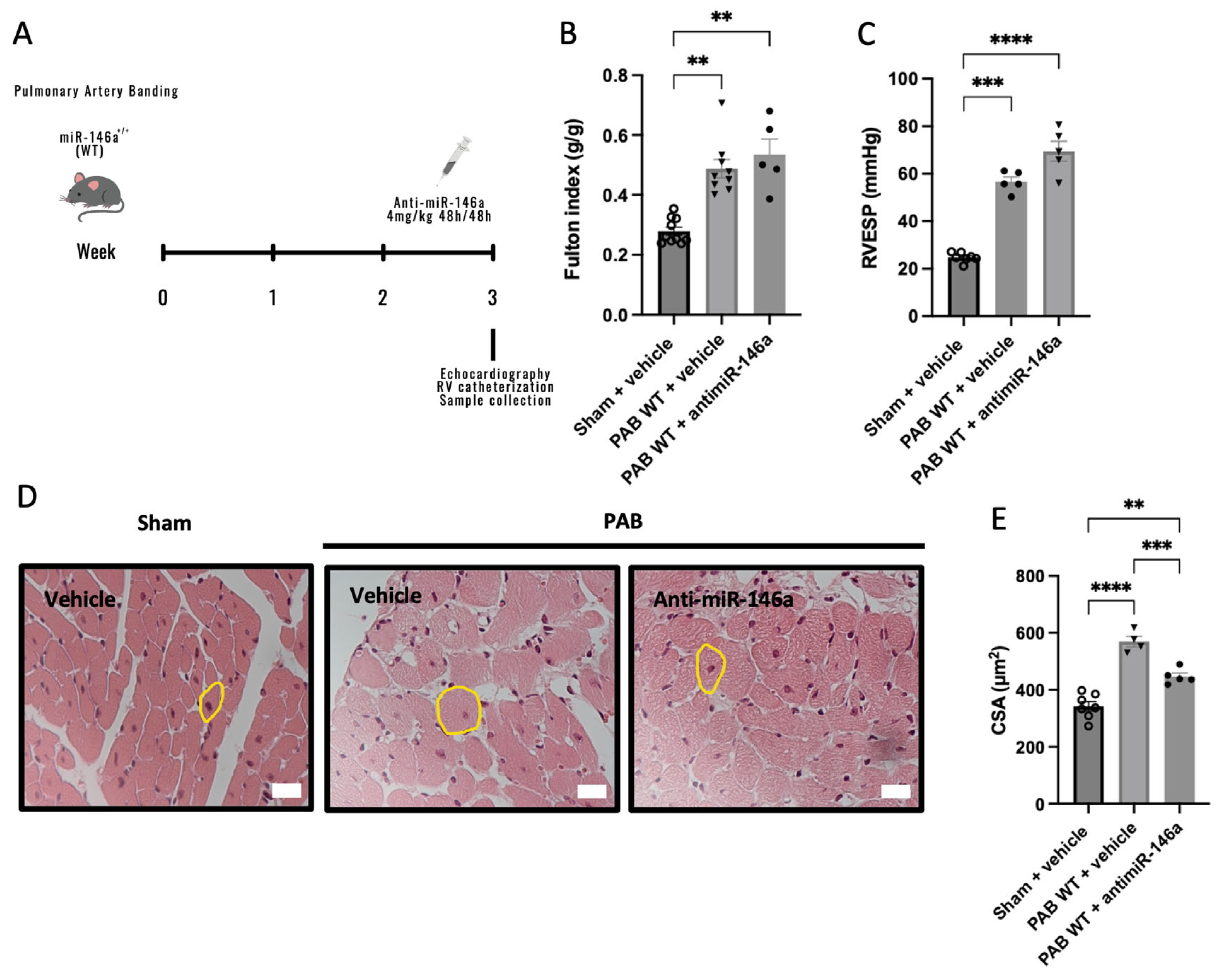
2.4. MiR-146a Inhibition Results in Decreased RV Remodeling in Response to Pressure Overload
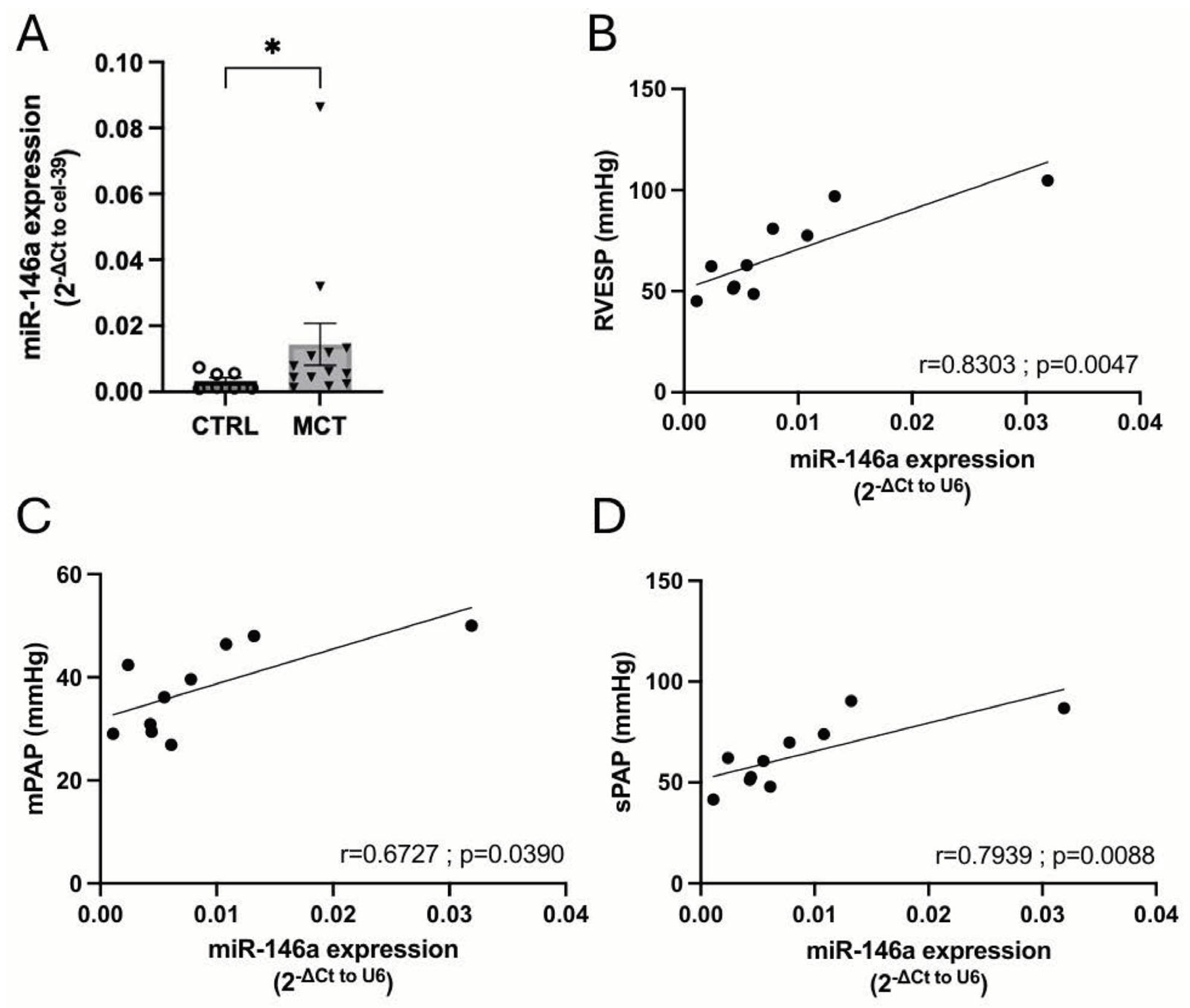
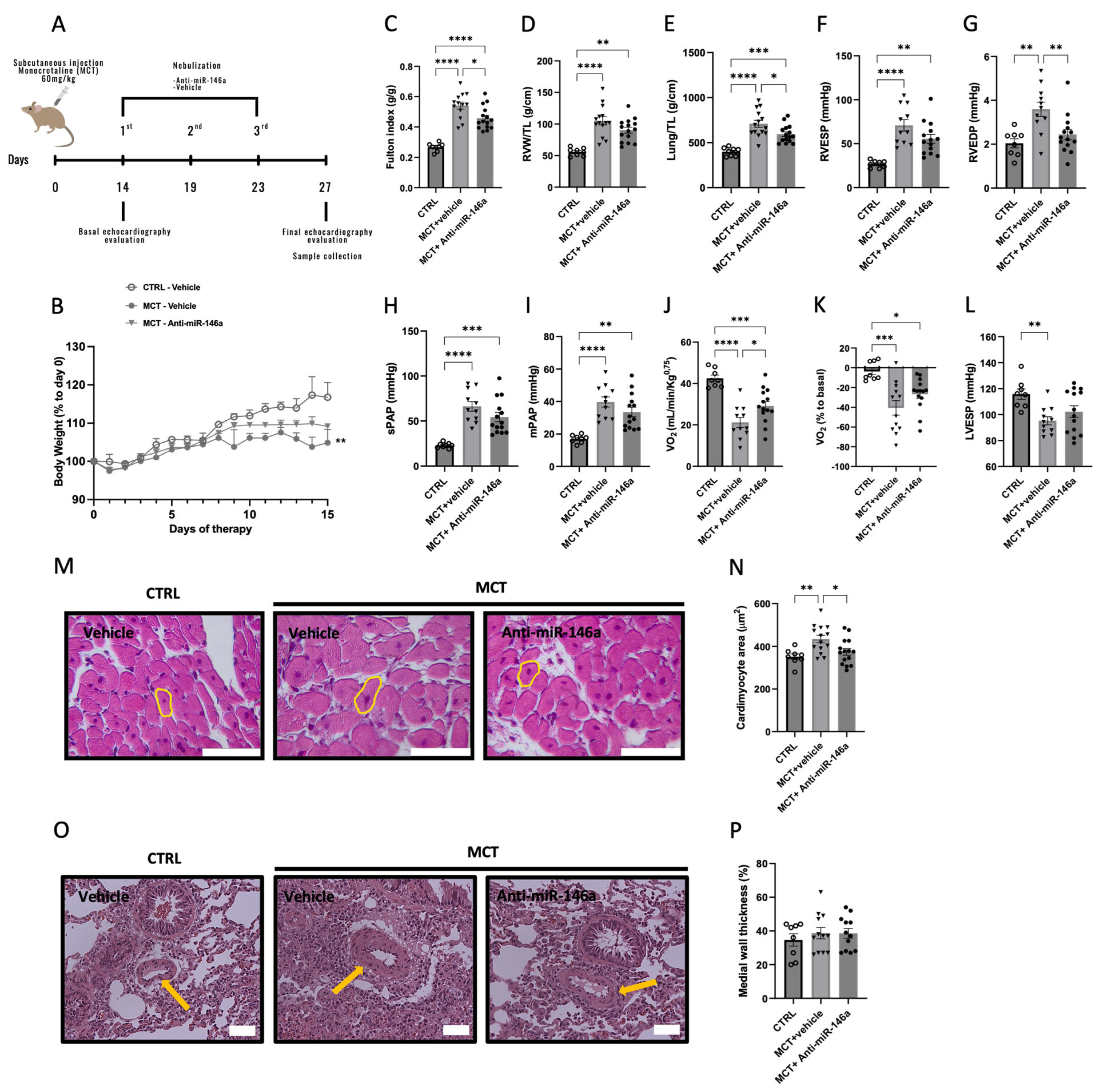
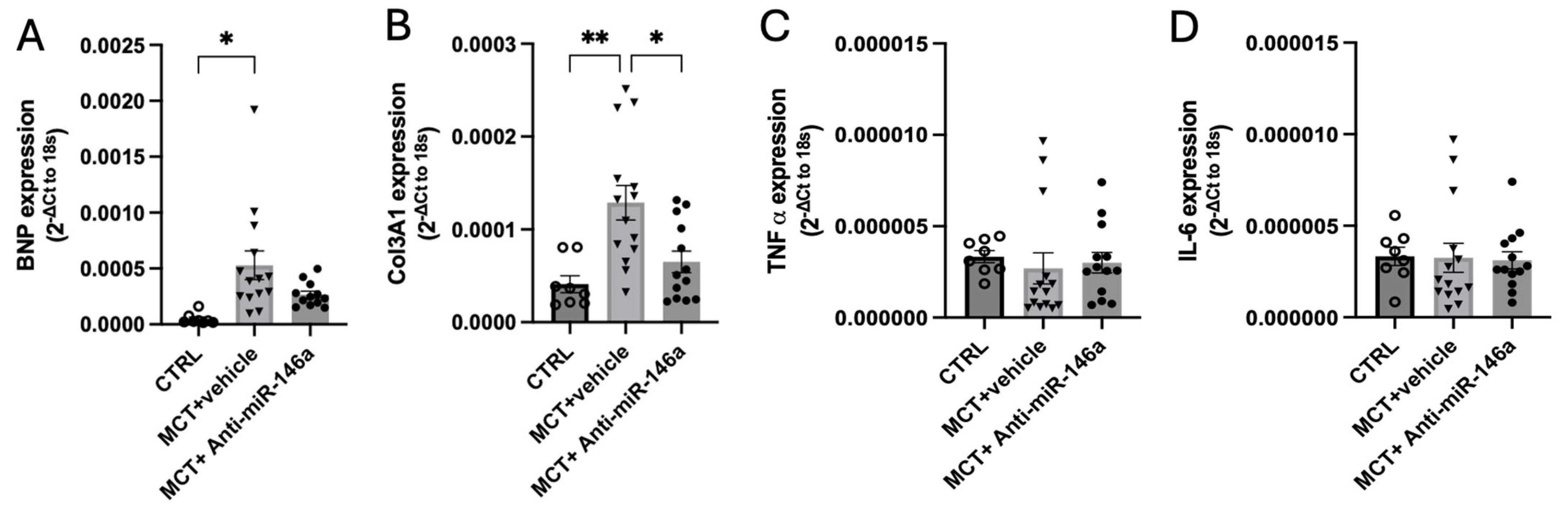
2.5. MiR-146a Inhibition Results in Significant Improvements in the Progression of PAH
2.6. Consequence of miR-146a Overexpression on hPASMCs
3. Discussion
4. Materials and Methods
4.1. Patients and Human Lung Samples
4.2. Animal Models
4.2.1. Sugen 5416/Hypoxia-Induced Pulmonary Hypertension
4.2.2. Pulmonary Artery Banding (PAB)-Induced Right Ventricle Hypertrophy
4.2.3. Monocrotaline (MCT)-Induced Pulmonary Arterial Hypertension
4.3. Echocardiography
4.4. Right Heart Catheterization
4.5. Histological Analysis
4.6. In Vitro
4.6.1. MiR-146a Transfection
4.6.2. hPASMCs Proliferation Measurement
4.6.3. Wound Healing Assay
4.6.4. hPASMCs Induced-Apoptosis Measurement
4.7. Quantitative RT-PCR
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic Definitions and Updated Clinical Classification of Pulmonary Hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension. Eur. Heart J. 2022, 43, 3618–3731. [Google Scholar] [CrossRef] [PubMed]
- Santos-Ribeiro, D.; Mendes-Ferreira, P.; Maia-Rocha, C.; Adão, R.; Leite-Moreira, A.F.; Brás-Silva, C. Pulmonary Arterial Hypertension: Basic Knowledge for Clinicians. Arch. Cardiovasc. Dis. 2016, 109, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.M.T.; Giannoulatou, E.; Celermajer, D.S.; Humbert, M. Epidemiology and Treatment of Pulmonary Arterial Hypertension. Nat. Rev. Cardiol. 2017, 14, 603–614. [Google Scholar] [CrossRef]
- Balsa, A.; Adão, R.; Brás-Silva, C. Therapeutic Approaches in Pulmonary Arterial Hypertension with Beneficial Effects on Right Ventricular Function-Preclinical Studies. Int. J. Mol. Sci. 2023, 24, 15539. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Lau, E.M.T.; Montani, D.; Jaïs, X.; Sitbon, O.; Simonneau, G. Advances in Therapeutic Interventions for Patients with Pulmonary Arterial Hypertension. Circulation 2014, 130, 2189–2208. [Google Scholar] [CrossRef]
- Humbert, M.; Sitbon, O.; Yaïci, A.; Montani, D.; O’Callaghan, D.S.; Jaïs, X.; Parent, F.; Savale, L.; Natali, D.; Günther, S.; et al. Survival in Incident and Prevalent Cohorts of Patients with Pulmonary Arterial Hypertension. Eur. Respir. J. 2010, 36, 549–555. [Google Scholar] [CrossRef]
- Small, E.M.; Olson, E.N. Pervasive Roles of MicroRNAs in Cardiovascular Biology. Nature 2011, 469, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, E.; Bayraktar, R.; Oztatlici, H.; Lopez-Berestein, G.; Amero, P.; Rodriguez-Aguayo, C. Targeting MiRNAs and Other Non-Coding RNAs as a Therapeutic Approach: An Update. Non-Coding RNA 2023, 9, 27. [Google Scholar] [CrossRef]
- Di Leva, G.; Croce, C.M. MiRNA Profiling of Cancer. Curr. Opin. Genet. Dev. 2013, 23, 3–11. [Google Scholar] [CrossRef]
- Kroh, E.M.; Parkin, R.K.; Mitchell, P.S.; Tewari, M. Analysis of Circulating MicroRNA Biomarkers in Plasma and Serum Using Quantitative Reverse Transcription-PCR (QRT-PCR). Methods 2010, 50, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, F.S.; Mardi, S.; Mohammadi, S.; Ansari, S.; Yaslianifard, S.; Fallah, P.; Mozhgani, S.-H. MicroRNA-146: Biomarker and Mediator of Cardiovascular Disease. Dis. Markers 2022, 2022, 7767598. [Google Scholar] [CrossRef]
- Fang, L.; Ellims, A.H.; Moore, X.-L.; White, D.A.; Taylor, A.J.; Chin-Dusting, J.; Dart, A.M. Circulating MicroRNAs as Biomarkers for Diffuse Myocardial Fibrosis in Patients with Hypertrophic Cardiomyopathy. J. Transl. Med. 2015, 13, 314. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, R.; Adão, R.; Leite-Moreira, A.F.; Mâncio, J.; Brás-Silva, C. Candidate MicroRNAs as Prognostic Biomarkers in Heart Failure: A Systematic Review. Rev. Port. Cardiol. 2022, 41, 865–885. [Google Scholar] [CrossRef] [PubMed]
- Boucherat, O.; Potus, F.; Bonnet, S. MicroRNA and Pulmonary Hypertension. Adv. Exp. Med. Biol. 2015, 888, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi-Jahromi, S.S.; Aslani, M.; Mirshafiey, A. A Comprehensive Review on MiR-146a Molecular Mechanisms in a Wide Spectrum of Immune and Non-Immune Inflammatory Diseases. Immunol. Lett. 2020, 227, 8–27. [Google Scholar] [CrossRef]
- Boldin, M.P.; Taganov, K.D.; Rao, D.S.; Yang, L.; Zhao, J.L.; Kalwani, M.; Garcia-Flores, Y.; Luong, M.; Devrekanli, A.; Xu, J.; et al. MiR-146a Is a Significant Brake on Autoimmunity, Myeloproliferation, and Cancer in Mice. J. Exp. Med. 2011, 208, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Horie, T.; Ono, K.; Nishi, H.; Nagao, K.; Kinoshita, M.; Watanabe, S.; Kuwabara, Y.; Nakashima, Y.; Takanabe-Mori, R.; Nishi, E.; et al. Acute Doxorubicin Cardiotoxicity Is Associated with MiR-146a-Induced Inhibition of the Neuregulin-ErbB Pathway. Cardiovasc. Res. 2010, 87, 656–664. [Google Scholar] [CrossRef]
- Heggermont, W.; Carai, P.; Van Aelst, L.; Verhesen, W.; Corsten, M.; Carmliet, P.; Van de Werf, F.; Schroen, B.; Papageorgiou, A.; Heymans, S. Micro-RNA 146a: A New Kid on the Block in the Pathophysiology of Cardiac Hypertrophy and Hypertensive Heart Failure, and a Promising Therapeutic Target. Eur. Heart J. Suppl. 2012, 33, 115. [Google Scholar]
- Oh, J.G.; Watanabe, S.; Lee, A.; Gorski, P.A.; Lee, P.; Jeong, D.; Liang, L.; Liang, Y.; Baccarini, A.; Sahoo, S.; et al. MiR-146a Suppresses SUMO1 Expression and Induces Cardiac Dysfunction in Maladaptive Hypertrophy. Circ. Res. 2018, 123, 673–685. [Google Scholar] [CrossRef]
- Sun, S.G.; Zheng, B.; Han, M.; Fang, X.M.; Li, H.X.; Miao, S.B.; Su, M.; Han, Y.; Shi, H.J.; Wen, J.K. MiR-146a and Krüppel-like Factor 4 Form a Feedback Loop to Participate in Vascular Smooth Muscle Cell Proliferation. EMBO Rep. 2011, 12, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Oerlemans, M.I.F.J.; Mosterd, A.; Dekker, M.S.; de Vrey, E.A.; van Mil, A.; Pasterkamp, G.; Doevendans, P.A.; Hoes, A.W.; Sluijter, J.P.G. Early Assessment of Acute Coronary Syndromes in the Emergency Department: The Potential Diagnostic Value of Circulating MicroRNAs. EMBO Mol. Med. 2012, 4, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.I.; Bogaard, H.J.; Mizuno, S.; Clifton, B.; Xie, B.; Gao, Y.; Dumur, C.I.; Fawcett, P.; Voelkel, N.F.; Natarajan, R. Molecular Signature of a Right Heart Failure Program in Chronic Severe Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 Controls Mitochondrial Metabolism during Hypoxia by Repressing the Iron-Sulfur Cluster Assembly Proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Kheyfets, V.O.; Sucharov, C.C.; Truong, U.; Dunning, J.; Hunter, K.; Ivy, D.; Miyamoto, S.; Shandas, R. Circulating MiRNAs in Pediatric Pulmonary Hypertension Show Promise as Biomarkers of Vascular Function. Oxid. Med. Cell. Longev. 2017, 2017, 4957147. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Chen, S.; McArthur, K.; Wu, Y.; Sen, S.; Ding, Q.; Feldman, R.D.; Chakrabarti, S. MiR-146a-Mediated Extracellular Matrix Protein Production in Chronic Diabetes Complications. Diabetes 2011, 60, 2975–2984. [Google Scholar] [CrossRef]
- Kookli, K.; Soleimani, K.T.; Amr, E.F.; Ehymayed, H.M.; Zabibah, R.S.; Daminova, S.B.; Saadh, M.J.; Alsaikhan, F.; Adil, M.; Ali, M.S.; et al. Role of MicroRNA-146a in Cancer Development by Regulating Apoptosis. Pathol. Res. Pract. 2024, 254, 155050. [Google Scholar] [CrossRef]
- Mamazhakypov, A.; Sommer, N.; Assmus, B.; Tello, K.; Schermuly, R.T.; Kosanovic, D.; Sarybaev, A.S.; Weissmann, N.; Pak, O. Novel Therapeutic Targets for the Treatment of Right Ventricular Remodeling: Insights from the Pulmonary Artery Banding Model. Int. J. Environ. Res. Public Health 2021, 18, 8297. [Google Scholar] [CrossRef]
- Boucherat, O.; Agrawal, V.; Lawrie, A.; Bonnet, S. The Latest in Animal Models of Pulmonary Hypertension and Right Ventricular Failure. Circ. Res. 2022, 130, 1466–1486. [Google Scholar] [CrossRef] [PubMed]
- Rafikova, O.; James, J.; Eccles, C.A.; Kurdyukov, S.; Niihori, M.; Varghese, M.V.; Rafikov, R. Early Progression of Pulmonary Hypertension in the Monocrotaline Model in Males Is Associated with Increased Lung Permeability. Biol. Sex. Differ. 2020, 11, 11. [Google Scholar] [CrossRef]
- Sydykov, A.; Luitel, H.; Mamazhakypov, A.; Wygrecka, M.; Pradhan, K.; Pak, O.; Petrovic, A.; Kojonazarov, B.; Weissmann, N.; Seeger, W.; et al. Genetic Deficiency and Pharmacological Stabilization of Mast Cells Ameliorate Pressure Overload-Induced Maladaptive Right Ventricular Remodeling in Mice. Int. J. Mol. Sci. 2020, 21, 9099. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arroyo, J.G.; Farkas, L.; Alhussaini, A.A.; Farkas, D.; Kraskauskas, D.; Voelkel, N.F.; Bogaard, H.J. The Monocrotaline Model of Pulmonary Hypertension in Perspective. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L363–L369. [Google Scholar] [CrossRef] [PubMed]
- Hye, T.; Hossain, M.R.; Saha, D.; Foyez, T.; Ahsan, F. Emerging Biologics for the Treatment of Pulmonary Arterial Hypertension. J. Drug Target. 2023, 31, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, X.; Li, T.; Wang, L.; Wu, X.; Liu, J.; Xu, Y.; Wei, W. Multiple Roles of Microrna-146a in Immune Responses and Hepatocellular Carcinoma (Review). Oncol. Lett. 2019, 18, 5033–5042. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Frid, M.G.; Yeager, M.; Li, M.; Riddle, S.; McKinsey, T.; El Kasmi, K.C. Targeting the Adventitial Microenvironment in Pulmonary Hypertension: A Potential Approach to Therapy That Considers Epigenetic Change. Pulm. Circ. 2012, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.S.; Preston, I.R.; Roberts, K.E. Inhaled Therapies for Pulmonary Hypertension. Respir. Care 2015, 60, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.W.; Chang, K.H.; Woo, C.J.; Kim, H.C.; Kwak, B.S.; Park, B.J.; Nam, K.C. Evaluation of Antibody Drug Delivery Efficiency via Nebulizer in Various Airway Models and Breathing Patterns. BMC Pharmacol. Toxicol. 2023, 24, 70. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.H.; Ma, J.L.; Ding, D.; Ma, Y.J.; Wei, Y.P.; Jing, Z.C. Experimental Animal Models of Pulmonary Hypertension: Development and Challenges. Animal Model. Exp. Med. 2022, 5, 207–216. [Google Scholar] [CrossRef]
- Heuslein, J.L.; McDonnell, S.P.; Song, J.; Annex, B.H.; Price, R.J. MicroRNA-146a Regulates Perfusion Recovery in Response to Arterial Occlusion via Arteriogenesis. Front. Bioeng. Biotechnol. 2018, 6, 325999. [Google Scholar] [CrossRef]
- Ntelios, D.; Efthimiadis, G.; Zegkos, T.; Didagelos, M.; Katopodi, T.; Meditskou, S.; Parcharidou, D.; Karvounis, H.; Tzimagiorgis, G. Correlation of MiR-146a-5p Plasma Levels and Rs2910164 Polymorphism with Left Ventricle Outflow Tract Obstruction in Hypertrophic Cardiomyopathy. Hell. J. Cardiol. 2021, 62, 349–354. [Google Scholar] [CrossRef]
- Heggermont, W.A.; Papageorgiou, A.P.; Quaegebeur, A.; Deckx, S.; Carai, P.; Verhesen, W.; Eelen, G.; Schoors, S.; Van Leeuwen, R.; Alekseev, S.; et al. Inhibition of MicroRNA-146a and Overexpression of Its Target Dihydrolipoyl Succinyltransferase Protect Against Pressure Overload-Induced Cardiac Hypertrophy and Dysfunction. Circulation 2017, 136, 747–761. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Li, C.; Zhang, H.; Qiu, S.; Fu, T.; Xu, Y. Downregulation of MiR-146a Contributes to Cardiac Dysfunction Induced by the Tyrosine Kinase Inhibitor Sunitinib. Front. Pharmacol. 2019, 10, 914. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Lu, Y.; Song, X.; Gong, X.; Li, Y. Inhibition of MicroRNA-146a Attenuated Heart Failure in Myocardial Infarction Rats. Biosci. Rep. 2019, 39, BSR20191732. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-KappaB-Dependent Induction of MicroRNA MiR-146, an Inhibitor Targeted to Signaling Proteins of Innate Immune Responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed]
- Tahamtan, A.; Teymoori-Rad, M.; Nakstad, B.; Salimi, V. Anti-Inflammatory MicroRNAs and Their Potential for Inflammatory Diseases Treatment. Front. Immunol. 2018, 9, 1377. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Mehta, A.; Zhao, J.L.; Lee, K.; Marinov, G.K.; Garcia-Flores, Y.; Baltimore, D. An NF-ΚB-MicroRNA Regulatory Network Tunes Macrophage Inflammatory Responses. Nat. Commun. 2017, 8, 851. [Google Scholar] [CrossRef]
- Wang, X.P.; Luoreng, Z.M.; Zan, L.-S.; Li, F.; Li, N. Bovine MiR-146a Regulates Inflammatory Cytokines of Bovine Mammary Epithelial Cells via Targeting the TRAF6 Gene. J. Dairy. Sci. 2017, 100, 7648–7658. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Zheng, R.; Shao, G. Mechanisms and Application Strategies of MiRNA-146a Regulating Inflammation and Fibrosis at Molecular and Cellular Levels (Review). Int. J. Mol. Med. 2023, 51, 7. [Google Scholar] [CrossRef]
- Hou, J.; Deng, Q.; Deng, X.; Zhong, W.; Liu, S.; Zhong, Z. MicroRNA-146a-5p Alleviates Lipopolysaccharide-Induced NLRP3 Inflammasome Injury and pro-Inflammatory Cytokine Production via the Regulation of TRAF6 and IRAK1 in Human Umbilical Vein Endothelial Cells (HUVECs). Ann. Transl. Med. 2021, 9, 1433. [Google Scholar] [CrossRef]
- Jang, S.Y.; Chae, M.K.; Lee, J.H.; Lee, E.J.; Yoon, J.S. Role of MiR-146a in the Regulation of Inflammation in an In Vitro Model of Graves’ Orbitopathy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4027–4034. [Google Scholar] [CrossRef]
- Bhargava, A.; Kumar, A.; Yuan, N.; Gewitz, M.H.; Mathew, R. Monocrotaline Induces Interleukin-6 MRNA Expression in Rat Lungs. Heart Dis. 1999, 1, 126–132. [Google Scholar]
- Pan, Q.; Liu, H.; Zheng, C.; Zhao, Y.; Liao, X.; Wang, Y.; Chen, Y.; Zhao, B.; Lazartigues, E.; Yang, Y.; et al. Microvesicles Derived from Inflammation-Challenged Endothelial Cells Modulate Vascular Smooth Muscle Cell Functions. Front. Physiol. 2017, 7, 692. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Xiong, W.; Yuan, J.; Li, J.; Liu, J.; Xu, X. MiRNA-146a Regulates the Maturation and Differentiation of Vascular Smooth Muscle Cells by Targeting NF-ΚB Expression. Mol. Med. Rep. 2013, 8, 407–412. [Google Scholar] [CrossRef]
- Thenappan, T.; Chan, S.Y.; Weir, E.K. Role of Extracellular Matrix in the Pathogenesis of Pulmonary Arterial Hypertension. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1322–H1331. [Google Scholar] [CrossRef]
- Cheng, H.S.; Sivachandran, N.; Lau, A.; Boudreau, E.; Zhao, J.L.; Baltimore, D.; Delgado-Olguin, P.; Cybulsky, M.I.; Fish, J.E. MicroRNA-146 Represses Endothelial Activation by Inhibiting pro-Inflammatory Pathways. EMBO Mol. Med. 2013, 5, 1017–1034. [Google Scholar] [CrossRef]
- Ma, S.; Tian, X.Y.; Zhang, Y.; Mu, C.; Shen, H.; Bismuth, J.; Pownall, H.J.; Huang, Y.; Wong, W.T. E-Selectin-Targeting Delivery of MicroRNAs by Microparticles Ameliorates Endothelial Inflammation and Atherosclerosis. Sci. Rep. 2016, 6, 22910. [Google Scholar] [CrossRef]
- Sánchez-Gloria, J.L.; Carbó, R.; Buelna-Chontal, M.; Osorio-Alonso, H.; Henández-Díazcouder, A.; de la Fuente-León, R.L.; Sandoval, J.; Sánchez, F.; Rubio-Gayosso, I.; Sánchez-Muñoz, F. Cold Exposure Aggravates Pulmonary Arterial Hypertension through Increased MiR-146a-5p, MiR-155-5p and Cytokines TNF-α, IL-1β, and IL-6. Life Sci. 2021, 287, 120091. [Google Scholar] [CrossRef]
- Qiu, M.; Li, T.; Wang, B.; Gong, H.; Huang, T. MiR-146a-5p Regulated Cell Proliferation and Apoptosis by Targeting SMAD3 and SMAD4. Protein Pept. Lett. 2020, 27, 411–418. [Google Scholar] [CrossRef]
- Zabini, D.; Granton, E.; Hu, Y.; Miranda, M.Z.; Weichelt, U.; Bonnet, S.B.; Bonnet, S.; Morrell, N.W.; Connelly, K.A.; Provencher, S.; et al. Loss of SMAD3 Promotes Vascular Remodeling in Pulmonary Arterial Hypertension via MRTF Disinhibition. Am. J. Respir. Crit. Care Med. 2018, 197, 244–260. [Google Scholar] [CrossRef]
- Shi, L.; Kojonazarov, B.; Elgheznawy, A.; Popp, R.; Dahal, B.K.; Böhm, M.; Pullamsetti, S.S.; Ghofrani, H.A.; Gödecke, A.; Jungmann, A.; et al. MiR-223-IGF-IR Signalling in Hypoxia- and Load-Induced Right-Ventricular Failure: A Novel Therapeutic Approach. Cardiovasc. Res. 2016, 111, 184–193. [Google Scholar] [CrossRef]
- Mendes-Ferreira, P.; Santos-Ribeiro, D.; Adão, R.; Maia-Rocha, C.; Mendes-Ferreira, M.; Sousa-Mendes, C.; Leite-Moreira, A.F.; Brás-Silva, C. Distinct Right Ventricle Remodeling in Response to Pressure Overload in the Rat. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H85–H95. [Google Scholar] [CrossRef] [PubMed]
- Adão, R.; Mendes-Ferreira, P.; Maia-Rocha, C.; Santos-Ribeiro, D.; Rodrigues, P.G.; Vidal-Meireles, A.; Monteiro-Pinto, C.; Pimentel, L.D.; Falcão-Pires, I.; De Keulenaer, G.W.; et al. Neuregulin-1 Attenuates Right Ventricular Diastolic Stiffness in Experimental Pulmonary Hypertension. Clin. Exp. Pharmacol. Physiol. 2019, 46, 255–265. [Google Scholar] [CrossRef]
- Adão, R.; Mendes-Ferreira, P.; Santos-Ribeiro, D.; Maia-Rocha, C.; Pimentel, L.D.; Monteiro-Pinto, C.; Mulvaney, E.P.; Reid, H.M.; Kinsella, B.T.; Potus, F.; et al. Urocortin-2 Improves Right Ventricular Function and Attenuates Pulmonary Arterial Hypertension. Cardiovasc. Res. 2018, 114, 1165–1177. [Google Scholar] [CrossRef]
- Mendes-Ferreira, P.; Maia-Rocha, C.; Adão, R.; Mendes, M.J.; Santos-Ribeiro, D.; Alves, B.S.; Cerqueira, R.J.; Castro-Chaves, P.; Lourenço, A.P.; De Keulenaer, G.W.; et al. Neuregulin-1 Improves Right Ventricular Function and Attenuates Experimental Pulmonary Arterial Hypertension. Cardiovasc. Res. 2016, 109, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Sankhe, S.; Manousakidi, S.; Antigny, F.; Arthur Ataam, J.; Bentebbal, S.; Ruchon, Y.; Lecerf, F.; Sabourin, J.; Price, L.; Fadel, E.; et al. T-Type Ca2+ Channels Elicit pro-Proliferative and Anti-Apoptotic Responses through Impaired PP2A/Akt1 Signaling in PASMCs from Patients with Pulmonary Arterial Hypertension. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1631–1641. [Google Scholar] [CrossRef]
- Lambert, M.; Capuano, V.; Boet, A.; Tesson, L.; Bertero, T.; Nakhleh, M.K.; Remy, S.; Anegon, I.; Pechoux, C.; Hautefort, A.; et al. Characterization of Kcnk3-Mutated Rat, a Novel Model of Pulmonary Hypertension. Circ. Res. 2019, 125, 678–695. [Google Scholar] [CrossRef]
- Le Ribeuz, H.; Masson, B.; Capuano, V.; Dutheil, M.; Gooroochurn, H.; Boet, A.; Ghigna, M.R.; De Montpreville, V.; Girerd, B.; Lambert, M.; et al. SUR1 As a New Therapeutic Target for Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2022, 66, 539–554. [Google Scholar] [CrossRef]
- Le Ribeuz, H.; Masson, B.; Dutheil, M.; Boët, A.; Beauvais, A.; Sabourin, J.; De Montpreville, V.T.; Capuano, V.; Mercier, O.; Humbert, M.; et al. Involvement of SUR2/Kir6.1 Channel in the Physiopathology of Pulmonary Arterial Hypertension. Front. Cardiovasc. Med. 2023, 9, 1066047. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos-Gomes, J.; Mendes-Ferreira, P.; Adão, R.; Maia-Rocha, C.; Rego, B.; Poels, M.; Saint-Martin Willer, A.; Masson, B.; Provencher, S.; Bonnet, S.; et al. Unraveling the Impact of miR-146a in Pulmonary Arterial Hypertension Pathophysiology and Right Ventricular Function. Int. J. Mol. Sci. 2024, 25, 8054. https://doi.org/10.3390/ijms25158054
Santos-Gomes J, Mendes-Ferreira P, Adão R, Maia-Rocha C, Rego B, Poels M, Saint-Martin Willer A, Masson B, Provencher S, Bonnet S, et al. Unraveling the Impact of miR-146a in Pulmonary Arterial Hypertension Pathophysiology and Right Ventricular Function. International Journal of Molecular Sciences. 2024; 25(15):8054. https://doi.org/10.3390/ijms25158054
Chicago/Turabian StyleSantos-Gomes, Joana, Pedro Mendes-Ferreira, Rui Adão, Carolina Maia-Rocha, Beatriz Rego, Manu Poels, Anaïs Saint-Martin Willer, Bastien Masson, Steeve Provencher, Sébastien Bonnet, and et al. 2024. "Unraveling the Impact of miR-146a in Pulmonary Arterial Hypertension Pathophysiology and Right Ventricular Function" International Journal of Molecular Sciences 25, no. 15: 8054. https://doi.org/10.3390/ijms25158054