
An In Vivo Model of Propionic Acid-Rich Diet-Induced Gliosis and Neuro-Inflammation in Mice (FVB/N-Tg(GFAPGFP)14Mes/J): A Potential Link to Autism Spectrum Disorder



Abstract
:1. Introduction
2. Results
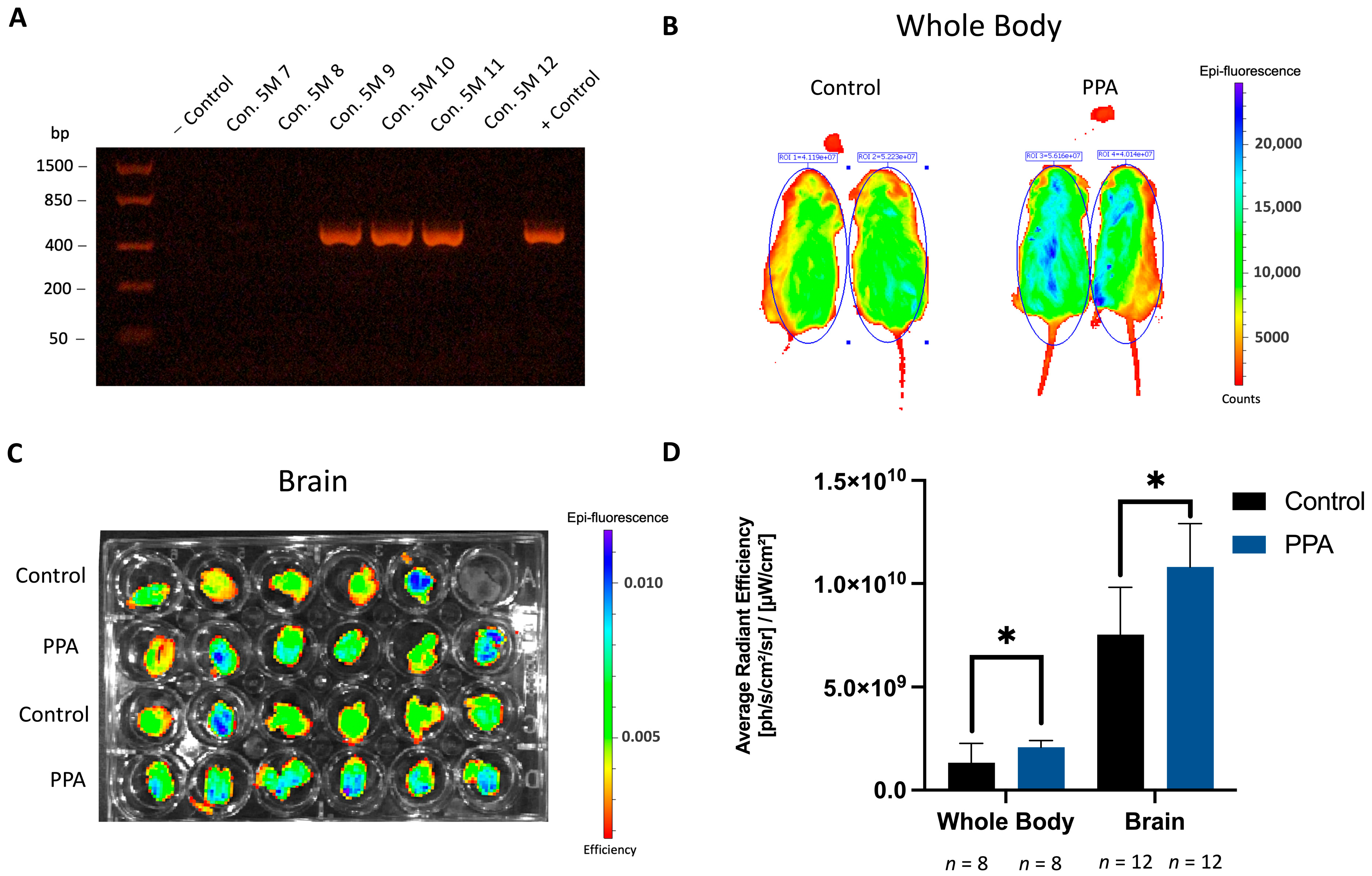
2.1. Fluorescence from the GFAP-GFP Construct Is Significantly Elevated in the PPA-Rich Diet Group
2.2. GFAP Is Significantly Elevated in the Brain of Animals in the PPA-Rich Diet Group While Tubulin III β Is Decreased
2.3. A PPA-Rich Diet Correlates with Elevated GPR41 and Phosphorylated Akt and Decreased PTEN Expression and Protein Concentration in Offspring Brains
2.4. A Maternal PPA-Rich Diet and Post-Natal Exposure Increases Expression of Offspring Pro-Inflammatory Cytokines and Decreases Expression of Anti-Inflammatory Cytokines
3. Discussion
4. Materials and Methods
4.1. Animals and Diet Establishment
4.2. Genotyping Using Touchdown PCR to Detect the GFAP/GFP Construct
4.3. In Vivo and Ex Vivo IVIS Imaging
4.4. Gene Expression
4.5. Protein Levels
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lagod, P.P.; Naser, S.A. The Role of Short-Chain Fatty Acids and Altered Microbiota Composition in Autism Spectrum Disorder: A Comprehensive Literature Review. Int. J. Mol. Sci. 2023, 24, 17432. [Google Scholar] [CrossRef]
- Vahia, V.N. Diagnostic and statistical manual of mental disorders 5: A quick glance. Indian. J. Psychiatry 2013, 55, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Maenner, M.J.; Shaw, K.A.; Bakian, A.V.; Bilder, D.A.; Durkin, M.S.; Esler, A.; Furnier, S.M.; Hallas, L.; Hall-Lande, J.; Hudson, A.; et al. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2018. MMWR Surveill. Summ. 2021, 70, 1–16. [Google Scholar] [CrossRef]
- Abdelli, L.S.; Samsam, A.; Naser, S.A. Propionic Acid Induces Gliosis and Neuro-inflammation through Modulation of PTEN/AKT Pathway in Autism Spectrum Disorder. Sci. Rep. 2019, 9, 8824. [Google Scholar] [CrossRef] [PubMed]
- Towairqi, W.A.; Alosaimi, W.; Zaidi, S.A.; Helmy, F.F.; Sherif, E.A. Depression among mothers of autistic spectral disorder children. Int. J. Contemp. Pediatr. 2015, 2, 119–126. [Google Scholar] [CrossRef]
- Gilbert, J.; Man, H.Y. Fundamental Elements in Autism: From Neurogenesis and Neurite Growth to Synaptic Plasticity. Front. Cell Neurosci. 2017, 11, 359. [Google Scholar] [CrossRef]
- Tsai, C.H.; Chen, K.L.; Li, H.J.; Chen, K.H.; Hsu, C.W.; Lu, C.H.; Hsieh, K.Y.; Huang, C.Y. The symptoms of autism including social communication deficits and repetitive and restricted behaviors are associated with different emotional and behavioral problems. Sci. Rep. 2020, 10, 20509. [Google Scholar] [CrossRef]
- Maenner, M.J.; Warren, Z.; Williams, A.R.; Amoakohene, E.; Bakian, A.V.; Bilder, D.A.; Durkin, M.S.; Fitzgerald, R.T.; Furnier, S.M.; Hughes, M.M.; et al. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2020. MMWR Surveill. Summ. 2023, 72, 1–14. [Google Scholar] [CrossRef]
- Reichow, B.; Barton, E.E.; Boyd, B.A.; Hume, K. Early intensive behavioral intervention (EIBI) for young children with autism spectrum disorders (ASD). Cochrane Database Syst. Rev. 2012, 10, Cd009260. [Google Scholar] [CrossRef]
- Weitlauf, A.S.; McPheeters, M.L.; Peters, B.; Sathe, N.; Travis, R.; Aiello, R.; Williamson, E.; Veenstra-VanderWeele, J.; Krishnaswami, S.; Jerome, R.; et al. AHRQ Comparative Effectiveness Reviews. In Therapies for Children with Autism Spectrum Disorder: Behavioral Interventions Update; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2014. [Google Scholar]
- Frye, R.E.; Nankova, B.; Bhattacharyya, S.; Rose, S.; Bennuri, S.C.; MacFabe, D.F. Modulation of Immunological Pathways in Autistic and Neurotypical Lymphoblastoid Cell Lines by the Enteric Microbiome Metabolite Propionic Acid. Front. Immunol. 2017, 8, 1670. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. A review of research trends in physiological abnormalities in autism spectrum disorders: Immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol. Psychiatry 2012, 17, 389–401. [Google Scholar] [CrossRef] [PubMed]
- El-Ansary, A.; Bhat, R.S.; Al-Daihan, S.; Al Dbass, A.M. The neurotoxic effects of ampicillin-associated gut bacterial imbalances compared to those of orally administered propionic acid in the etiology of persistent autistic features in rat pups: Effects of various dietary regimens. Gut Pathog. 2015, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Yenkoyan, K.; Mkhitaryan, M.; Bjørklund, G. Environmental Risk Factors in Autism Spectrum Disorder: A Narrative Review. Curr. Med. Chem. 2024, 31, 2345–2360. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Gong, X.; Hu, B.; Lin, L.; Lin, X.; Gong, W.; Zhang, B.; Cao, M.; Xu, Y.; Xia, R.; et al. Altered Gut Microbiota and Short-chain Fatty Acids in Chinese Children with Constipated Autism Spectrum Disorder. Sci. Rep. 2023, 13, 19103. [Google Scholar] [CrossRef] [PubMed]
- Coretti, L.; Paparo, L.; Riccio, M.P.; Amato, F.; Cuomo, M.; Natale, A.; Borrelli, L.; Corrado, G.; Comegna, M.; Buommino, E.; et al. Gut Microbiota Features in Young Children with Autism Spectrum Disorders. Front. Microbiol. 2018, 9, 3146. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.; Piccolo, M.; Vannini, L.; Siragusa, S.; De Giacomo, A.; Serrazzanetti, D.I.; Cristofori, F.; Guerzoni, M.E.; Gobbetti, M.; Francavilla, R. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS ONE 2013, 8, e76993. [Google Scholar] [CrossRef] [PubMed]
- Mehra, A.; Arora, G.; Sahni, G.; Kaur, M.; Singh, H.; Singh, B.; Kaur, S. Gut microbiota and Autism Spectrum Disorder: From pathogenesis to potential therapeutic perspectives. J. Tradit. Complement. Med. 2022, 13, 135–149. [Google Scholar] [CrossRef]
- Strati, F.; Cavalieri, D.; Albanese, D.; De Felice, C.; Donati, C.; Hayek, J.; Jousson, O.; Leoncini, S.; Renzi, D.; Calabrò, A.; et al. New evidences on the altered gut microbiota in autism spectrum disorders. Microbiome 2017, 5, 24. [Google Scholar] [CrossRef]
- Finegold, S.M. State of the art; microbiology in health and disease. Intestinal bacterial flora in autism. Anaerobe 2011, 17, 367–368. [Google Scholar] [CrossRef]
- Li, Q.; Han, Y.; Dy, A.B.C.; Hagerman, R.J. The Gut Microbiota and Autism Spectrum Disorders. Front. Cell Neurosci. 2017, 11, 120. [Google Scholar] [CrossRef]
- Macfabe, D.F. Short-chain fatty acid fermentation products of the gut microbiome: Implications in autism spectrum disorders. Microb. Ecol. Health Dis. 2012, 23, 19260. [Google Scholar] [CrossRef] [PubMed]
- David, M.M.; Tataru, C.; Daniels, J.; Schwartz, J.; Keating, J.; Hampton-Marcell, J.; Gottel, N.; Gilbert, J.A.; Wall, D.P. Children with Autism and Their Typically Developing Siblings Differ in Amplicon Sequence Variants and Predicted Functions of Stool-Associated Microbes. mSystems 2021, 6, e00193-20. [Google Scholar] [CrossRef] [PubMed]
- Jendraszak, M.; Gałęcka, M.; Kotwicka, M.; Regdos, A.; Pazgrat-Patan, M.; Andrusiewicz, M. Commercial microbiota test revealed differences in the composition of intestinal microorganisms between children with autism spectrum disorders and neurotypical peers. Sci. Rep. 2021, 11, 24274. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Food additives and Nutrient Sources added to Food (ANS). Scientific Opinion on the re-evaluation of propionic acid (E 280), sodium propionate (E 281), calcium propionate (E 282) and potassium propionate (E 283) as food additives. EFSA J. 2014, 12, 3779. [Google Scholar] [CrossRef]
- Rahman, M.; Islam, R.; Hasan, S.; Zzaman, W.; Rana, M.R.; Ahmed, S.; Roy, M.; Sayem, A.; Matin, A.; Raposo, A.; et al. A Comprehensive Review on Bio-Preservation of Bread: An Approach to Adopt Wholesome Strategies. Foods 2022, 11, 319. [Google Scholar] [CrossRef]
- Meeking, M.M.; MacFabe, D.F.; Mepham, J.R.; Foley, K.A.; Tichenoff, L.J.; Boon, F.H.; Kavaliers, M.; Ossenkopp, K.P. Propionic acid induced behavioural effects of relevance to autism spectrum disorder evaluated in the hole board test with rats. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 97, 109794. [Google Scholar] [CrossRef]
- Witters, P.; Debbold, E.; Crivelly, K.; Vande Kerckhove, K.; Corthouts, K.; Debbold, B.; Andersson, H.; Vannieuwenborg, L.; Geuens, S.; Baumgartner, M.; et al. Autism in patients with propionic acidemia. Mol. Genet. Metab. 2016, 119, 317–321. [Google Scholar] [CrossRef]
- Forny, P.; Hörster, F.; Ballhausen, D.; Chakrapani, A.; Chapman, K.A.; Dionisi-Vici, C.; Dixon, M.; Grünert, S.C.; Grunewald, S.; Haliloglu, G.; et al. Guidelines for the diagnosis and management of methylmalonic acidaemia and propionic acidaemia: First revision. J. Inherit. Metab. Dis. 2021, 44, 566–592. [Google Scholar] [CrossRef] [PubMed]
- Tajima, G.; Kagawa, R.; Sakura, F.; Nakamura-Utsunomiya, A.; Hara, K.; Yuasa, M.; Hasegawa, Y.; Sasai, H.; Okada, S. Current Perspectives on Neonatal Screening for Propionic Acidemia in Japan: An Unexpectedly High Incidence of Patients with Mild Disease Caused by a Common PCCB Variant. Int. J. Neonatal Screen. 2021, 7, 35. [Google Scholar] [CrossRef]
- Shultz, S.R.; MacFabe, D.F.; Ossenkopp, K.P.; Scratch, S.; Whelan, J.; Taylor, R.; Cain, D.P. Intracerebroventricular injection of propionic acid, an enteric bacterial metabolic end-product, impairs social behavior in the rat: Implications for an animal model of autism. Neuropharmacology 2008, 54, 901–911. [Google Scholar] [CrossRef]
- Foley, K.A.; Ossenkopp, K.P.; Kavaliers, M.; Macfabe, D.F. Pre- and neonatal exposure to lipopolysaccharide or the enteric metabolite, propionic acid, alters development and behavior in adolescent rats in a sexually dimorphic manner. PLoS ONE 2014, 9, e87072. [Google Scholar] [CrossRef] [PubMed]
- Lobzhanidze, G.; Japaridze, N.; Lordkipanidze, T.; Rzayev, F.; MacFabe, D.; Zhvania, M. Behavioural and brain ultrastructural changes following the systemic administration of propionic acid in adolescent male rats. Further development of a rodent model of autism. Int. J. Dev. Neurosci. 2020, 80, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Than, U.T.T.; Nguyen, L.T.; Nguyen, P.H.; Nguyen, X.H.; Trinh, D.P.; Hoang, D.H.; Nguyen, P.A.T.; Dang, V.D. Inflammatory mediators drive neuroinflammation in autism spectrum disorder and cerebral palsy. Sci. Rep. 2023, 13, 22587. [Google Scholar] [CrossRef]
- Chez, M.G.; Dowling, T.; Patel, P.B.; Khanna, P.; Kominsky, M. Elevation of tumor necrosis factor-alpha in cerebrospinal fluid of autistic children. Pediatr. Neurol. 2007, 36, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Ricci, S.; Businaro, R.; Ippoliti, F.; Lo Vasco, V.R.; Massoni, F.; Onofri, E.; Troili, G.M.; Pontecorvi, V.; Morelli, M.; Rapp Ricciardi, M.; et al. Altered cytokine and BDNF levels in autism spectrum disorder. Neurotox. Res. 2013, 24, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Saghazadeh, A.; Ataeinia, B.; Keynejad, K.; Abdolalizadeh, A.; Hirbod-Mobarakeh, A.; Rezaei, N. Anti-inflammatory cytokines in autism spectrum disorders: A systematic review and meta-analysis. Cytokine 2019, 123, 154740. [Google Scholar] [CrossRef] [PubMed]
- Vakilzadeh, G.; Martinez-Cerdeño, V. Pathology and Astrocytes in Autism. Neuropsychiatr. Dis. Treat. 2023, 19, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Edmonson, C.; Ziats, M.N.; Rennert, O.M. Altered glial marker expression in autistic post-mortem prefrontal cortex and cerebellum. Mol. Autism 2014, 5, 3. [Google Scholar] [CrossRef]
- Crawford, J.D.; Chandley, M.J.; Szebeni, K.; Szebeni, A.; Waters, B.; Ordway, G.A. Elevated GFAP Protein in Anterior Cingulate Cortical White Matter in Males with Autism Spectrum Disorder. Autism Res. 2015, 8, 649–657. [Google Scholar] [CrossRef]
- Cavaliere, C.; Tramontano, L.; Fiorenza, D.; Alfano, V.; Aiello, M.; Salvatore, M. Gliosis and Neurodegenerative Diseases: The Role of PET and MR Imaging. Front. Cell Neurosci. 2020, 14, 75. [Google Scholar] [CrossRef]
- Alvarez-Buylla, A.; García-Verdugo, J.M.; Tramontin, A.D. A unified hypothesis on the lineage of neural stem cells. Nat. Rev. Neurosci. 2001, 2, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Higashimori, H.; Morel, L. Developmental maturation of astrocytes and pathogenesis of neurodevelopmental disorders. J. Neurodev. Disord. 2013, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Amlerova, Z.; Chmelova, M.; Anderova, M.; Vargova, L. Reactive gliosis in traumatic brain injury: A comprehensive review. Front. Cell Neurosci. 2024, 18, 1335849. [Google Scholar] [CrossRef] [PubMed]
- Mora, P.; Chapouly, C. Astrogliosis in multiple sclerosis and neuro-inflammation: What role for the notch pathway? Front. Immunol. 2023, 14, 1254586. [Google Scholar] [CrossRef] [PubMed]
- Simone, M.; De Giacomo, A.; Palumbi, R.; Palazzo, C.; Lucisano, G.; Pompamea, F.; Micella, S.; Pascali, M.; Gabellone, A.; Marzulli, L.; et al. Serum Neurofilament Light Chain and Glial Fibrillary Acidic Protein as Potential Diagnostic Biomarkers in Autism Spectrum Disorders: A Preliminary Study. Int. J. Mol. Sci. 2023, 24, 3057. [Google Scholar] [CrossRef] [PubMed]
- Laurence, J.A.; Fatemi, S.H. Glial fibrillary acidic protein is elevated in superior frontal, parietal and cerebellar cortices of autistic subjects. Cerebellum 2005, 4, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, E.; Pierce, K.; Schumann, C.M.; Redcay, E.; Buckwalter, J.A.; Kennedy, D.P.; Morgan, J. Mapping early brain development in autism. Neuron 2007, 56, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Ang, Z.; Ding, J.L. GPR41 and GPR43 in Obesity and Inflammation - Protective or Causative? Front. Immunol. 2016, 7, 28. [Google Scholar] [CrossRef]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef]
- Rademacher, S.; Eickholt, B.J. PTEN in Autism and Neurodevelopmental Disorders. Cold Spring Harb. Perspect. Med. 2019, 9, a036780. [Google Scholar] [CrossRef] [PubMed]
- Skelton, P.D.; Stan, R.V.; Luikart, B.W. The Role of PTEN in Neurodevelopment. Mol. Neuropsychiatry 2020, 5, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M.; Dowd, S.E.; Gontcharova, V.; Liu, C.; Henley, K.E.; Wolcott, R.D.; Youn, E.; Summanen, P.H.; Granpeesheh, D.; Dixon, D.; et al. Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe 2010, 16, 444–453. [Google Scholar] [CrossRef]
- MacFabe, D.F.; Cain, N.E.; Boon, F.; Ossenkopp, K.P.; Cain, D.P. Effects of the enteric bacterial metabolic product propionic acid on object-directed behavior, social behavior, cognition, and neuroinflammation in adolescent rats: Relevance to autism spectrum disorder. Behav. Brain Res. 2011, 217, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Modabbernia, A.; Velthorst, E.; Reichenberg, A. Environmental risk factors for autism: An evidence-based review of systematic reviews and meta-analyses. Mol. Autism 2017, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Bölte, S.; Girdler, S.; Marschik, P.B. The contribution of environmental exposure to the etiology of autism spectrum disorder. Cell Mol. Life Sci. 2019, 76, 1275–1297. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M. Imaging In Mice with Fluorescent Proteins: From Macro to Subcellular. Sensors 2008, 8, 1157–1173. [Google Scholar] [CrossRef] [PubMed]
- Fattorusso, A.; Di Genova, L.; Dell’Isola, G.B.; Mencaroni, E.; Esposito, S. Autism Spectrum Disorders and the Gut Microbiota. Nutrients 2019, 11, 521. [Google Scholar] [CrossRef] [PubMed]
- Sandler, R.H.; Finegold, S.M.; Bolte, E.R.; Buchanan, C.P.; Maxwell, A.P.; Väisänen, M.L.; Nelson, M.N.; Wexler, H.M. Short-term benefit from oral vancomycin treatment of regressive-onset autism. J. Child. Neurol. 2000, 15, 429–435. [Google Scholar] [CrossRef]
- Frye, R.E.; Slattery, J.; MacFabe, D.F.; Allen-Vercoe, E.; Parker, W.; Rodakis, J.; Adams, J.B.; Krajmalnik-Brown, R.; Bolte, E.; Kahler, S.; et al. Approaches to studying and manipulating the enteric microbiome to improve autism symptoms. Microb. Ecol. Health Dis. 2015, 26, 26878. [Google Scholar] [CrossRef]
- Veleva-Rotse, B.O.; Barnes, A.P. Brain patterning perturbations following PTEN loss. Front. Mol. Neurosci. 2014, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Gary, D.S.; Mattson, M.P. PTEN regulates Akt kinase activity in hippocampal neurons and increases their sensitivity to glutamate and apoptosis. Neuromolecular Med. 2002, 2, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, L.; Sun, B.; Zhang, C.L.; Fine, A.; Chiu, S.Y.; Messing, A. Live astrocytes visualized by green fluorescent protein in transgenic mice. Dev. Biol. 1997, 187, 36–42. [Google Scholar] [CrossRef]
- AlQasrawi, D.; Naser, E.; Naser, S.A. Nicotine Increases Macrophage Survival through α7nAChR/NF-κB Pathway in Mycobacterium avium paratuberculosis Infection. Microorganisms 2021, 9, 1086. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product # | Control (Cat# D12020102) | PPA (Cat# D19071504) | ||
|---|---|---|---|---|
| Research Diets, Inc. | ||||
| gm% | kcal% | gm% | kcal% | |
| Protein | 19.9 | 20 | 19.8 | 20 |
| Carbohydrate | 63.8 | 64 | 63.4 | 64 |
| Fat | 6.9 | 16 | 6.8 | 26 |
| Total | 100 | 100 | ||
| kcal/gm | 3.96 | 3.94 | ||
| Ingredient | gm | kcal | gm | kcal |
| Casein | 200 | 800 | 200 | 8000 |
| L-Cystine | 3 | 12 | ||
| Corn Starch | 346 | 1384 | 346 | 1384 |
| Maltodextrin 10 | 45 | 180 | 45 | 180 |
| Dextrose | 250 | 1000 | 250 | 1000 |
| Cellulose, BW200 | 50 | 0 | 50 | 0 |
| Soybean Oil | 70 | 630 | 70 | 630 |
| Mineral Mix S10026 | 10 | 0 | 10 | 0 |
| Dicalcium Phosphate | 13 | 0 | 13 | 0 |
| Calcium Carbonate | 5.5 | 0 | 5.5 | 0 |
| Potassium Citrate, 1 H2O | 16.5 | 0 | 16.5 | 0 |
| Vitamin Mix V10001 | 10 | 40 | 10 | 40 |
| Choline Bitartrate | 2 | 0 | 2 | 0 |
| Sodium Propionate | 0 | 0 | 5.14 | 0 |
| Yellow Dye #5, FD&C | 0.05 | 0 | 0.025 | 0 |
| Blue Dye #1, FD&C | 0 | 0.025 | 0 | |
| Total | 1021.05 | 4046 | 1026.19 | 4046 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lagod, P.P.; Abdelli, L.S.; Naser, S.A. An In Vivo Model of Propionic Acid-Rich Diet-Induced Gliosis and Neuro-Inflammation in Mice (FVB/N-Tg(GFAPGFP)14Mes/J): A Potential Link to Autism Spectrum Disorder. Int. J. Mol. Sci. 2024, 25, 8093. https://doi.org/10.3390/ijms25158093
Lagod PP, Abdelli LS, Naser SA. An In Vivo Model of Propionic Acid-Rich Diet-Induced Gliosis and Neuro-Inflammation in Mice (FVB/N-Tg(GFAPGFP)14Mes/J): A Potential Link to Autism Spectrum Disorder. International Journal of Molecular Sciences. 2024; 25(15):8093. https://doi.org/10.3390/ijms25158093
Chicago/Turabian StyleLagod, Piotr P., Latifa S. Abdelli, and Saleh A. Naser. 2024. "An In Vivo Model of Propionic Acid-Rich Diet-Induced Gliosis and Neuro-Inflammation in Mice (FVB/N-Tg(GFAPGFP)14Mes/J): A Potential Link to Autism Spectrum Disorder" International Journal of Molecular Sciences 25, no. 15: 8093. https://doi.org/10.3390/ijms25158093
APA StyleLagod, P. P., Abdelli, L. S., & Naser, S. A. (2024). An In Vivo Model of Propionic Acid-Rich Diet-Induced Gliosis and Neuro-Inflammation in Mice (FVB/N-Tg(GFAPGFP)14Mes/J): A Potential Link to Autism Spectrum Disorder. International Journal of Molecular Sciences, 25(15), 8093. https://doi.org/10.3390/ijms25158093