
Association of KRAS Mutation and Gene Pathways in Colorectal Carcinoma: A Transcriptome- and Methylome-Wide Study and Potential Implications for Therapy
 ,
,
Abstract
1. Introduction
2. Results
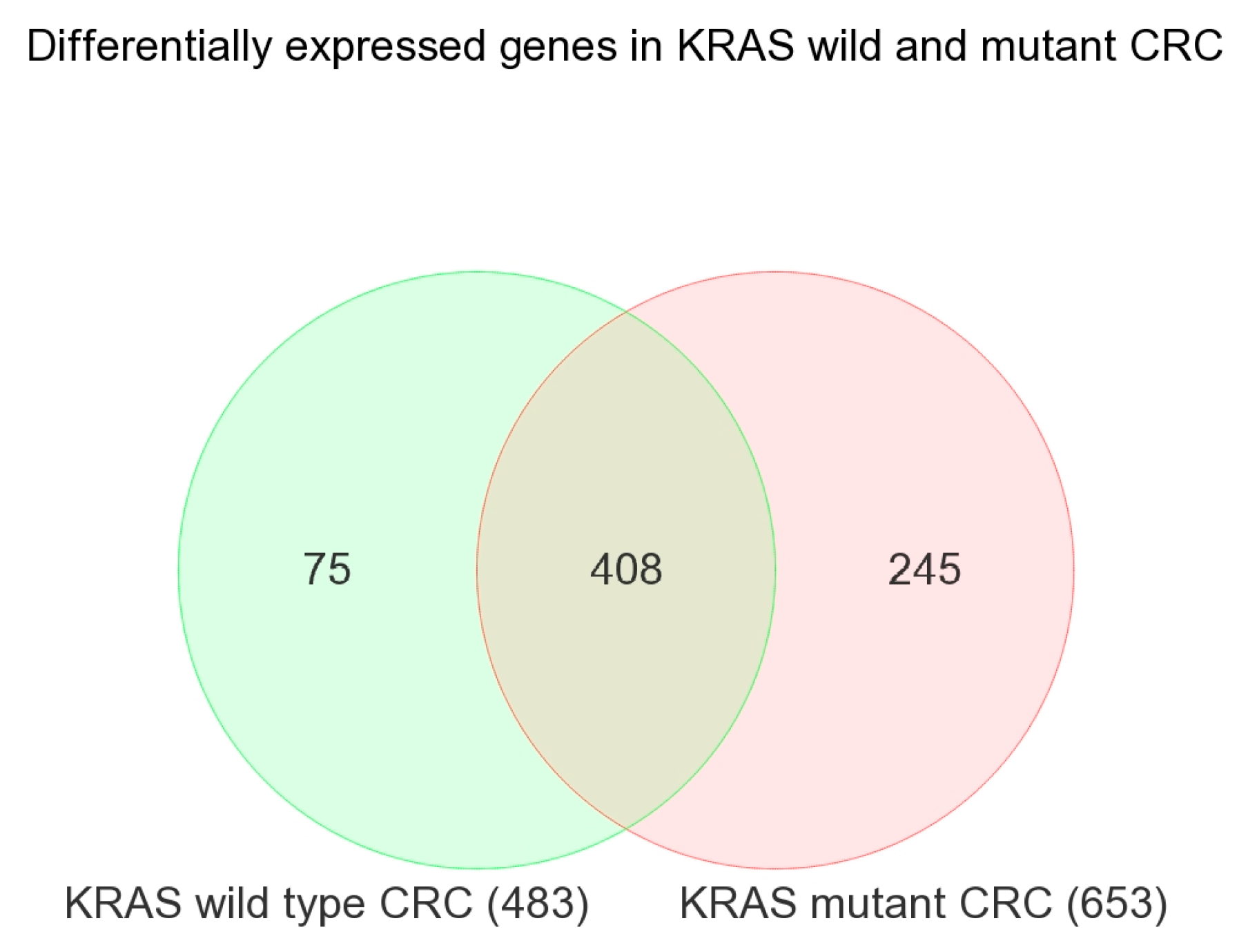
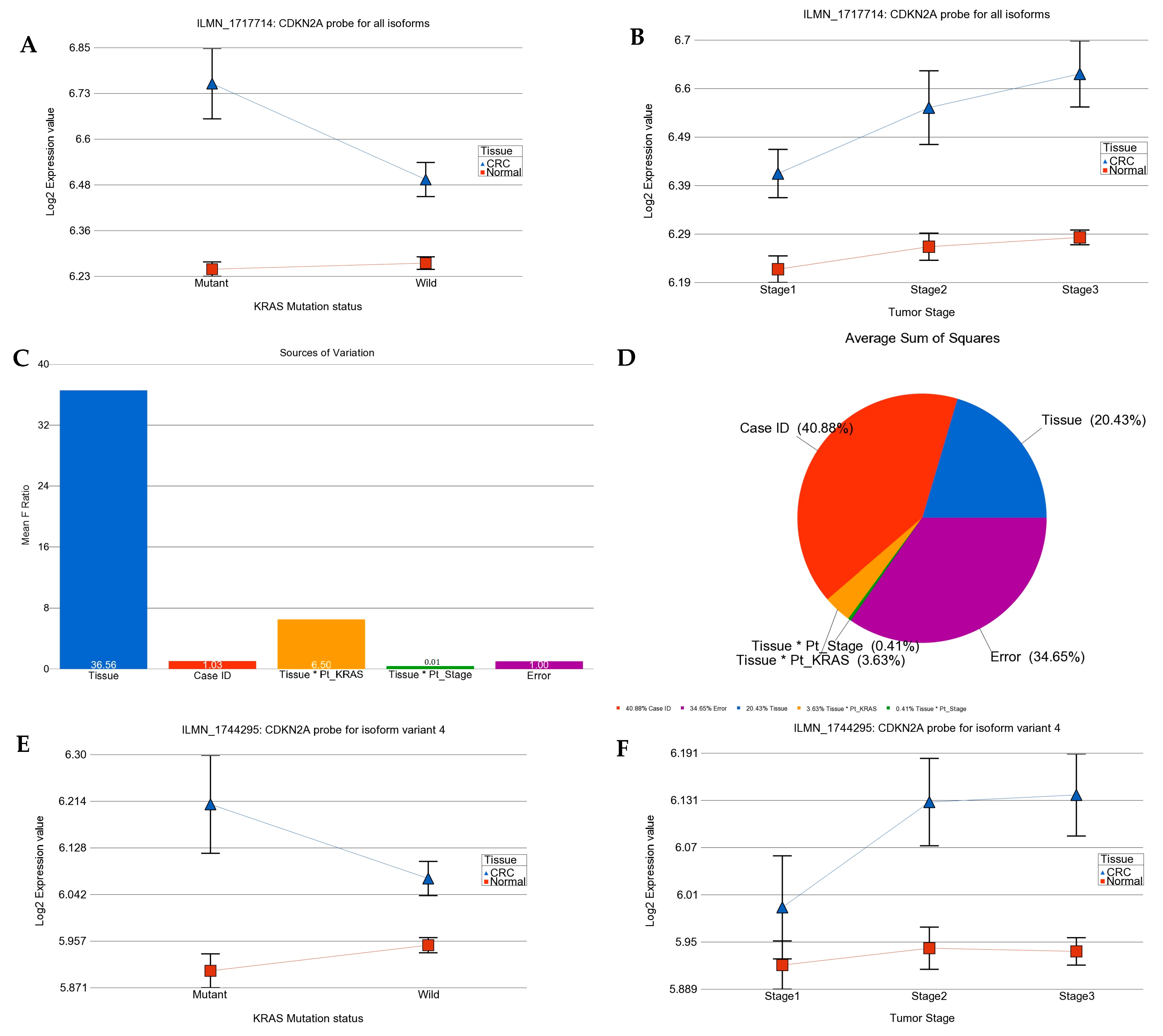
2.1. Transcriptome-Wide Analyses at Gene Level
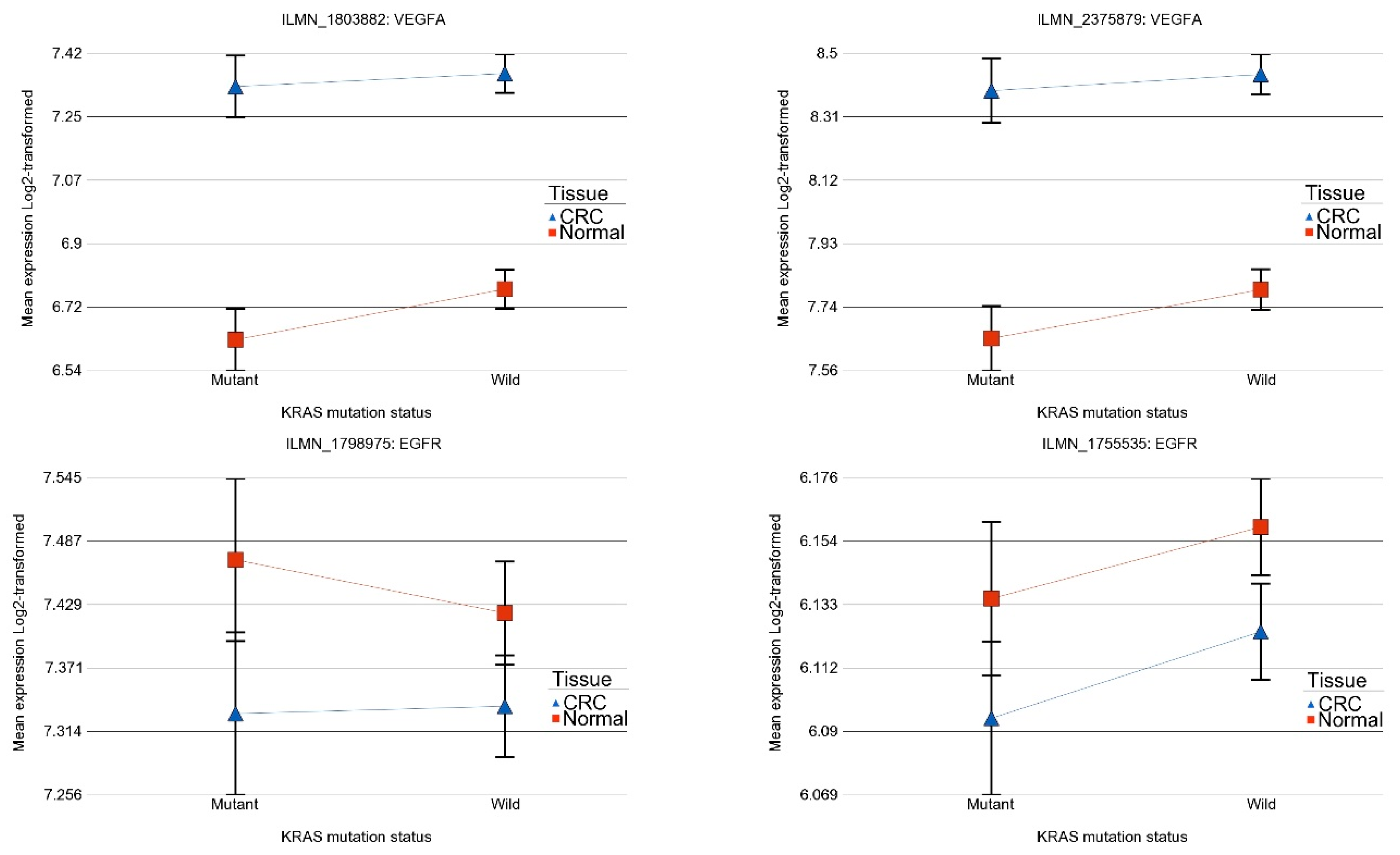
2.2. Association of KRAS Mutation and Genes Related to Tyrosine Kinase Inhibitor Targets and Other Angiogenic Targets
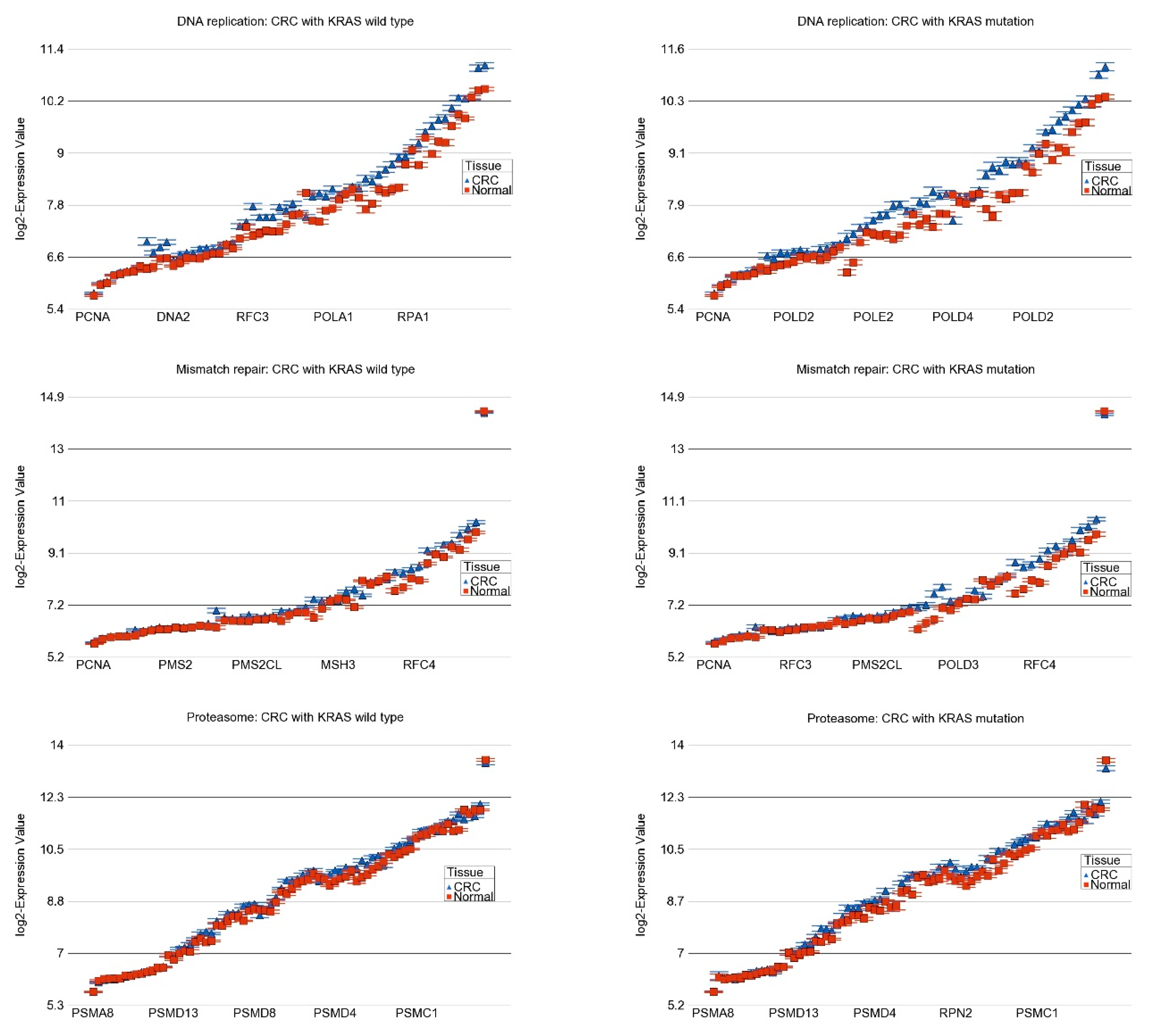
2.3. Transcriptome-Wide Analyses at Gene Pathway Level
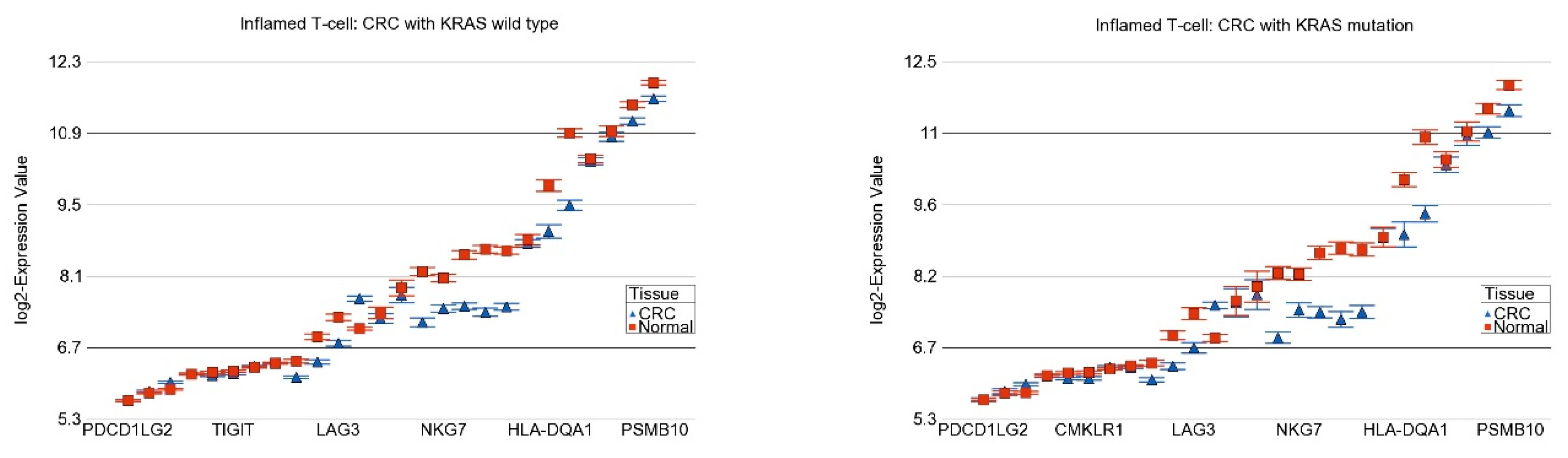
2.4. Association of KRAS Mutation and Genes Related to Immune Checkpoint Inhibitors (ICIs)
2.5. Effect of Interaction of KRAS Mutation and MSI Status on Differential Expression of Gene Pathways
2.6. Differential DNA Promoter Methylation of Gene Pathways
3. Discussion
4. Materials and Methods
4.1. DNA Extraction and Quality Control
4.2. Gene Expression
4.3. DNA Methylation
4.4. Mutation Detection
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 233–254. [Google Scholar] [CrossRef] [PubMed]
- Marcellinaro, R.; Spoletini, D.; Grieco, M.; Avella, P.; Cappuccio, M.; Troiano, R.; Lisi, G.; Garbarino, G.M.; Carlini, M. Colorectal Cancer: Current Updates and Future Perspectives. J. Clin. Med. 2023, 13, 40. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dinu, D.; Dobre, M.; Panaitescu, E.; Bîrlă, R.; Iosif, C.; Hoara, P.; Caragui, A.; Boeriu, M.; Constantinoiu, S.; Ardeleanu, C. Prognostic significance of KRAS gene mutations in colorectal cancer—Preliminary study. J. Med. Life 2014, 7, 581–587. [Google Scholar] [PubMed] [PubMed Central]
- Jancík, S.; Drábek, J.; Radzioch, D.; Hajdúch, M. Clinical relevance of KRAS in human cancers. J. Biomed. Biotechnol. 2010, 2010, 150960. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Ros, J.; Vaghi, C.; Baraibar, I.; Saoudi González, N.; Rodríguez-Castells, M.; García, A.; Alcaraz, A.; Salva, F.; Tabernero, J.; Elez, E. Targeting KRAS G12C Mutation in Colorectal Cancer, A Review: New Arrows in the Quiver. Int. J. Mol. Sci. 2024, 25, 3304. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, D.; Lai, X.; Xu, F.; Li, Y.; Jiang, W.; Ma, D. Prognosis and clinical characteristics of colorectal cancer patients with KRAS gene mutation: A 5-year follow-up study. Int. J. Clin. Exp. Pathol. 2019, 12, 409–418. [Google Scholar] [PubMed] [PubMed Central]
- Zeissig, M.N.; Ashwood, L.M.; Kondrashova, O.; Sutherland, K.D. Next batter up! Targeting cancers with KRAS-G12D mutations. Trends Cancer 2023, 9, 955–967. [Google Scholar] [CrossRef] [PubMed]
- El Agy, F.; El Bardai, S.; El Otmani, I.; Benbrahim, Z.; Karim, M.H.; Mazaz, K.; Benjelloun, E.B.; Ousadden, A.; El Abkari, M.; Ibrahimi, S.A.; et al. Mutation status and prognostic value of KRAS and NRAS mutations in Moroccan colon cancer patients: A first report. PLoS ONE 2021, 16, e0248522. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jasmine, F.; Haq, Z.; Kamal, M.; Raza, M.; da Silva, G.; Gorospe, K.; Paul, R.; Strzempek, P.; Ahsan, H.; Kibriya, M.G. Interaction between Microsatellite Instability (MSI) and Tumor DNA Methylation in the Pathogenesis of Colorectal Carcinoma. Cancers 2021, 13, 4956. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kibriya, M.G.; Jasmine, F.; Khamkevych, Y.; Raza, M.; Kamal, M.; Bissonnette, M.; Ahsan, H. Association of Microsatellite Instability and Gene Expression Profile in Colorectal Carcinoma and Potential Implications for Therapy. Medicina 2024, 60, 348. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wilson, P.M.; Labonte, M.J.; Lenz, H.J. Molecular markers in the treatment of metastatic colorectal cancer. Cancer J. 2010, 16, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Nelson, V.M.; Benson, A.B., 3rd. Status of targeted therapies in the adjuvant treatment of colon cancer. J. Gastrointest. Oncol. 2013, 4, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Barresi, V.; Castorina, S.; Musso, N.; Capizzi, C.; Luca, T.; Privitera, G.; Condorelli, D.F. Chromosomal instability analysis and regional tumor heterogeneity in colon cancer. Cancer Genet. 2017, 210, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Jasmine, F.; Rahaman, R.; Dodsworth, C.; Roy, S.; Paul, R.; Raza, M.; Paul-Brutus, R.; Kamal, M.; Ahsan, H.; Kibriya, M.G. A genome-wide study of cytogenetic changes in colorectal cancer using SNP microarrays: Opportunities for future personalized treatment. PLoS ONE 2012, 7, e31968. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Diao, Z.; Han, Y.; Chen, Y.; Zhang, R.; Li, J. The clinical utility of microsatellite instability in colorectal cancer. Crit. Rev. Oncol. Hematol. 2021, 157, 103171. [Google Scholar] [CrossRef] [PubMed]
- Mei, W.J.; Mi, M.; Qian, J.; Xiao, N.; Yuan, Y.; Ding, P.R. Clinicopathological characteristics of high microsatellite instability/mismatch repair-deficient colorectal cancer: A narrative review. Front. Immunol. 2022, 13, 1019582. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fatemi, N.; Tierling, S.; Es, H.A.; Varkiani, M.; Mojarad, E.N.; Aghdaei, H.A.; Walter, J.; Totonchi, M. DNA methylation biomarkers in colorectal cancer: Clinical applications for precision medicine. Int. J. Cancer 2022, 151, 2068–2081. [Google Scholar] [CrossRef] [PubMed]
- Kibriya, M.G.; Raza, M.; Jasmine, F.; Roy, S.; Paul-Brutus, R.; Rahaman, R.; Dodsworth, C.; Rakibuz-Zaman, M.; Kamal, M.; Ahsan, H. A genome-wide DNA methylation study in colorectal carcinoma. BMC Med. Genom. 2011, 4, 50. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Müller, D.; Győrffy, B. DNA methylation-based diagnostic, prognostic, and predictive biomarkers in colorectal cancer. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188722. [Google Scholar] [CrossRef] [PubMed]
- Ewing, A.D.; Gacita, A.; Wood, L.D.; Ma, F.; Xing, D.; Kim, M.S.; Manda, S.S.; Abril, G.; Pereira, G.; Makohon-Moore, A.; et al. Widespread somatic L1 retrotransposition occurs early during gastrointestinal cancer evolution. Genome Res. 2015, 25, 1536–1545. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Formica, V.; Sera, F.; Cremolini, C.; Riondino, S.; Morelli, C.; Arkenau, H.T.; Roselli, M. KRAS and BRAF Mutations in Stage II and III Colon Cancer: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2022, 114, 517–527. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Frattini, M.; Balestra, D.; Suardi, S.; Oggionni, M.; Alberici, P.; Radice, P.; Costa, A.; Daidone, M.G.; Leo, E.; Pilotti, S.; et al. Different genetic features associated with colon and rectal carcinogenesis. Clin. Cancer Res. 2004, 10, 4015–4021. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Choi, S.J.; Park, J.S. Correlations among KRAS Mutation, Microsatellite Instability, and 18F-FDG Uptake in Colon Cancer. Asian Pac. J. Cancer Prev. 2022, 23, 3501–3506. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Negri, F.; Bottarelli, L.; de’Angelis, G.L.; Gnetti, L. KRAS: A Druggable Target in Colon Cancer Patients. Int. J. Mol. Sci. 2022, 23, 4120. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kibriya, M.G.; Raza, M.; Kamal, M.; Haq, Z.; Paul, R.; Mareczko, A.; Pierce, B.L.; Ahsan, H.; Jasmine, F. Relative Telomere Length Change in Colorectal Carcinoma and Its Association with Tumor Characteristics, Gene Expression and Microsatellite Instability. Cancers 2022, 14, 2250. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pauleck, S.; Sinnott, J.A.; Zheng, Y.L.; Gadalla, S.M.; Viskochil, R.; Haaland, B.; Cawthon, R.M.; Hoffmeister, A.; Hardikar, S. Association of Telomere Length with Colorectal Cancer Risk and Prognosis: A Systematic Review and Meta-Analysis. Cancers 2023, 15, 1159. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, K.; Prescott, J.; Hazra, A.; Meyerhardt, J.A.; Zhang, X.; De Vivo, I.; Chan, A.T.; Du, M.; Giovannucci, E.L.; Nan, H. Pre-diagnostic telomere length and colorectal cancer risk. Cancer Epidemiol. 2022, 77, 102100. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, H.; Deng, J.; Hou, T.W.; Shan, Y.Q. Villosol reverses 5-FU resistance in colorectal cancer by inhibiting the CDKN2A gene regulated TP53-PI3K/Akt signaling axis. J. Ethnopharmacol. 2024, 325, 117907. [Google Scholar] [CrossRef] [PubMed]
- Shima, K.; Nosho, K.; Baba, Y.; Cantor, M.; Meyerhardt, J.A.; Giovannucci, E.L.; Fuchs, C.S.; Ogino, S. Prognostic significance of CDKN2A (p16) promoter methylation and loss of expression in 902 colorectal cancers: Cohort study and literature review. Int. J. Cancer 2011, 128, 1080–1094. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guan, R.J.; Fu, Y.; Holt, P.R.; Pardee, A.B. Association of K-ras mutations with p16 methylation in human colon cancer. Gastroenterology 1999, 116, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Al-Ahwal, M.; Gomaa, W.; Emam, E.; Qari, Y.; Buhmeida, A.; Radwi, S.; Al-Maghrabi, B.; Al-Qahtani, M.; Al-Maghrabi, J. p16 protein is upregulated in a stepwise fashion in colorectal adenoma and colorectal carcinoma. Saudi J. Gastroenterol. 2016, 22, 435–440. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Watanabe, T.; Kobunai, T.; Yamamoto, Y.; Matsuda, K.; Ishihara, S.; Nozawa, K.; Iinuma, H.; Ikeuchi, H.; Eshima, K. Differential gene expression signatures between colorectal cancers with and without KRAS mutations: Crosstalk between the KRAS pathway and other signalling pathways. Eur. J. Cancer 2011, 47, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Adnan, M.; Jairajpuri, D.S.; Chaddha, M.; Khan, M.S.; Yadav, D.K.; Mohammad, T.; Elasbali, A.M.; Abu Al-Soud, W.; Hussain Alharethi, S.; Hassan, M.I. Discovering Tuberosin and Villosol as Potent and Selective Inhibitors of AKT1 for Therapeutic Targeting of Oral Squamous Cell Carcinoma. J. Pers. Med. 2022, 12, 1083. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Parsons, B.L.; Myers, M.B. Personalized cancer treatment and the myth of KRAS wild-type colon tumors. Discov. Med. 2013, 15, 259–267. [Google Scholar] [PubMed]
- Kumar, S.S.; Price, T.J.; Mohyieldin, O.; Borg, M.; Townsend, A.; Hardingham, J.E. KRAS G13D Mutation and Sensitivity to Cetuximab or Panitumumab in a Colorectal Cancer Cell Line Model. Gastrointest. Cancer Res. 2014, 7, 23–26. [Google Scholar] [PubMed] [PubMed Central]
- Duerr, E.M.; Mizukami, Y.; Moriichi, K.; Gala, M.; Jo, W.S.; Kikuchi, H.; Xavier, R.J.; Chung, D.C. Oncogenic KRAS regulates BMP4 expression in colon cancer cell lines. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1223–G1230. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brzozowa-Zasada, M.; Matysiak, N.; Piecuch, A.; Gawełek, E.; Michalski, M.; Kucharzewski, M.; Łos, M.J. The Prognostic Significance of Apoptotic Protease Activating Factor (Apaf-1) Protein Expression in Colon Adenocarcinoma Tissue-Preliminary Report. Front. Biosci. 2023, 28, 29. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Talty, R.; Jain, A.; Cai, Y.; Zheng, J.; Shen, X.; Muca, E.; Paty, P.B.; Bosenberg, M.W.; Khan, S.A.; et al. Discovery of decreased ferroptosis in male colorectal cancer patients with KRAS mutations. Redox Biol. 2023, 62, 102699. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, P.; Li, X.; Zhang, R.; Liu, S.; Xiang, Y.; Zhang, M.; Chen, X.; Pan, T.; Yan, L.; Feng, J.; et al. Combinative treatment of β-elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial-mesenchymal transformation. Theranostics 2020, 10, 5107–5119. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hassan, R.T.; Al Hassawi, B.; Alkazzaz, M. The Clinicopathological Correlation of KRAS Mutation and PTEN Expression Status in Primary and Metastatic Colorectal Carcinoma. Cureus 2024, 16, e53884. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ribeiro, K.B.; da Silva Zanetti, J.; Ribeiro-Silva, A.; Rapatoni, L.; de Oliveira, H.F.; da Cunha Tirapelli, D.P.; Garcia, S.B.; Feres, O.; da Rocha, J.J.; Peria, F.M. KRAS mutation associated with CD44/CD166 immunoexpression as predictors of worse outcome in metastatic colon cancer. Cancer Biomark 2016, 16, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Zhang, H.; Li, Q.; Guan, N.; Yun, C.; Zhao, H.; Zhang, F.; Zeng, Z.; Hu, Y.; Li, Q.; et al. A novel prognostic signature contributes to precision treatment in colon adenocarcinoma with KRAS mutation. Eur. J. Cancer Prev. 2023, 32, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Wang, X.; Duanmu, J.; Li, T.; Jiang, Q. KRAS mutations are negatively correlated with immunity in colon cancer. Aging 2020, 13, 750–768. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ilm, K.; Kemmner, W.; Osterland, M.; Burock, S.; Koch, G.; Herrmann, P.; Schlag, P.M.; Stein, U. High MACC1 expression in combination with mutated KRAS G13 indicates poor survival of colorectal cancer patients. Mol. Cancer 2015, 14, 38. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, J.; Huang, X.; Liu, H.; Wei, C.; Ru, H.; Qin, H.; Lai, H.; Meng, Y.; Wu, G.; Xie, W.; et al. Immune landscape and prognostic immune-related genes in KRAS-mutant colorectal cancer patients. J. Transl. Med. 2021, 19, 27. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shaykevich, A.; Chae, D.; Silverman, I.; Bassali, J.; Louloueian, N.; Siegman, A.; Bandyopadhyaya, G.; Goel, S.; Maitra, R. Impact of carbamazepine on SMARCA4 (BRG1) expression in colorectal cancer: Modulation by KRAS mutation status. Investig. New Drugs 2024, 42, 229–239. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Peng, L.; Li, J.; Wu, J.; Xu, B.; Wang, Z.; Giamas, G.; Stebbing, J.; Yu, Z. A Pan-Cancer Analysis of SMARCA4 Alterations in Human Cancers. Front. Immunol. 2021, 12, 762598. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pu, W.; Qian, F.; Liu, J.; Shao, K.; Xiao, F.; Jin, Q.; Liu, Q.; Jiang, S.; Zhang, R.; Zhang, J.; et al. Targeted Bisulfite Sequencing Reveals DNA Methylation Changes in Zinc Finger Family Genes Associated With KRAS Mutated Colorectal Cancer. Front. Cell Dev. Biol. 2021, 9, 759813. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nagasaka, T.; Sasamoto, H.; Notohara, K.; Cullings, H.M.; Takeda, M.; Kimura, K.; Kambara, T.; MacPhee, D.G.; Young, J.; Leggett, B.A.; et al. Colorectal cancer with mutation in BRAF, KRAS, and wild-type with respect to both oncogenes showing different patterns of DNA methylation. J. Clin. Oncol. 2004, 22, 4584–4594. [Google Scholar] [CrossRef] [PubMed]
- Hinoue, T.; Weisenberger, D.J.; Lange, C.P.; Shen, H.; Byun, H.M.; Van Den Berg, D.; Malik, S.; Pan, F.; Noushmehr, H.; van Dijk, C.M.; et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012, 22, 271–282. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nagasaka, M.; Li, Y.; Sukari, A.; Ou, S.I.; Al-Hallak, M.N.; Azmi, A.S. KRAS G12C Game of Thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treat. Rev. 2020, 84, 101974. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bekaii-Saab, T.S.; Yaeger, R.; Spira, A.I.; Pelster, M.S.; Sabari, J.K.; Hafez, N.; Barve, M.; Velastegui, K.; Yan, X.; Shetty, A.; et al. Adagrasib in Advanced Solid Tumors Harboring a KRAS(G12C) Mutation. J. Clin. Oncol. 2023, 41, 4097–4106. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ooi, Z.S.; Pang, S.W.; Teow, S.Y. RAS and BRAF genes as biomarkers and target for personalised colorectal cancer therapy: An update. Malays. J. Pathol. 2022, 44, 415–428. [Google Scholar] [PubMed]
- Desai, J.; Alonso, G.; Kim, S.H.; Cervantes, A.; Karasic, T.; Medina, L.; Shacham-Shmueli, E.; Cosman, R.; Falcon, A.; Gort, E.; et al. Divarasib plus cetuximab in KRAS G12C-positive colorectal cancer: A phase 1b trial. Nat. Med. 2024, 30, 271–278. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sacher, A.; LoRusso, P.; Patel, M.R.; Miller, W.H., Jr.; Garralda, E.; Forster, M.D.; Santoro, A.; Falcon, A.; Kim, T.W.; Paz-Ares, L.; et al. Single-Agent Divarasib (GDC-6036) in Solid Tumors with a KRAS G12C Mutation. N. Engl. J. Med. 2023, 389, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Kibriya, M.G.; Raza, M.; Quinn, A.; Kamal, M.; Ahsan, H.; Jasmine, F. A Transcriptome and Methylome Study Comparing Tissues of Early and Late Onset Colorectal Carcinoma. Int. J. Mol. Sci. 2022, 23, 14261. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gonzalez-Bosquet, J.; Calcei, J.; Wei, J.S.; Garcia-Closas, M.; Sherman, M.E.; Hewitt, S.; Vockley, J.; Lissowska, J.; Yang, H.P.; Khan, J.; et al. Detection of somatic mutations by high-resolution DNA melting (HRM) analysis in multiple cancers. PLoS ONE 2011, 6, e14522. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO Description | Fold Change | (95% CI) | p-Value |
|---|---|---|---|
| DNA replication | 1.23 | (1.22–1.25) | 0.00 × 100 |
| Ribosome biogenesis in eukaryotes | 1.18 | (1.17–1.19) | 0.00 × 100 |
| RNA polymerase | 1.16 | (1.15–1.17) | 4.82 × 10−178 |
| Mismatch repair | 1.16 | (1.14–1.17) | 8.37 × 10−165 |
| Bladder cancer | 1.14 | (1.12–1.16) | 4.01 × 10−61 |
| ECM-receptor interaction | 1.14 | (1.13–1.15) | 4.91 × 10−147 |
| One carbon pool by folate | 1.13 | (1.12–1.15) | 1.08 × 10−51 |
| Nucleotide excision repair | 1.13 | (1.12–1.14) | 1.84 × 10−187 |
| Vitamin B6 metabolism | 1.12 | (1.08–1.16) | 4.13 × 10−10 |
| Proteasome | 1.11 | (1.11–1.12) | 1.56 × 10−128 |
| Malaria | 1.11 | (1.09–1.13) | 9.13 × 10−28 |
| Propanoate metabolism | −1.11 | (−1.12–−1.09) | 5.43 × 10−60 |
| Gastric acid secretion | −1.11 | (−1.12–−1.1) | 2.11 × 10−117 |
| Antigen processing and presentation | −1.11 | (−1.12–−1.1) | 3.98 × 10−92 |
| Collecting duct acid secretion | −1.11 | (−1.13–−1.09) | 5.59 × 10−31 |
| Type I diabetes mellitus | −1.11 | (−1.14–−1.09) | 4.77 × 10−27 |
| Fat digestion and absorption | −1.11 | (−1.13–−1.1) | 2.35 × 10−40 |
| Galactose metabolism | −1.12 | (−1.13–−1.1) | 3.49 × 10−52 |
| Butanoate metabolism | −1.12 | (−1.14–−1.1) | 9.13 × 10−48 |
| Ether lipid metabolism | −1.12 | (−1.14–−1.11) | 3.41 × 10−69 |
| Citrate cycle (TCA cycle) | −1.12 | (−1.14–−1.11) | 6.91 × 10−79 |
| Other glycan degradation | −1.12 | (−1.14–−1.1) | 2.98 × 10−31 |
| Bile secretion | −1.13 | (−1.14–−1.12) | 3.50 × 10−91 |
| Valine, leucine, and isoleucine degradation | −1.13 | (−1.15–−1.12) | 1.72 × 10−82 |
| Porphyrin and chlorophyll metabolism | −1.14 | (−1.16–−1.12) | 3.21 × 10−55 |
| Autoimmune thyroid disease | −1.14 | (−1.16–−1.13) | 3.50 × 10−74 |
| Metabolism of xenobiotics by cytochrome P450 | −1.15 | (−1.16–−1.13) | 7.19 × 10−86 |
| Primary immunodeficiency | −1.16 | (−1.17–−1.14) | 1.20 × 10−137 |
| Chemical carcinogenesis | −1.16 | (−1.17–−1.15) | 4.30 × 10−135 |
| Mineral absorption | −1.17 | (−1.18–−1.15) | 3.03 × 10−117 |
| Steroid hormone biosynthesis | −1.17 | (−1.18–−1.15) | 7.99 × 10−91 |
| Graft-versus-host disease | −1.17 | (−1.2–−1.15) | 5.87 × 10−60 |
| Fatty acid degradation | −1.18 | (−1.19–−1.16) | 3.96 × 10−149 |
| Drug metabolism-cytochrome P450 | −1.19 | (−1.21–−1.17) | 1.07 × 10−116 |
| Aldosterone-regulated sodium reabsorption | −1.21 | (−1.23–−1.2) | 1.55 × 10−172 |
| Retinol metabolism | −1.22 | (−1.24–−1.2) | 6.74 × 10−139 |
| Ascorbate and aldarate metabolism | −1.22 | (−1.25–−1.2) | 2.50 × 10−61 |
| Synthesis and degradation of ketone bodies | −1.23 | (−1.27–−1.19) | 2.16 × 10−30 |
| Asthma | −1.23 | (−1.26–−1.2) | 2.52 × 10−55 |
| Allograft rejection | −1.23 | (−1.26–−1.21) | 3.88 × 10−82 |
| Intestinal immune network for IgA production | −1.25 | (−1.27–−1.22) | 8.48 × 10−134 |
| Caffeine metabolism | −1.26 | (−1.31–−1.22) | 2.71 × 10−34 |
| Pentose and glucoronate interconversions | −1.27 | (−1.29–−1.24) | 3.03 × 10−93 |
| Sulfur metabolism | −1.29 | (−1.33–−1.25) | 1.75 × 10−52 |
| Proximal tubule bicarbonate reclamation | −1.34 | (−1.37–−1.31) | 1.67 × 10−120 |
| Nitrogen metabolism | −1.72 | (−1.81–−1.63) | 2.45 × 10−89 |
| GO Description | KRAS Wild-Type | KRAS-Mutant | Interaction-p | ||
|---|---|---|---|---|---|
| Fold Change | (95% CI) | Fold Change | (95% CI) | ||
| DNA replication | 1.21 | (1.19–1.22) | 1.27 | (1.25–1.3) | 1.86 × 10−05 |
| Ribosome biogenesis in eukaryotes | 1.16 | (1.15–1.17) | 1.21 | (1.19–1.23) | 4.35 × 10−07 |
| Mismatch repair | 1.13 | (1.12–1.15) | 1.19 | (1.17–1.22) | 5.39 × 10−06 |
| RNA polymerase | 1.15 | (1.13–1.16) | 1.19 | (1.17–1.21) | 4.25 × 10−03 |
| One carbon pool by folate | 1.11 | (1.09–1.13) | 1.18 | (1.15–1.22) | 3.10 × 10−03 |
| Nucleotide excision repair | 1.12 | (1.11–1.13) | 1.15 | (1.14–1.17) | 5.67 × 10−04 |
| Proteasome | 1.10 | (1.09–1.11) | 1.14 | (1.12–1.16) | 9.07 × 10−11 |
| Aminoacyl-tRNA biosynthesis | 1.09 | (1.07–1.1) | 1.14 | (1.12–1.16) | 7.85 × 10−06 |
| Base excision repair | 1.09 | (1.07–1.1) | 1.13 | (1.11–1.15) | 2.49 × 10−04 |
| ECM-receptor interaction | 1.14 | (1.13–1.15) | 1.12 | (1.1–1.14) | 8.17 × 10−05 |
| Steroid biosynthesis | 1.06 | (1.03–1.09) | 1.11 | (1.07–1.15) | 1.31 × 10−04 |
| Homologous recombination | 1.07 | (1.06–1.08) | 1.10 | (1.08–1.12) | 5.15 × 10−03 |
| Inflammatory bowel disease (IBD) | −1.07 | (−1.09–−1.06) | −1.10 | (−1.13–−1.08) | 1.55 × 10−02 |
| Staphylococcus aureus infection | −1.09 | (−1.11–−1.08) | −1.11 | (−1.13–−1.08) | 3.95 × 10−03 |
| Antigen processing and presentation | −1.11 | (−1.12–−1.1) | −1.12 | (−1.14–−1.09) | 2.81 × 10−05 |
| Renin secretion | −1.09 | (−1.1–−1.08) | −1.12 | (−1.14–−1.1) | 7.59 × 10−03 |
| Viral myocarditis | −1.09 | (−1.11–−1.08) | −1.12 | (−1.15–−1.1) | 9.38 × 10−04 |
| Tyrosine metabolism | −1.08 | (−1.1–−1.06) | −1.12 | (−1.16–−1.09) | 9.63 × 10−04 |
| Type I diabetes mellitus | −1.11 | (−1.13–−1.08) | −1.13 | (−1.18–−1.09) | 2.41 × 10−03 |
| Autoimmune thyroid disease | −1.13 | (−1.15–−1.11) | −1.17 | (−1.2–−1.14) | 4.30 × 10−04 |
| Primary immunodeficiency | −1.15 | (−1.16–−1.13) | −1.19 | (−1.21–−1.16) | 1.19 × 10−02 |
| Graft-versus-host disease | −1.16 | (−1.19–−1.14) | −1.20 | (−1.25–−1.16) | 8.67 × 10−04 |
| Mineral absorption | −1.15 | (−1.17–−1.13) | −1.20 | (−1.23–−1.18) | 5.52 × 10−03 |
| Allograft rejection | −1.21 | (−1.24–−1.18) | −1.28 | (−1.33–−1.23) | 1.60 × 10−04 |
| Asthma | −1.21 | (−1.25–−1.17) | −1.29 | (−1.36–−1.23) | 9.46 × 10−03 |
| Intestinal immune network for IgA production | −1.23 | (−1.26–−1.21) | −1.29 | (−1.33–−1.25) | 7.53 × 10−03 |
| DNA Replication Pathway | Proteasome Pathway | Mismatch Repair Pathway | ||||||
|---|---|---|---|---|---|---|---|---|
| KRAS Mut − ve | KRAS Mut + ve | KRAS Mut − ve | KRAS Mut + ve | KRAS Mut − ve | KRAS Mut + ve | |||
| MSI Status | MSI | FC | 1.19 | 1.27 | 1.06 | 1.18 | 1.12 | 1.20 |
| 95% CI | (1.17–1.22) | (1.22–1.32) | (1.04–1.08) | (1.14–1.22) | (1.09–1.14) | (1.15–1.25) | ||
| MSS | FC | 1.22 | 1.27 | 1.12 | 1.12 | 1.14 | 1.19 | |
| 95% CI | (1.2–1.23) | (1.24–1.3) | (1.1–1.13) | (1.1–1.15) | (1.13–1.16) | (1.16–1.22) | ||
| Interaction-p | 7.51 × 10−16 | 4.71 × 10−29 | 2.17 × 10−10 | |||||
| GO Description | KRAS Wild-Type | KRAS-Mutant | Interaction-p | ||
|---|---|---|---|---|---|
| p-Value | Delta Beta | p-Value | Delta Beta | ||
| Steroid biosynthesis | 3.12 × 10−11 | −0.0149 | 2.43 × 10−10 | −0.0232 | 5.72 × 10−02 |
| RNA polymerase | 4.19 × 10−16 | −0.0090 | 1.34 × 10−13 | −0.0134 | 8.67 × 10−04 |
| One carbon pool by folate | 6.67 × 10−05 | −0.0056 | 7.73 × 10−06 | −0.0104 | 1.75 × 10−04 |
| Aminoacyl-tRNA biosynthesis | 2.00 × 10−49 | −0.0084 | 6.50 × 10−20 | −0.0085 | 1.48 × 10−09 |
| Proteasome | 2.33 × 10−36 | −0.0082 | 1.48 × 10−11 | −0.0072 | 1.58 × 10−12 |
| Base excision repair | 7.55 × 10−13 | −0.0071 | 1.34 × 10−05 | −0.0070 | 2.99 × 10−11 |
| Mismatch repair | 9.67 × 10−06 | −0.0054 | 1.81 × 10−02 | −0.0047 | 3.11 × 10−08 |
| DNA replication | 1.28 × 10−16 | −0.0067 | 9.83 × 10−04 | −0.0044 | 6.96 × 10−15 |
| Homologous recombination | 3.71 × 10−25 | −0.0094 | 3.76 × 10−03 | −0.0043 | 2.62 × 10−18 |
| Nucleotide excision repair | 1.25 × 10−04 | −0.0031 | 6.99 × 10−01 | −0.0005 | 2.72 × 10−18 |
| Ribosome biogenesis in eukaryotes | 2.46 × 10−02 | −0.0016 | 2.88 × 10−04 | 0.0043 | 2.92 × 10−27 |
| ECM-receptor interaction | 6.05 × 10−23 | 0.0201 | 9.36 × 10−19 | 0.0295 | 7.49 × 10−10 |
| GO Description | KRAS Wild-Type | KRAS-Mutant | Interaction p | ||
|---|---|---|---|---|---|
| p-Value | Delta Beta | p-Value | Delta Beta | ||
| Asthma | 8.16 × 10−26 | 0.072 | 2.73 × 10−16 | 0.090 | 2.88 × 10−02 |
| Intestinal immune network for IgA production | 3.34 × 10−30 | 0.030 | 7.54 × 10−39 | 0.057 | 8.17 × 10−23 |
| Type I diabetes mellitus | 5.50 × 10−194 | 0.045 | 6.78 × 10−106 | 0.054 | 3.50 × 10−09 |
| Allograft rejection | 5.50 × 10−194 | 0.045 | 6.78 × 10−106 | 0.054 | 3.50 × 10−09 |
| Graft-versus-host disease | 5.50 × 10−194 | 0.045 | 6.78 × 10−106 | 0.054 | 3.50 × 10−09 |
| Autoimmune thyroid disease | 5.88 × 10−171 | 0.043 | 3.43 × 10−92 | 0.051 | 7.91 × 10−08 |
| Antigen processing and presentation | 1.01 × 10−187 | 0.031 | 1.14 × 10−98 | 0.036 | 5.64 × 10−11 |
| Primary immunodeficiency | 7.71 × 10−16 | 0.015 | 3.21 × 10−24 | 0.030 | 5.23 × 10−18 |
| Inflammatory bowel disease (IBD) | 1.34 × 10−33 | 0.019 | 8.04 × 10−31 | 0.029 | 1.56 × 10−11 |
| Viral myocarditis | 1.03 × 10−93 | 0.017 | 4.71 × 10−53 | 0.021 | 3.51 × 10−15 |
| Renin secretion | 5.43 × 10−46 | 0.014 | 1.56 × 10−10 | 0.010 | 8.34 × 10−04 |
| Tyrosine metabolism | 4.23 × 10−01 | 0.001 | 4.78 × 10−01 | 0.002 | 6.32 × 10−09 |
| Mineral absorption | 1.12 × 10−05 | −0.005 | 6.48 × 10−01 | −0.001 | 2.28 × 10−07 |
| Staphylococcus aureus infection | 9.65 × 10−07 | −0.017 | 1.48 × 10−02 | −0.014 | 2.10 × 10−02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jasmine, F.; Almazan, A.; Khamkevych, Y.; Bissonnette, M.; Ahsan, H.; Kibriya, M.G. Association of KRAS Mutation and Gene Pathways in Colorectal Carcinoma: A Transcriptome- and Methylome-Wide Study and Potential Implications for Therapy. Int. J. Mol. Sci. 2024, 25, 8094. https://doi.org/10.3390/ijms25158094
Jasmine F, Almazan A, Khamkevych Y, Bissonnette M, Ahsan H, Kibriya MG. Association of KRAS Mutation and Gene Pathways in Colorectal Carcinoma: A Transcriptome- and Methylome-Wide Study and Potential Implications for Therapy. International Journal of Molecular Sciences. 2024; 25(15):8094. https://doi.org/10.3390/ijms25158094
Chicago/Turabian StyleJasmine, Farzana, Armando Almazan, Yuliia Khamkevych, Marc Bissonnette, Habibul Ahsan, and Muhammad G. Kibriya. 2024. "Association of KRAS Mutation and Gene Pathways in Colorectal Carcinoma: A Transcriptome- and Methylome-Wide Study and Potential Implications for Therapy" International Journal of Molecular Sciences 25, no. 15: 8094. https://doi.org/10.3390/ijms25158094
APA StyleJasmine, F., Almazan, A., Khamkevych, Y., Bissonnette, M., Ahsan, H., & Kibriya, M. G. (2024). Association of KRAS Mutation and Gene Pathways in Colorectal Carcinoma: A Transcriptome- and Methylome-Wide Study and Potential Implications for Therapy. International Journal of Molecular Sciences, 25(15), 8094. https://doi.org/10.3390/ijms25158094