
Inline Raman Spectroscopy Provides Versatile Molecular Monitoring for Platelet Extracellular Vesicle Purification with Anion-Exchange Chromatography
 , ,
, ,  ,
,
Abstract
1. Introduction
2. Results
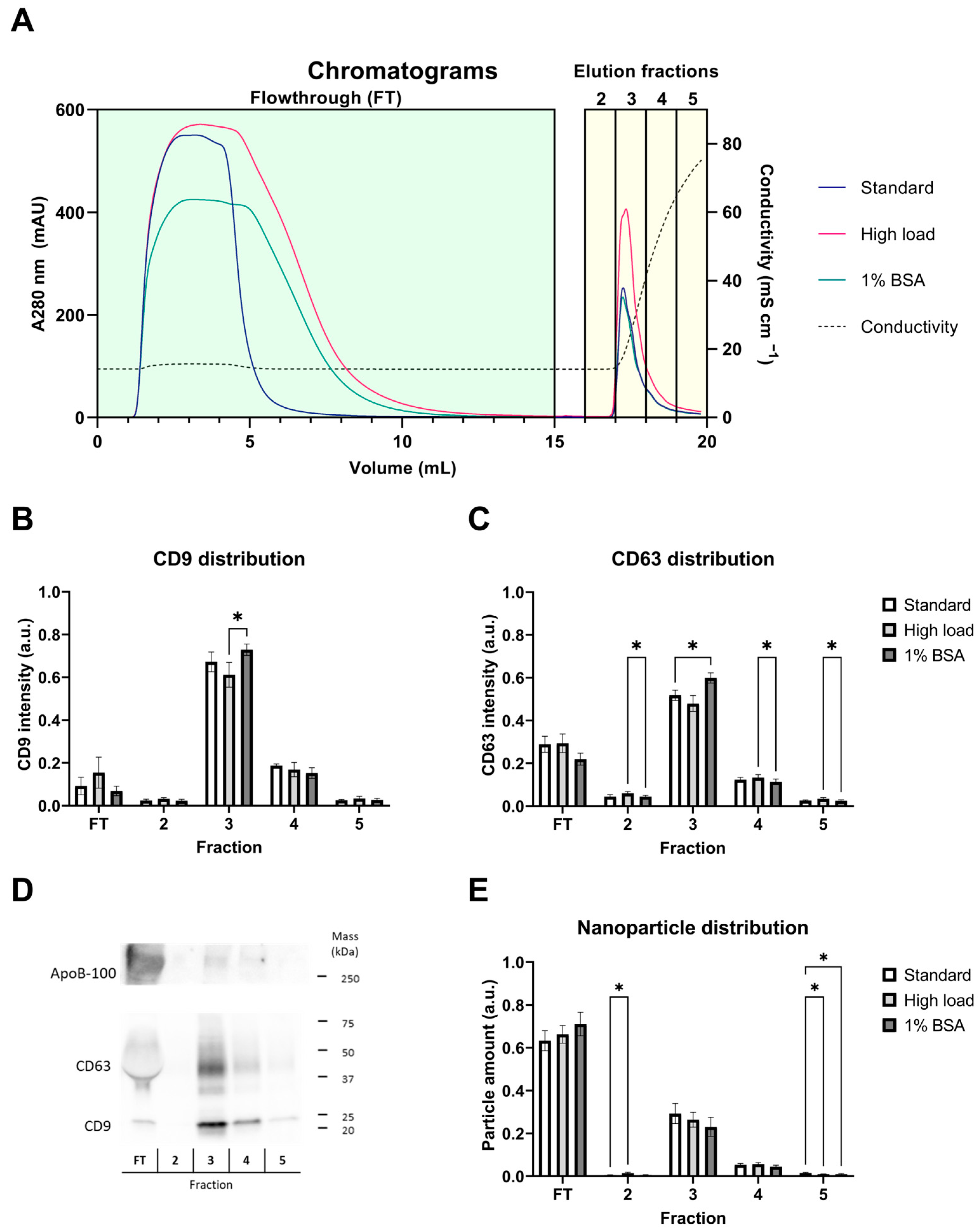
2.1. Anion-Exchange Chromatography Effectively Captures EVs from the Platelet Supernatant
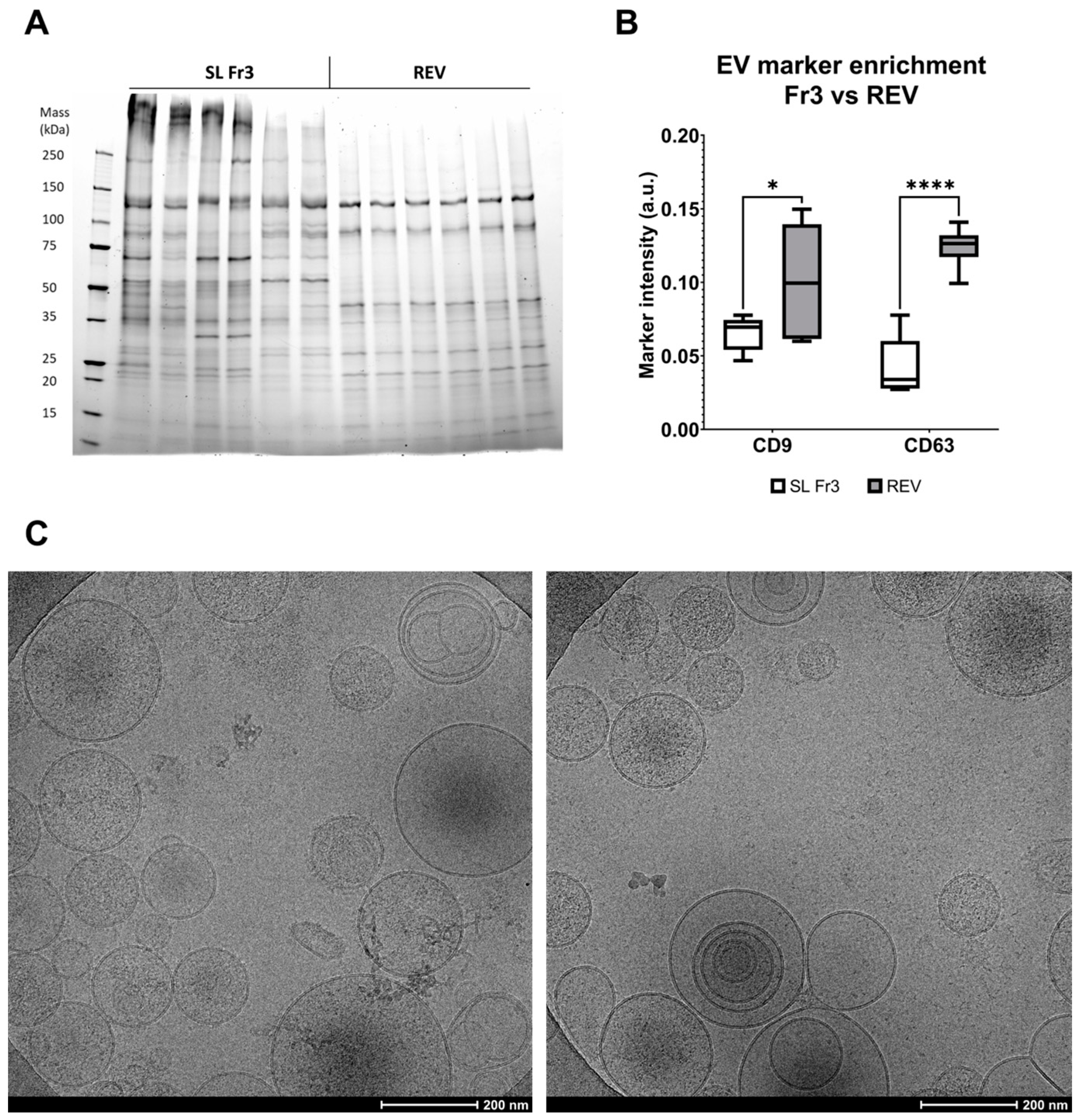
2.2. EVs Are Enriched in the Eluate and Purified during Anion-Exchange Chromatography
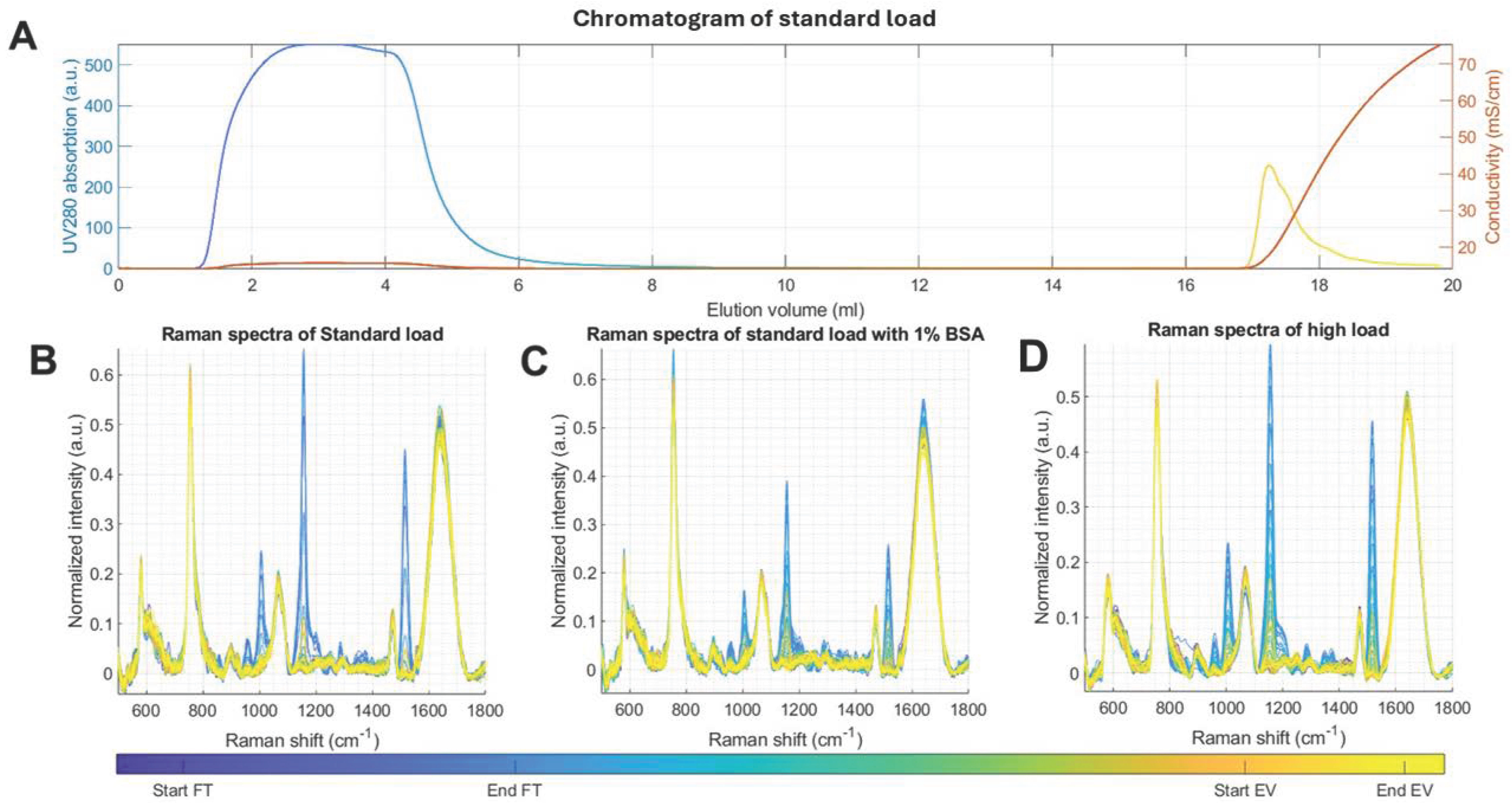
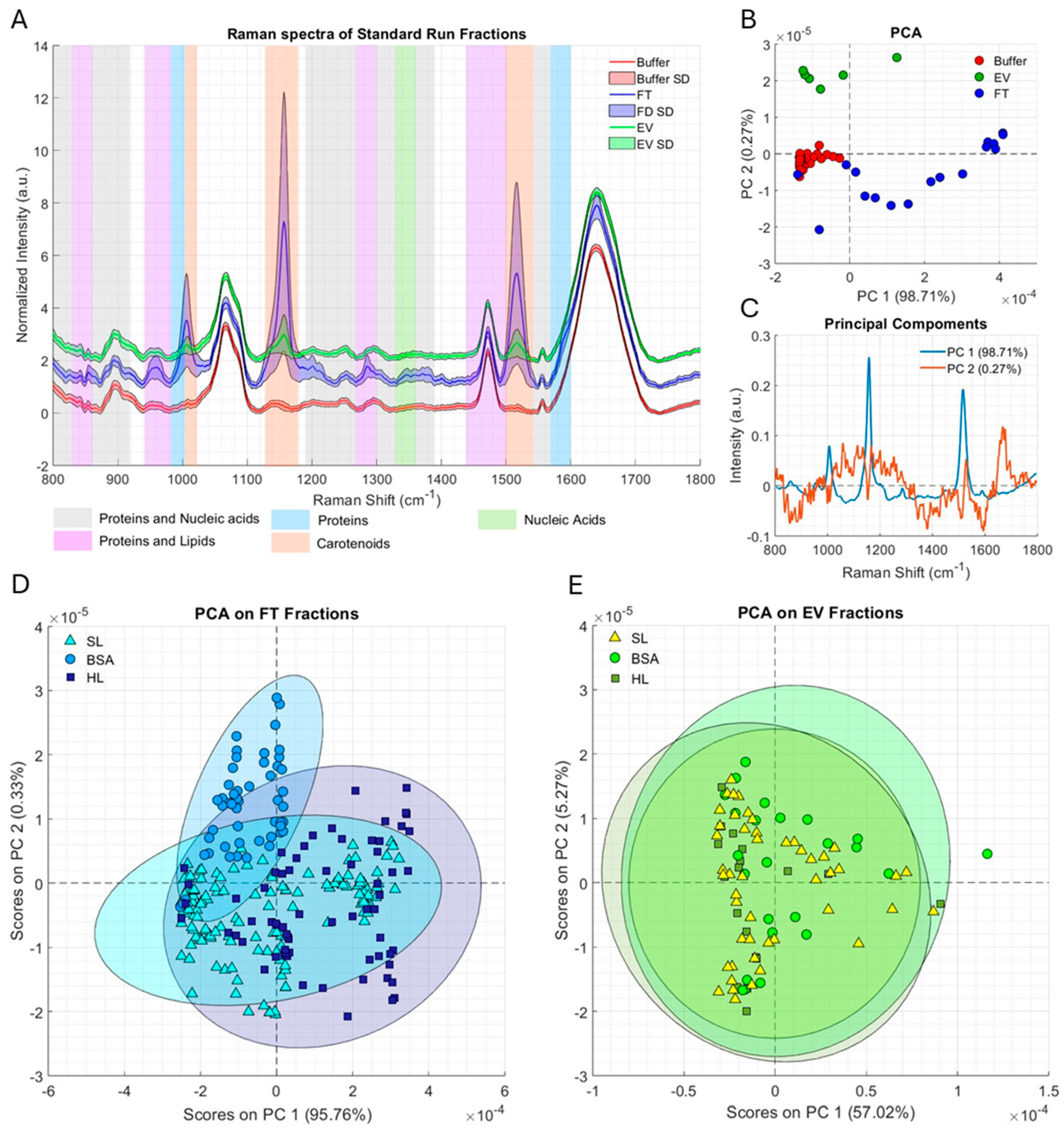
2.3. Inline Raman Spectroscopy Can Identify Separate Purification Phases and Their Deviations
3. Discussion
4. Materials and Methods
4.1. Platelet Supernatant Preparation
4.2. Anion-Exchange Chromatography
4.3. Reference EV (REV) Preparation with Multimodal Size-Exclusion Chromatography and Ultracentrifugation
4.4. Nanoparticle Tracking Analysis
4.5. SDS-PAGE and Western Blotting
4.6. Cryo-Transmission Electron Microscopy
4.7. Raman Measurement
4.8. Statistical Analysis
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herrmann, I.K.; Wood, M.J.A.; Fuhrmann, G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021, 16, 748–759. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; Carter, D.R.F.; Clayton, A.; Lambert, D.W.; Raposo, G.; Vader, P. Challenges and directions in studying cell–cell communication by extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2022, 23, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Bedina Zavec, A.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Wolfram, J.; Srivastava, S. Extracellular Vesicles in Cancer Detection: Hopes and Hypes. Trends Cancer 2021, 7, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Koch, L.F.; Best, T.; Wüstenhagen, E.; Adrian, K.; Rammo, O.; Saul, M.J. Novel insights into the isolation of extracellular vesicles by anion exchange chromatography. Front. Bioeng. Biotechnol. 2023, 11, 1298892. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrügger, U.; et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 2024, 13, e12404. [Google Scholar] [CrossRef] [PubMed]
- Watson, D.C.; Yung, B.C.; Bergamaschi, C.; Chowdhury, B.; Bear, J.; Stellas, D.; Morales-Kastresana, A.; Jones, J.C.; Felber, B.K.; Chen, X.; et al. Scalable, cGMP-compatible purification of extracellular vesicles carrying bioactive human heterodimeric IL-15/lactadherin complexes. J. Extracell. Vesicles 2018, 7, 1442088. [Google Scholar] [CrossRef] [PubMed]
- Multia, E.; Liangsupree, T.; Jussila, M.; Ruiz-Jimenez, J.; Kemell, M.; Riekkola, M.L. Automated On-Line Isolation and Fractionation System for Nanosized Biomacromolecules from Human Plasma. Anal. Chem. 2020, 92, 13058–13065. [Google Scholar] [CrossRef] [PubMed]
- Kaddour, H.; Lyu, Y.; Shouman, N.; Mohan, M.; Okeoma, C.M. Development of Novel High-Resolution Size-Guided Turbidimetry-Enabled Particle Purification Liquid Chromatography (PPLC): Extracellular Vesicles and Membraneless Condensates in Focus. Int. J. Mol. Sci. 2020, 21, 5361. [Google Scholar] [CrossRef]
- Kitchener, B.G.; Wainwright, J.; Parsons, A.J. A review of the principles of turbidity measurement. Prog. Phys. Geogr. Earth Environ. 2017, 41, 620–642. [Google Scholar] [CrossRef]
- Bachurski, D.; Schuldner, M.; Nguyen, P.H.; Malz, A.; Reiners, K.S.; Grenzi, P.C.; Babatz, F.; Schauss, A.C.; Hansen, H.P.; Hallek, M. Extracellular vesicle measurements with nanoparticle tracking analysis–An accuracy and repeatability comparison between NanoSight NS300 and ZetaView. J. Extracell. Vesicles 2019, 8, 1596016. [Google Scholar] [CrossRef] [PubMed]
- Normak, K.; Papp, M.; Ullmann, M.; Paganini, C.; Manno, M.; Bongiovanni, A.; Bergese, P.; Arosio, P. Multiparametric Orthogonal Characterization of Extracellular Vesicles by Liquid Chromatography Combined with In-Line Light Scattering and Fluorescence Detection. Anal. Chem. 2023, 95, 12443–12451. [Google Scholar] [CrossRef] [PubMed]
- Kitka, D.; Mihály, J.; Fraikin, J.-L.; Beke-Somfai, T.; Varga, Z. Detection and phenotyping of extracellular vesicles by size exclusion chromatography coupled with on-line fluorescence detection. Sci. Rep. 2019, 9, 19868. [Google Scholar] [CrossRef] [PubMed]
- Wiedmer, S.K.; Riekkola, M.L. Field-flow fractionation—An excellent tool for fractionation, isolation and/or purification of biomacromolecules. J. Chromatogr. A 2023, 1712, 464492. [Google Scholar] [CrossRef]
- Pittman, Z.A.; McCarthy, M.E.; Birtwistle, M.R.; Kitchens, C.L. Method for Improved Fluorescence Corrections for Molar Mass Characterization by Multi-Angle Light Scattering. Biomacromolecules 2022, 23, 3743. [Google Scholar] [CrossRef] [PubMed]
- Zini, J.; Saari, H.; Ciana, P.; Viitala, T.; Lõhmus, A.; Saarinen, J.; Yliperttula, M. Infrared and Raman spectroscopy for purity assessment of extracellular vesicles. Eur. J. Pharm. Sci. 2022, 172, 106135. [Google Scholar] [CrossRef] [PubMed]
- Gualerzi, A.; Kooijmans, S.A.A.; Niada, S.; Picciolini, S.; Brini, A.T.; Camussi, G.; Bedoni, M. Raman spectroscopy as a quick tool to assess purity of extracellular vesicle preparations and predict their functionality. J. Extracell. Vesicles 2019, 8, 1568780. [Google Scholar] [CrossRef] [PubMed]
- Imanbekova, M.; Suarasan, S.; Lu, Y.; Jurchuk, S.; Wachsmann-Hogiu, S. Recent advances in optical label-free characterization of extracellular vesicles. Nanophotonics 2022, 11, 2827–2863. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Ding, Q.; Li, C.; Chen, W.; Chen, X.; Lin, Q.; Wang, D.; Weng, Y.; Lin, D. Recent progress in surface-enhanced Raman spectroscopy-based biosensors for the detection of extracellular vesicles. Anal. Methods 2022, 14, 4161–4173. [Google Scholar] [CrossRef]
- Rautaniemi, K.; Zini, J.; Löfman, E.; Saari, H.; Haapalehto, I.; Laukka, J.; Vesamäki, S.; Efimov, A.; Yliperttula, M.; Laaksonen, T.; et al. Addressing challenges in the removal of unbound dye from passively labelled extracellular vesicles. Nanoscale Adv. 2021, 4, 226–240. [Google Scholar] [CrossRef]
- Saari, H.; Pusa, R.; Marttila, H.; Yliperttula, M.; Laitinen, S. Development of tandem cation exchange chromatography for high purity extracellular vesicle isolation: The effect of ligand steric availability. J. Chromatogr. A 2023, 1707, 464293. [Google Scholar] [CrossRef] [PubMed]
- Kosanović, M.; Milutinović, B.; Goč, S.; Mitić, N.; Janković, M. Ion-exchange chromatography purification of extracellular vesicles. Biotechniques 2017, 63, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Nishida, H.; An, S.Y.; Shetty, A.K.; Bartosh, T.J.; Prockop, D.J. Chromatographically isolated CD63+CD81+ extracellular vesicles from mesenchymal stromal cells rescue cognitive impairments after TBI. Proc. Natl. Acad. Sci. USA 2016, 113, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Sódar, B.W.; Kittel, Á.; Pálóczi, K.; Vukman, K.V.; Osteikoetxea, X.; Szabó-Taylor, K.; Németh, A.; Sperlágh, B.; Baranyai, T.; Giricz, Z.; et al. Low-density lipoprotein mimics blood plasma-derived exosomes and microvesicles during isolation and detection. Sci. Rep. 2016, 6, 24316. [Google Scholar] [CrossRef] [PubMed]
- Midekessa, G.; Godakumara, K.; Ord, J.; Viil, J.; Lättekivi, F.; Dissanayake, K.; Kopanchuk, S.; Rinken, A.; Andronowska, A.; Bhattacharjee, S.; et al. Zeta Potential of Extracellular Vesicles: Toward Understanding the Attributes that Determine Colloidal Stability. ACS Omega 2020, 5, 16701–16710. [Google Scholar] [CrossRef] [PubMed]
- Czamara, K.; Majzner, K.; Pacia, M.Z.; Kochan, K.; Kaczor, A.; Baranska, M. Raman spectroscopy of lipids: A review. J. Raman Spectrosc. 2015, 46, 4–20. [Google Scholar] [CrossRef]
- Rygula, A.; Majzner, K.; Marzec, K.M.; Kaczor, A.; Pilarczyk, M.; Baranska, M. Raman spectroscopy of proteins: A review. J. Raman Spectrosc. 2013, 44, 1061–1076. [Google Scholar] [CrossRef]
- Liangsupree, T.; Multia, E.; Saarinen, J.; Ruiz-Jimenez, J.; Kemell, M.; Riekkola, M.L. Raman spectroscopy combined with comprehensive gas chromatography for label-free characterization of plasma-derived extracellular vesicle subpopulations. Anal. Biochem. 2022, 647, 114672. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Zhao, T.; Song, N.; Pan, K.; Yang, Y.; Zhu, X.; Chen, P.; Zhang, J.; Xia, C. Platelets and platelet extracellular vesicles in drug delivery therapy: A review of the current status and future prospects. Front. Pharmacol. 2022, 13, 1026386. [Google Scholar] [CrossRef]
- Johnson, J.; Wu, Y.-W.; Blyth, C.; Lichtfuss, G.; Goubran, H.; Burnouf, T. Prospective Therapeutic Applications of Platelet Extracellular Vesicles. Trends Biotechnol. 2021, 39, 598–612. [Google Scholar] [CrossRef]
- Macernis, M.; Galzerano, D.; Sulskus, J.; Kish, E.; Kim, Y.-H.; Koo, S.; Valkunas, L.; Robert, B. Resonance Raman spectra of carotenoid molecules: Influence of methyl substitutions. J. Phys. Chem. A 2015, 119, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Desmarchelier, C.; Borel, P. Overview of carotenoid bioavailability determinants: From dietary factors to host genetic variations. Trends Food Sci. Technol. 2017, 69, 270–280. [Google Scholar] [CrossRef]
- Barkur, S.; Bankapur, A.; Chidangil, S.; Mathur, D. Effect of infrared light on live blood cells: Role of β-carotene. J. Photochem. Photobiol. B Biol. 2017, 171, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.K.; Cho, Y.K.; Lee, C.Y.; Lee, H.; Castro, C.M.; Lee, H. Characterization and modulation of surface charges to enhance extracellular vesicle isolation in plasma. Theranostics 2022, 12, 1988–1998. [Google Scholar] [CrossRef]
- Van Deun, J.; Jo, A.; Li, H.; Lin, H.; Weissleder, R.; Im, H.; Lee, H. Integrated Dual-Mode Chromatography to Enrich Extracellular Vesicles from Plasma. Adv. Biosyst. 2020, 4, 1900310. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Li, Z.; Wei, S.; Chi, X.; Yan, X.; Lv, H.; Zhao, L.; Zhao, L. Combination of size-exclusion chromatography and ion exchange adsorption for improving the proteomic analysis of plasma-derived extracellular vesicles. Proteomics 2023, 23, e2200364. [Google Scholar] [CrossRef]
- Rojalin, T.; Kurki, L.; Laaksonen, T.; Viitala, T.; Kostamovaara, J.; Gordon, K.C.; Galvis, L.; Wachsmann-Hogiu, S.; Strachan, C.J.; Yliperttula, M. Fluorescence-suppressed time-resolved Raman spectroscopy of pharmaceuticals using complementary metal-oxide semiconductor (CMOS) single-photon avalanche diode (SPAD) detector. Anal. Bioanal. Chem. 2016, 408, 761–774. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak Region (cm−1) | Group/Bond | Assignment |
|---|---|---|
| 750–870 | Tyr, Trp | Protein |
| 850–900 | C-O-O | Lipid |
| 810–900 | DNA backbone | Nucleic acid |
| 1000 | Phe | Protein |
| 1000 | CH3 | Carotenoid |
| 900–1050 | C-H | Lipid |
| 1060–1180 | C-C | Lipid/carotenoid |
| 1250–1280 | =CH | Lipid |
| 1230–1270 | Amide III | Protein |
| 1250 | CMP | Nucleic acid |
| 1300 | CH2 | Lipid |
| 1450 | CH2-CH3 | Protein/lipid |
| 1520 | Conjugated C=C-bonds | Carotenoid |
| 1580 | GMP, dTMP | Nucleic acid |
| 1610–1620 | Tyr, Trp, Phe | Protein |
| 1650–1670 | Amide I | Protein |
| Runs | Sensitivity | Specificity |
|---|---|---|
| Standard load | ||
| Buffer | 0.953 ± 0.002 | 0.896 ± 0.021 |
| FT | 0.875 ± 0.125 | 0.939 ± 0.041 |
| EV | 0.8435 ± 0.0315 | 0.983 ± 0.017 |
| High load | ||
| Buffer | 0.752 ± 0.122 | 0.913 ± 0.004 |
| FT | 0.897 ± 0.022 | 0.847 ± 0.082 |
| EV | 0.937 ± 0.062 | 0.898 ± 0.034 |
| 1% BSA | ||
| Buffer | 0.860 ± 0.139 | 0.8375 ± 0.037 |
| FT | 0.869 ± 0.005 | 0.910 ± 0.067 |
| EV | 0.875 ± 0.002 | 0.932 ± 0.068 |
| Step | Starting Point (mL) | Total Volume (mL) | A (%) | B (%) | Fractionation |
|---|---|---|---|---|---|
| Sample injection and wash | 0 | 15 | 100 | 0 | One 15 mL fraction (flowthrough, FT) |
| Elution | 15 | 5 | 50 | 50 | 4 × 1 mL fractions, starting after 1 mL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saari, H.; Marttila, H.; Poranen, M.M.; Oksanen, H.M.; Zini, J.; Laitinen, S. Inline Raman Spectroscopy Provides Versatile Molecular Monitoring for Platelet Extracellular Vesicle Purification with Anion-Exchange Chromatography. Int. J. Mol. Sci. 2024, 25, 8130. https://doi.org/10.3390/ijms25158130
Saari H, Marttila H, Poranen MM, Oksanen HM, Zini J, Laitinen S. Inline Raman Spectroscopy Provides Versatile Molecular Monitoring for Platelet Extracellular Vesicle Purification with Anion-Exchange Chromatography. International Journal of Molecular Sciences. 2024; 25(15):8130. https://doi.org/10.3390/ijms25158130
Chicago/Turabian StyleSaari, Heikki, Heli Marttila, Minna M. Poranen, Hanna M. Oksanen, Jacopo Zini, and Saara Laitinen. 2024. "Inline Raman Spectroscopy Provides Versatile Molecular Monitoring for Platelet Extracellular Vesicle Purification with Anion-Exchange Chromatography" International Journal of Molecular Sciences 25, no. 15: 8130. https://doi.org/10.3390/ijms25158130
APA StyleSaari, H., Marttila, H., Poranen, M. M., Oksanen, H. M., Zini, J., & Laitinen, S. (2024). Inline Raman Spectroscopy Provides Versatile Molecular Monitoring for Platelet Extracellular Vesicle Purification with Anion-Exchange Chromatography. International Journal of Molecular Sciences, 25(15), 8130. https://doi.org/10.3390/ijms25158130