
Genome-Wide Association and RNA-Seq Analyses Reveal a Potential Candidate Gene Related to Oil Content in Soybean Seeds
Abstract
:1. Introduction
2. Results
2.1. Statistical Analysis of Oil Content
2.2. Population Structure Analysis
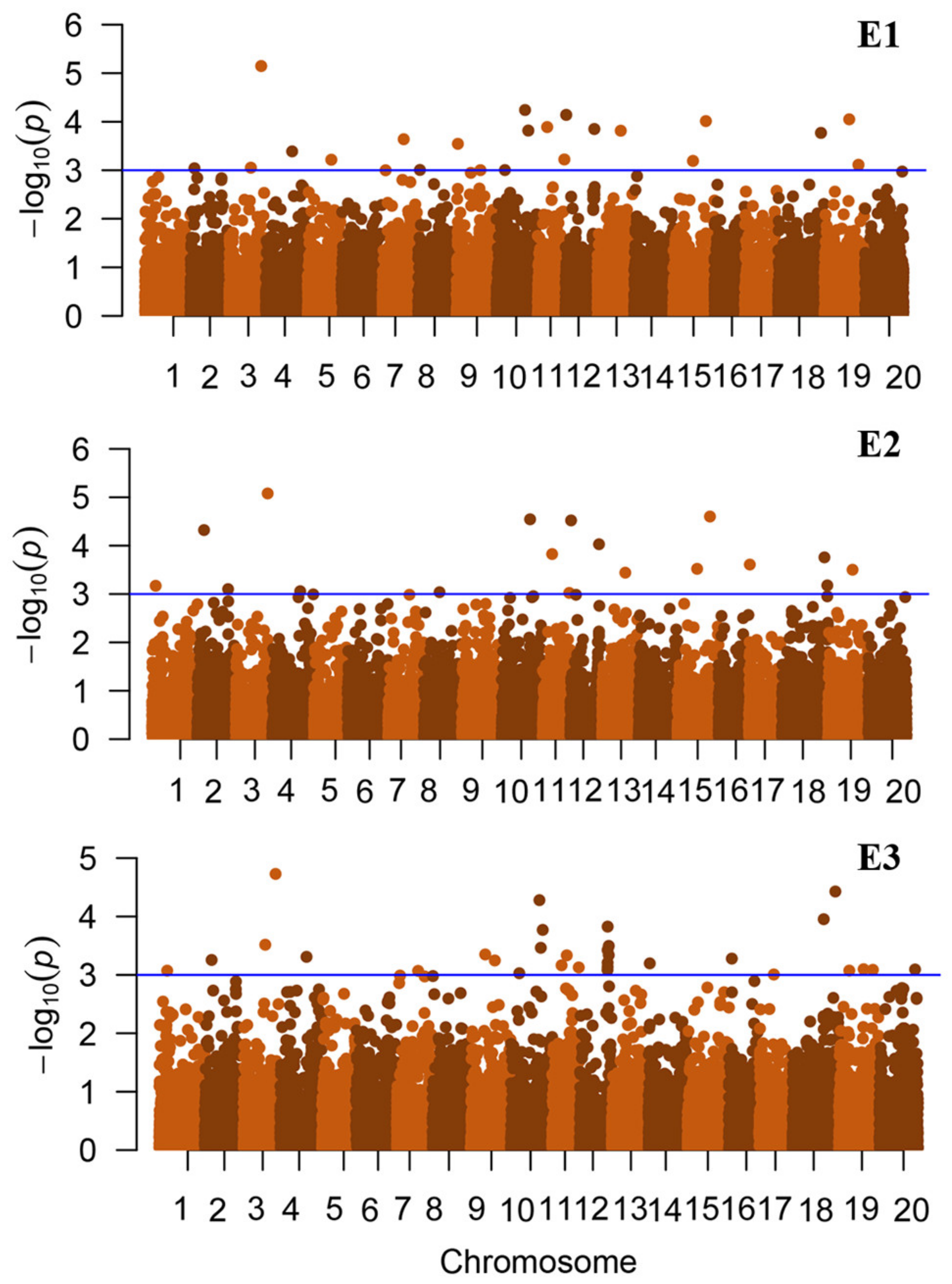
2.3. Genome-Wide Association Study
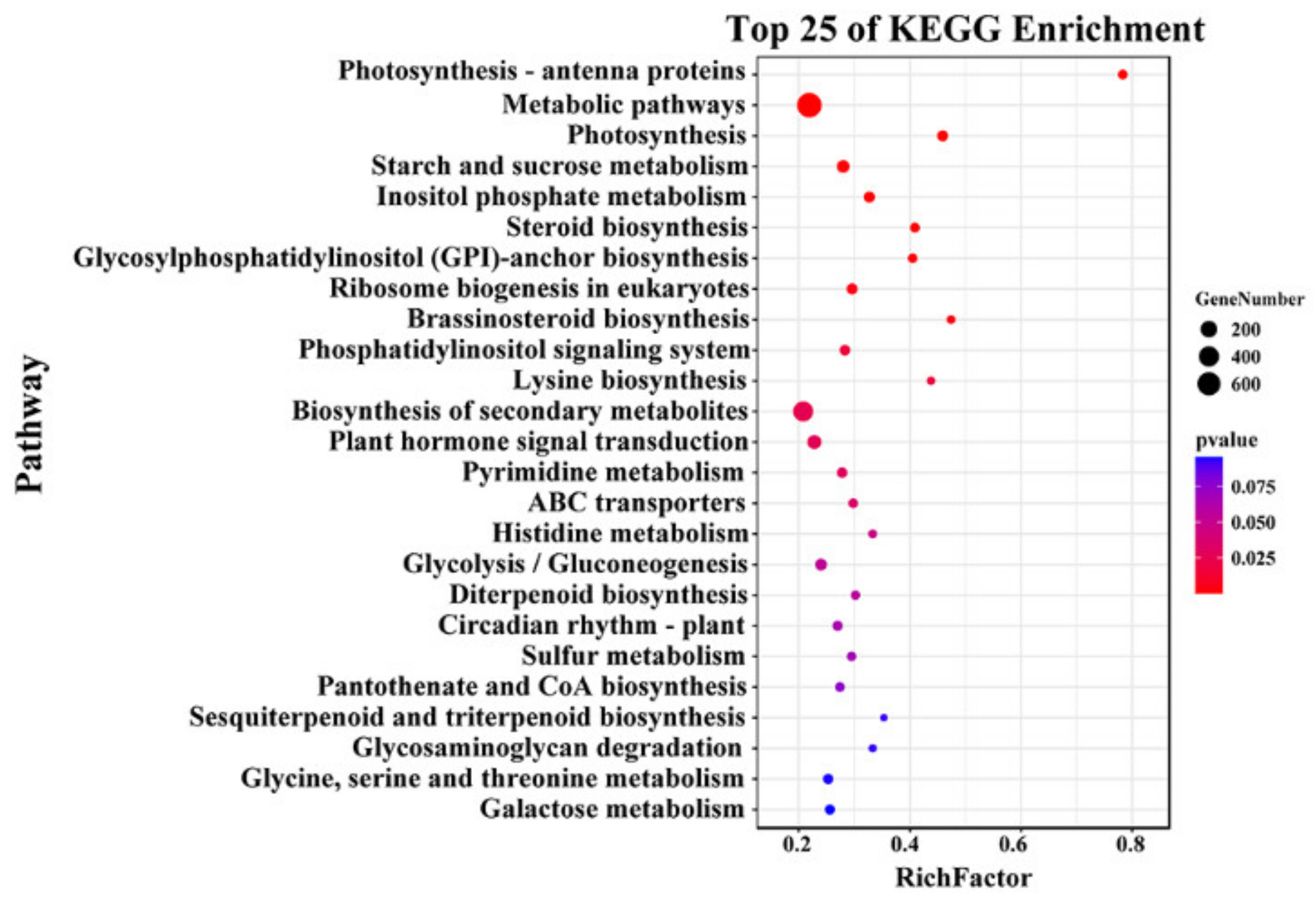
2.4. Functional Prediction of Candidate Genes by GWAS and RNA-Seq Analysis
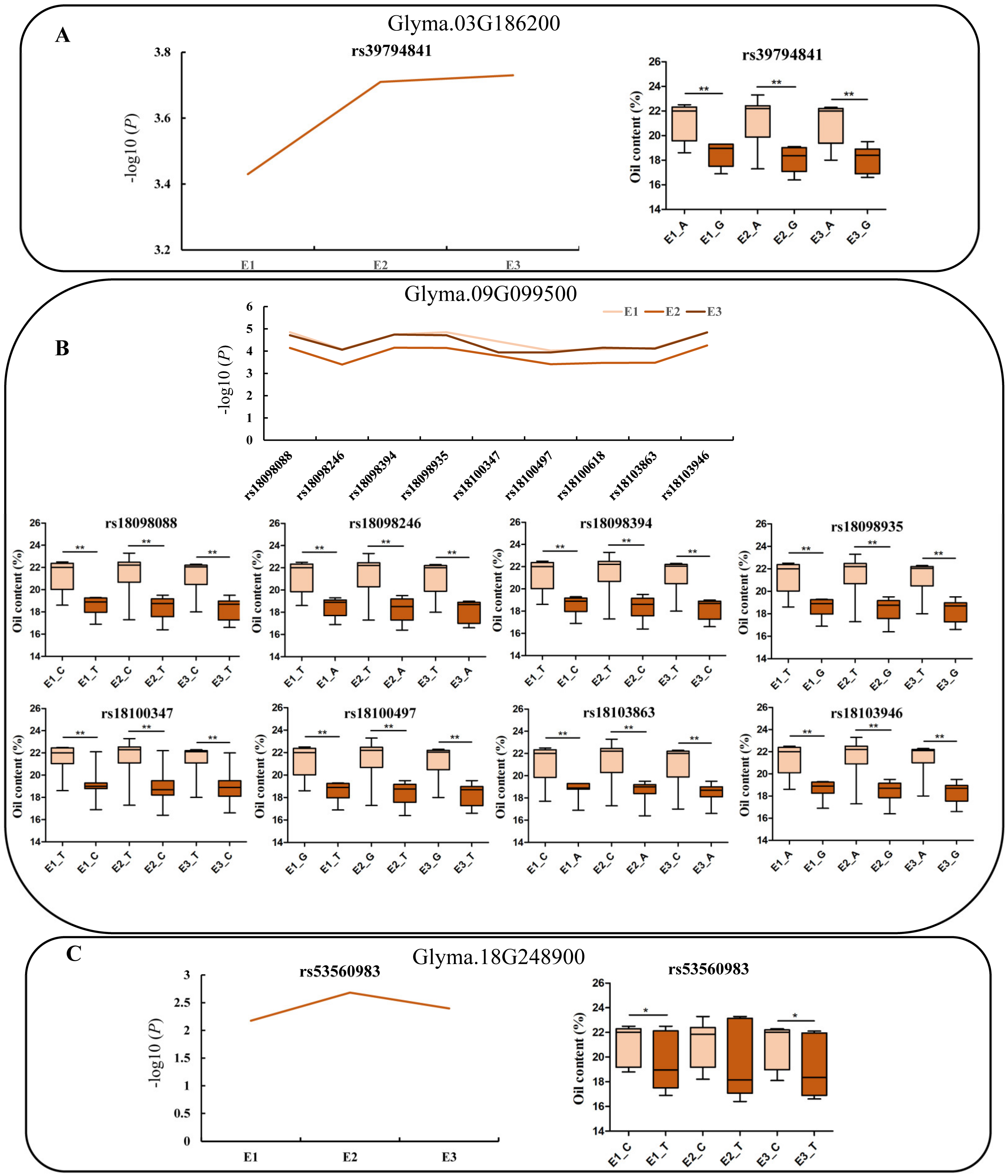
2.5. Gene-Based Association and Haplotype Analysis of Candidate Genes
2.6. Co–Expression Analysis of Transcription Factors and Candidate Genes
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Germplasm Population Genotype Analysis
4.3. Population Structure Evaluation, Linkage Disequilibrium, and Genome-Wide Association Analysis
4.4. Transcriptome Sequencing Analysis
4.5. Prediction of Candidate Genes
4.6. Co-Expression Analysis
4.7. Quantitative Real-Time PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, N.; Xu, C.; Li-Beisson, Y.; Philippar, K. Fatty acid and lipid transport in plant cells. Trends Plant Sci. 2016, 21, 145–158. [Google Scholar] [CrossRef]
- Yang, X.; Ma, H.; Zhang, P.; Yan, J.; Guo, Y.; Song, T.; Li, J. Characterization of QTL for oil content in maize kernel. Theor. Appl. Genet. 2012, 125, 1169–1179. [Google Scholar] [CrossRef]
- Jin, H.; Yang, X.; Zhao, H.; Song, X.; Tsvetkov, Y.D.; Wu, Y.; Gao, Q.; Zhang, R.; Zhang, J. Genetic analysis of protein content and oil content in soybean by genome-wide association study. Front. Plant Sci. 2023, 14, 1182771. [Google Scholar] [CrossRef]
- Xiao, Z.; Zhang, C.; Qu, C.; Wei, L.; Zhang, L.; Yang, B.; Lu, K.; Li, J. Identification of candidate genes regulating seed oil content by QTL mapping and transcriptome sequencing in Brassica napus. Front. Plant Sci. 2022, 13, 1067121. [Google Scholar] [CrossRef]
- Liu, N.; Huang, L.; Chen, W.; Wu, B.; Pandey, M.K.; Luo, H.; Zhou, X.; Guo, J.; Chen, H.; Huai, D.; et al. Dissection of the genetic basis of oil content in Chinese peanut cultivars through association mapping. BMC Genet. 2020, 21, 60. [Google Scholar] [CrossRef]
- Li, H.; Che, R.; Zhu, J.; Yang, X.; Li, J.; Fernie, A.R.; Yan, J. Multi-omics-driven advances in the understanding of triacylglycerol biosynthesis in oil seeds. Plant J. 2024, 117, 999–1017. [Google Scholar] [CrossRef]
- Gibellini, F.; Smith, T.K. The Kennedy pathway--De novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 2010, 62, 414–428. [Google Scholar] [CrossRef]
- Cao, J.; Li, J.; Li, D.; Tobin, J.F.; Gimeno, R.E. Molecular identification of microsomal acyl-CoA: Glycerol-3-phosphate acyltransferase, a key enzyme in de novo triacylglycerol synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 19695–19700. [Google Scholar] [CrossRef]
- Eastmond, P.J.; Quettier, A.L.; Kroon, J.T.; Craddock, C.; Adams, N.; Slabas, A.R. Phosphatidic acid phosphohydrolase 1 and 2 regulate phospholipid synthesis at the endoplasmic reticulum in Arabidopsis. Plant Cell 2010, 22, 2796–2811. [Google Scholar] [CrossRef]
- Cases, S.; Smith, S.J.; Zheng, Y.W.; Myers, H.M.; Lear, S.R.; Sande, E.; Novak, S.; Collins, C.; Welch, C.B.; Lusis, A.J.; et al. Identification of a gene encoding an acyl CoA: Diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. USA 1998, 95, 13018–13023. [Google Scholar] [CrossRef]
- Murphy, D.J. Storage lipid bodies in plants and other organisms. Prog. Lipid Res. 1990, 29, 299–324. [Google Scholar]
- Zheng, P.; Allen, W.B.; Roesler, K.; Williams, M.E.; Zhang, S.; Li, J.; Glassman, K.; Ranch, J.; Nubel, D.; Solawetz, W.; et al. A phenylalanine in DGAT is a key determinant of oil content and composition in maize. Nat. Genet. 2008, 40, 367–372. [Google Scholar] [CrossRef]
- Kim, W.J.; Kang, B.H.; Kang, S.; Shin, S.; Chowdhury, S.; Jeong, S.C.; Choi, M.S.; Park, S.K.; Moon, J.K.; Ryu, J.; et al. A genome-wide association study of protein, oil, and amino acid content in wild soybean (Glycine soja). Plants 2023, 12, 1665. [Google Scholar] [CrossRef]
- Xiao, Z.; Zhang, C.; Tang, F.; Yang, B.; Zhang, L.; Liu, J.; Huo, Q.; Wang, S.; Li, S.; Wei, L.; et al. Identification of candidate genes controlling oil content by combination of genome-wide association and transcriptome analysis in the oilseed crop Brassica napus. Biotechnol. Biofuels Bioprod. 2019, 12, 216. [Google Scholar] [CrossRef]
- Zhao, X.; Dong, H.; Chang, H.; Zhao, J.; Teng, W.; Qiu, L.; Li, W.; Han, Y. Genome wide association mapping and candidate gene analysis for hundred seed weight in soybean [Glycine max (L.) Merrill]. BMC Genom. 2019, 20, 648. [Google Scholar] [CrossRef]
- Liu, J.; Dong, L.; Duan, R.; Hu, L.; Zhao, Y.; Zhang, L.; Wang, X. Transcriptomic analysis reveals the regulatory networks and hub genes controlling the unsaturated fatty acid contents of developing seed in soybean. Front. Plant Sci. 2022, 13, 876371. [Google Scholar] [CrossRef]
- Wei, T.; He, Z.; Tan, X.; Liu, X.; Yuan, X.; Luo, Y.; Hu, S. An integrated RNA-Seq and network study reveals a complex regulation process of rice embryo during seed germination. Biochem. Biophys. Res. Commun. 2015, 464, 176–181. [Google Scholar] [CrossRef]
- Hyten, D.L.; Pantalone, V.R.; Sams, C.E.; Saxton, A.M.; Landau-Ellis, D.; Stefaniak, T.R.; Schmidt, M.E. Seed quality QTL in a prominent soybean population. Theor. Appl. Genet. 2004, 109, 552–561. [Google Scholar] [CrossRef]
- Kabelka, E.A.; Diers, B.W.; Fehr, W.R.; Leroy, A.R.; Baianu, I.C.; You, T.; Neece, D.J.; Nelson, R.L. Putative alleles for increased yield from soybean plant introductions. Crop Sci. 2004, 44, 784–791. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, G.; Green, M.; Scott, R.A.; Song, Q.; Hyten, D.L.; Cregan, P.B. Identification and validation of quantitative trait loci for seed yield, oil and protein contents in two recombinant inbred line populations of soybean. Mol. Genet. Genom. 2014, 289, 935–949. [Google Scholar] [CrossRef]
- Bachlava, E.; Dewey, R.E.; Burton, J.W.; Cardinal, A.J. Mapping and comparison of quantitative trait loci for oleic acid seed content in two segregating soybean populations. Crop Sci. 2009, 49, 433–442. [Google Scholar] [CrossRef]
- Qi, Z.; Wu, Q.; Han, X.; Sun, Y.; Du, X.; Liu, C.; Jiang, H.; Hu, G.; Chen, Q. Soybean oil content QTL mapping and integrating with meta-analysis method for mining genes. Euphytica 2011, 179, 499–514. [Google Scholar] [CrossRef]
- Mao, T.; Jiang, Z.; Han, Y.; Teng, W.; Zhao, X.; Li, W. Morris Identification of quantitative trait loci underlying seed protein and oil contents of soybean across multi-genetic backgrounds and environments. Plant Breed. 2013, 132, 630–641. [Google Scholar] [CrossRef]
- Mansur, L.M.; Lark, K.G.; Kross, H.; Oliveira, A. Interval mapping of quantitative trait loci for reproductive, morphological, and seed traits of soybean (Glycine max L.). Theor. Appl. Genet. 1993, 86, 907–913. [Google Scholar] [CrossRef]
- Li, H.; Zhao, T.; Wang, Y.; Yu, D.; Chen, S.; Zhou, R.; Gai, J. Genetic structure composed of additive QTL, epistatic QTL pairs and collective unmapped minor QTL conferring oil content and fatty acid components of soybeans. Euphytica 2011, 182, 117–132. [Google Scholar] [CrossRef]
- Diers, B.W.; Shoemaker, R.C. Restriction fragment length polymorphism analysis of soybean fatty acid content. J. Am. Oil Chem. Soc. 1992, 69, 1242–1244. [Google Scholar] [CrossRef]
- Ha, B.K.; Kim, H.J.; Velusamy, V.; Vuong, T.D.; Nguyen, H.T.; Shannon, J.G.; Lee, J.D. Identification of quantitative trait loci controlling linolenic acid concentration in PI483463 (Glycine soja). Theor. Appl. Genet. 2014, 127, 1501–1512. [Google Scholar] [CrossRef]
- Eskandari, M.; Cober, E.R.; Rajcan, I. Genetic control of soybean seed oil: I. QTL and genes associated with seed oil concentration in RIL populations derived from crossing moderately high-oil parents. Theor. Appl. Genet. 2013, 126, 483–495. [Google Scholar] [CrossRef]
- Panthee, D.R.; Pantalone, V.R.; West, D.R.; Saxton, A.M.; Sams, C.E. Quantitative trait loci for seed protein and oil concentration, and seed size in soybean. Crop Sci. 2005, 45, 2015–2022. [Google Scholar] [CrossRef]
- Specht, J.E.; Chase, K.; Macrander, M.; Graef, G.L.; Chung, J.; Markwell, J.P.; Germann, M.; Orf, J.H.; Lark, K.G. Soybean response to water: A QTL analysis of drought tolerance. Crop Sci. 2001, 41, 493–509. [Google Scholar] [CrossRef]
- Reinprecht, Y.; Poysa, V.W.; Yu, K.; Rajcan, I.; Ablett, G.R.; Pauls, K.P. Seed and agronomic QTL in low linolenic acid, lipoxygenase-free soybean (Glycine max (L.) Merrill) germplasm. Genome 2006, 49, 1510–1527. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, G.L.; Green, M.; Scott, R.A.; Hyten, D.L.; Cregan, P.B. Quantitative trait locus analysis of saturated fatty acids in a population of recombinant inbred lines of soybean. Mol. Breed. 2012, 30, 1163–1179. [Google Scholar] [CrossRef]
- Zhou, Y.; Cai, H.; Xiao, J.; Li, X.; Zhang, Q.; Lian, X. Over-expression of aspartate aminotransferase genes in rice resulted in altered nitrogen metabolism and increased amino acid content in seeds. Theor. Appl. Genet. 2009, 118, 1381–1390. [Google Scholar] [CrossRef]
- Menon, D.; Bhapkar, A.; Manchandia, B.; Charak, G.; Rathore, S.; Jha, R.M.; Nahak, A.; Mondal, M.; Omrane, M.; Bhaskar, A.K.; et al. ARL8B mediates lipid droplet contact and delivery to lysosomes for lipid remobilization. Cell Rep. 2023, 42, 113203. [Google Scholar] [CrossRef]
- Luo, Q.; Zhu, H.; Wang, C.; Li, Y.; Zou, X.; Hu, Z. A U-Box type E3 ubiquitin ligase Prp19-Like protein negatively regulates lipid accumulation and cell size in Chlamydomonas reinhardtii. Front. Microbiol. 2022, 13, 860024. [Google Scholar] [CrossRef]
- Jia, G.; Huang, X.; Zhi, H.; Zhao, Y.; Zhao, Q.; Li, W.; Chai, Y.; Yang, L.; Liu, K.; Lu, H.; et al. A haplotype map of genomic variations and genome-wide association studies of agronomic traits in foxtail millet (Setaria italica). Nat. Genet. 2013, 45, 957–961. [Google Scholar] [CrossRef]
- Li, H.; Peng, Z.; Yang, X.; Wang, W.; Fu, J.; Wang, J.; Han, Y.; Chai, Y.; Guo, T.; Yang, N.; et al. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 2013, 45, 43–50. [Google Scholar] [CrossRef]
- Liang, Q.; Chen, L.; Yang, X.; Yang, H.; Liu, S.; Kou, K.; Fan, L.; Zhang, Z.; Duan, Z.; Yuan, Y.; et al. Natural variation of Dt2 determines branching in soybean. Nat. Commun. 2022, 13, 6429. [Google Scholar] [CrossRef]
- Duan, Z.; Zhang, M.; Zhang, Z.; Liang, S.; Fan, L.; Yang, X.; Yuan, Y.; Pan, Y.; Zhou, G.; Liu, S.; et al. Natural allelic variation of GmST05 controlling seed size and quality in soybean. Plant Biotechnol. J. 2022, 20, 1807–1818. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Dong, L.; Fang, C.; Liu, S.; Kong, L.; Cheng, Q.; Chen, L.; Su, T.; Nan, H.; Zhang, D.; et al. Stepwise selection on homeologous PRR genes controlling flowering and maturity during soybean domestication. Nat. Genet. 2020, 52, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ma, J.; Yang, X.; Liu, Z.; Liu, Y.; Liu, X.; Liang, S.; Duan, Z.; Wang, Z.; Yang, X.; et al. Natural allelic diversities of GmPrx16 confer drought tolerance in soybean. Plant Biotechnol. J. 2024, 22, 535–537. [Google Scholar] [CrossRef]
- Karikari, B.; Li, S.; Bhat, J.A.; Cao, Y.; Kong, J.; Yang, J.; Gai, J.; Zhao, T. Genome-wide ddetection of major and epistatic effect QTLs for seed protein and oil content in soybean under multiple environments using high-density bin map. Int. J. Mol. Sci. 2019, 20, 979. [Google Scholar] [CrossRef]
- Tian, X.; Zhang, K.; Liu, S.; Sun, X.; Li, X.; Song, J.; Qi, Z.; Wang, Y.; Fang, Y.; Wang, J.; et al. Quantitative trait locus aanalysis of protein and oil content in response to planting density in soybean (Glycine max [L.] Merri.) Seeds Based SNP Link. Mapp. Front. Genet. 2020, 11, 563. [Google Scholar] [CrossRef]
- Yang, Y.; Zhu, X.; Cui, R.; Wang, R.; Li, H.; Wang, J.; Chen, H.; Zhang, D. Identification of soybean phosphorous efficiency QTLs and genes using chlorophyll fluorescence parameters through GWAS and RNA-seq. Planta 2021, 254, 110. [Google Scholar] [CrossRef]
- Song, J.; Xu, R.; Guo, Q.; Wu, C.; Li, Y.; Wang, X.; Wang, J.; Qiu, L. An omics strategy increasingly improves the discovery of genetic loci and genes for seed-coat color formation in soybean. Mol. Breed. 2023, 43, 71. [Google Scholar] [CrossRef]
- Ge, S.; Zhang, R.; Wang, Y.; Sun, P.; Chu, J.; Li, J.; Sun, P.; Wang, J.; Hetherington, A.M.; Liang, Y. The Arabidopsis Rab protein RABC1 affects stomatal development by regulating lipid droplet dynamics. Plant Cell 2022, 34, 4274–4292. [Google Scholar] [CrossRef]
- Khatoon, U.; Prasad, V.; Sawant, S.V. Expression dynamics and a loss-of-function of Arabidopsis RabC1 GTPase unveil its role in plant growth and seed development. Planta 2023, 257, 89. [Google Scholar] [CrossRef]
- Eroglu, S.; Giehl, R.F.H.; Meier, B.; Takahashi, M.; Terada, Y.; Ignatyev, K.; Andresen, E.; Küpper, H.; Peiter, E.; von Wirén, N. Metal tolerance protein 8 mediates manganese homeostasis and iron reallocation during seed development and germination. Plant Physiol. 2017, 174, 1633–1647. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, X.; Zhao, F.; Li, Q.; Niu, S.; Wei, W.; Zhang, W.; Ma, B.; Chen, S.; Zhang, J. Soybean GmDREBL increases lipid content in seeds of transgenic Arabidopsis. Sci. Rep. 2016, 6, 34307. [Google Scholar] [CrossRef]
- Su, Y.; Liang, W.; Liu, Z.; Wang, Y.; Zhao, Y.; Ijaz, B.; Hua, J. Overexpression of GhDof1 improved salt and cold tolerance and seed oil content in Gossypium hirsutum. J. Plant Physiol. 2017, 218, 222–234. [Google Scholar] [CrossRef]
- Blatti, J.L.; Beld, J.; Behnke, C.A.; Mendez, M.; Mayfield, S.P.; Burkart, M.D. Manipulating fatty acid biosynthesis in microalgae for biofuel through protein-protein interactions. PLoS ONE 2012, 7, e42949. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Env | Locus Name | Chr. | Pos | −Log10(p) | Known QTLs |
|---|---|---|---|---|---|
| E2 | rs4477127 | 1 | 4,477,127 | 3.17 | [18] |
| E3 | rs9691814 | 1 | 9,691,814 | 3.07 | [18] |
| E2 | rs38563127 | 2 | 38,563,127 | 3.1 | |
| E1 | rs4964805 | 2 | 4,964,805 | 3.04 | |
| E3/E2 | rs8406199 | 2 | 8,406,199 | 3.26/4.32 | [19] |
| E3/E1 | rs26910547 | 3 | 26,910,547 | 3.51/3.05 | [20] |
| E3/E2/E1 | rs39790117 | 3 | 39,790,117 | 4.73/5.08/5.15 | [21] |
| E3/E1 | rs32815793 | 4 | 32,815,793 | 3.31/3.39 | |
| E2 | rs35005741 | 4 | 35,005,741 | 3.06 | |
| E1 | rs29415473 | 5 | 29,415,473 | 3.22 | |
| E3/E1 | rs26521346 | 7 | 26,521,346 | 3.07/3.64 | |
| E2 | rs19738745 | 8 | 19,738,745 | 3.04 | |
| E1 | rs2147095 | 8 | 2,147,095 | 3.01 | [22] |
| E3 | rs18054299 | 9 | 18,054,299 | 3.35 | [23] |
| E1 | rs1843998 | 9 | 1,843,998 | 3.54 | [23,24] |
| E3/E1 | rs29978290 | 9 | 29,978,290 | 3.25/3 | [23] |
| E3/E1 | rs10499458 | 10 | 10,499,458 | 3.03/3 | [25] |
| E3/E2/E1 | rs35631359 | 10 | 35,631,359 | 4.28/4.55/4.24 | [23] |
| E3 | rs37347397 | 10 | 37,347,397 | 3.46 | [23] |
| E3/E1 | rs39714349 | 10 | 39,714,349 | 3.77/3.82 | [23] |
| E3/E2/E1 | rs11758049 | 11 | 11,758,049 | 3.16/3.83/3.89 | [26] |
| E3 | rs18218600 | 11 | 18,218,600 | 3.33 | [26] |
| E3/E2/E1 | rs33193693 | 11 | 33,193,693 | 3.13/3.02/3.22 | [25] |
| E3 | rs34470363 | 12 | 34,470,363 | 3.08 | [27] |
| E3 | rs34585918 | 12 | 34,585,918 | 3.43 | [27] |
| E3 | rs34600639 | 12 | 34,600,639 | 3.14 | [27] |
| E3 | rs34601911 | 12 | 34,601,911 | 3.21 | [27] |
| E3 | rs34654632 | 12 | 34,654,632 | 3.83 | [27] |
| E3/E2/E1 | rs35979450 | 12 | 35,979,450 | 3.34/4.03/3.85 | [28,29] |
| E3 | rs36025778 | 12 | 36,025,778 | 3.5 | [28,29] |
| E2/E1 | rs917378 | 12 | 917,378 | 4.52/4.14 | |
| E2/E1 | rs28963005 | 13 | 28,963,005 | 3.44/3.81 | [28,30] |
| E3 | rs1731411 | 14 | 1,731,411 | 3.2 | |
| E2/E1 | rs24670157 | 15 | 24,670,157 | 3.52/3.19 | [29,31] |
| E2/E1 | rs40773008 | 15 | 40,773,008 | 4.6/4.01 | [32] |
| E3 | rs3534128 | 16 | 3,534,128 | 3.28 | [19] |
| Env | SNP | Gene ID | Log2FC | Arabidopsis | Description |
|---|---|---|---|---|---|
| E3 | rs29978290 | Glyma.09G123900 | 3.11 | / | / |
| E3 | rs18218600 | Glyma.11G170100 | 3.16 | AT1G01040 | Dicer-like 1 |
| E3 | rs39790117 | Glyma.03G186200 | 3.19 | AT5G03530 | RAB GTPase homolog C2A |
| E2 | rs19738745 | Glyma.08G235400 | 3.23 | AT1G73260 | Kunitz trypsin inhibitor 1 |
| E3 | rs35979450 | Glyma.12G198700 | 3.25 | AT5G62990 | Ubiquitin carboxyl-terminal hydrolase family protein |
| E1 | rs1843998 | Glyma.09G022000 | 3.3 | AT3G54950 | Patatin-like protein 6 |
| E3 | rs37347397 | Glyma.10G139700 | 3.72 | AT3G62020 | Germin-like protein 10 |
| E3 | rs53468862 | Glyma.18G246800 | 3.96 | AT3G25500 | Formin homology 1 |
| E3 | rs35979450 | Glyma.12G199300 | 4.06 | AT4G03270 | Cyclin D6 |
| E3 | rs29978290 | Glyma.09G124200 | 4.21 | AT1G70670 | Caleosin-related family protein |
| E3 | rs53468862 | Glyma.18G248900 | 5 | / | / |
| E2 | rs24670157 | Glyma.15G201500 | 5.12 | / | / |
| E3 | rs3534128 | Glyma.16G037600 | 5.4 | AT5G39890 | Protein of unknown function |
| E2 | rs4477127 | Glyma.01G040600 | 8.12 | AT1G10350 | DNAJ heat shock family protein |
| E2 | rs57089675 | Glyma.18G293300 | 8.35 | AT2G04090 | MATE efflux family protein |
| E2 | rs1251586 | Glyma.17G016900 | 8.7 | AT3G06140 | RING/U-box superfamily protein |
| E1 | rs4964805 | Glyma.02G054400 | 9.04 | AT5G66440 | / |
| E2 | rs24670157 | Glyma.15G201000 | −7.71 | AT1G21280 | / |
| E3 | rs44446658 | Glyma.20G208400 | −6.94 | AT1G62510 | Bifunctional inhibitor/lipid-transfer protein/seed storage 2S albumin superfamily protein |
| E3 | rs33193693 | Glyma.11G238300 | −3.64 | AT2G22250 | Aspartate aminotransferase |
| E3 | rs18054299 | Glyma.09G099500 | −3.54 | AT1G16310 | Cation efflux family protein |
| E3 | rs18196571 | Glyma.17G174700 | −3.07 | AT5G09910 | Ras-related small GTP-binding family protein |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, H.; Han, D.; Yan, X.; Zhang, L.; Liang, J.; Lu, W. Genome-Wide Association and RNA-Seq Analyses Reveal a Potential Candidate Gene Related to Oil Content in Soybean Seeds. Int. J. Mol. Sci. 2024, 25, 8134. https://doi.org/10.3390/ijms25158134
Jia H, Han D, Yan X, Zhang L, Liang J, Lu W. Genome-Wide Association and RNA-Seq Analyses Reveal a Potential Candidate Gene Related to Oil Content in Soybean Seeds. International Journal of Molecular Sciences. 2024; 25(15):8134. https://doi.org/10.3390/ijms25158134
Chicago/Turabian StyleJia, Hongchang, Dezhi Han, Xiaofei Yan, Lei Zhang, Jili Liang, and Wencheng Lu. 2024. "Genome-Wide Association and RNA-Seq Analyses Reveal a Potential Candidate Gene Related to Oil Content in Soybean Seeds" International Journal of Molecular Sciences 25, no. 15: 8134. https://doi.org/10.3390/ijms25158134