
The Complete Mitogenome of Apostasia fujianica Y.Li & S.Lan and Comparative Analysis of Mitogenomes across Orchidaceae



Abstract
1. Introduction
2. Results
2.1. Mitogenome Characteristics
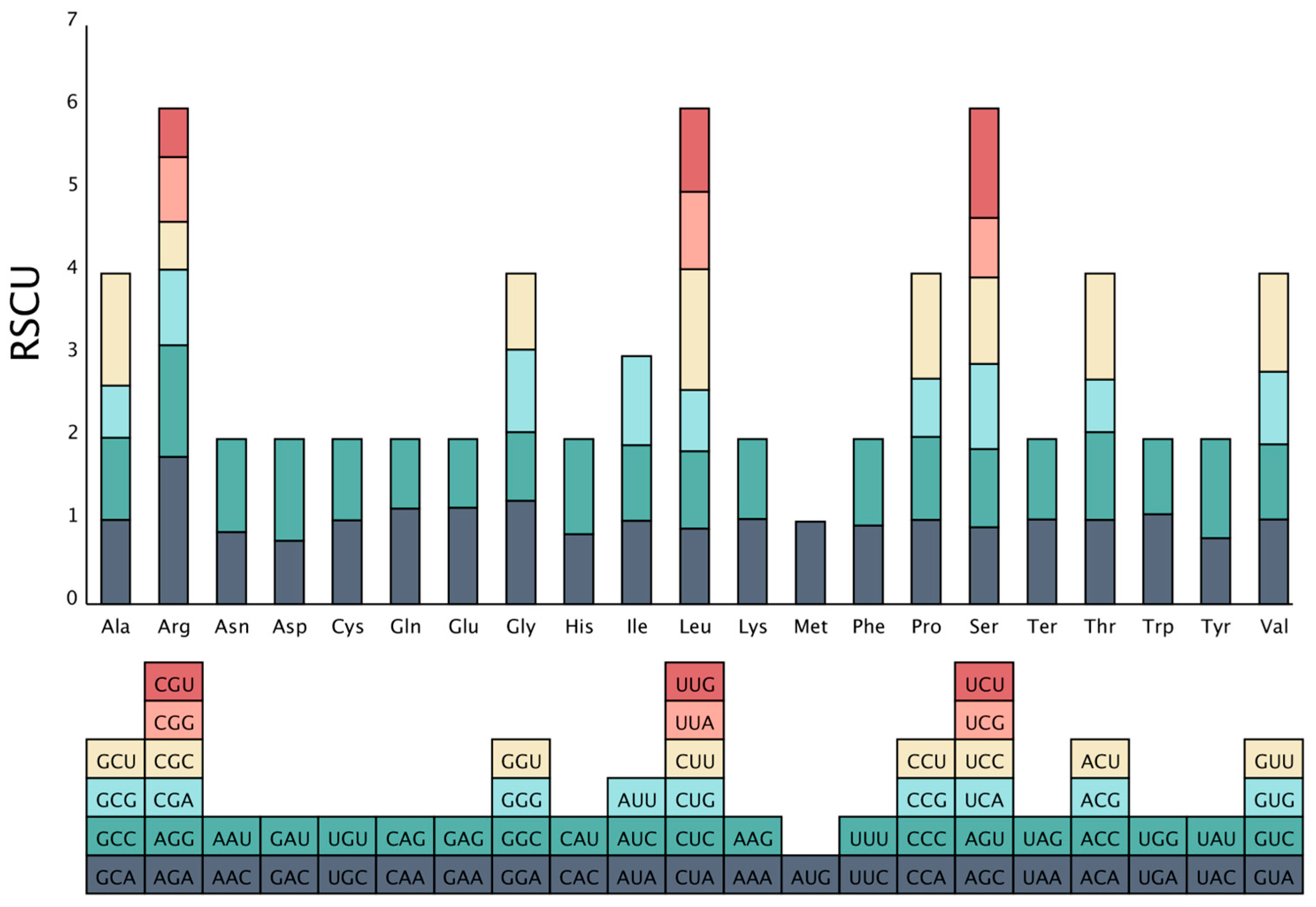
2.2. Codon Preference Analysis
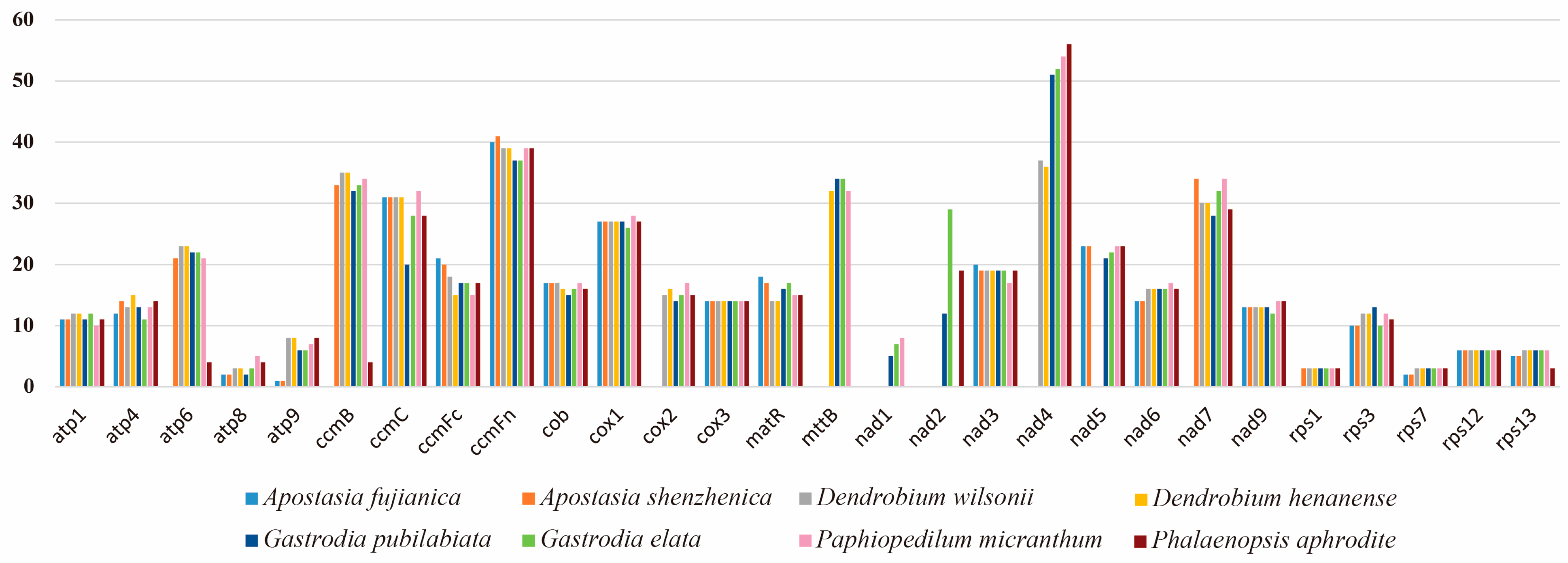
2.3. Repeat Sequence and Ka/Ks Analysis
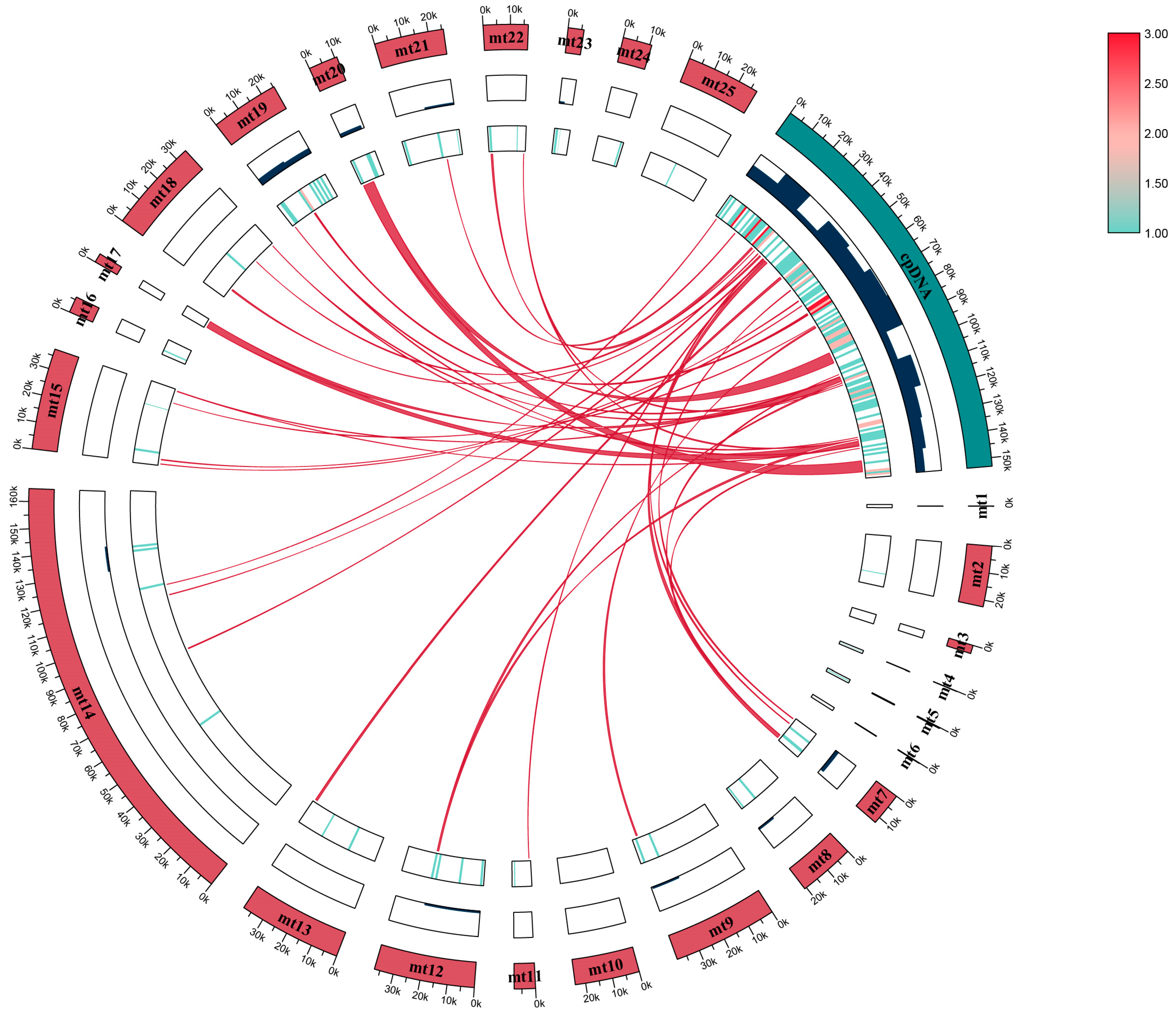
2.4. Gene Transfer between Organelle Genomes
2.5. RNA Editing Site Prediction and Phylogenetic Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Material, DNA Extraction and Sequencing
4.2. Assembly, Annotation and Condon Usage Analysis
4.3. Repeat Sequence and Selective Pressure Analysis
4.4. Gene Transfer, RNA Editing Sites Prediction, and Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gray, M.W.; Burger, G.; Lang, B.F. Mitochondrial evolution. Science 1999, 283, 1476–1481. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.W. Mitochondrial evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P.; Sloan, D.B.; Alverson, A.J. Plant mitochondrial genome diversity: The genomics revolution. In Plant Genome Diversity Volume 1: Plant Genomes, Their Residents, and Their Evolutionary Dynamics; Springer: Berlin/Heidelberg, Germany, 2012; pp. 123–144. [Google Scholar]
- Ward, B.L.; Anderson, R.S.; Bendich, A.J. The mitochondrial genome is large and variable in a family of plants (Cucurbitaceae). Cell 1981, 25, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, N.; Li, S.; Grover, C.E.; Nie, H.; Wendel, J.F.; Hua, J. Plant mitochondrial genome evolution and cytoplasmic male sterility. Crit. Rev. Plant Sci. 2017, 36, 55–69. [Google Scholar] [CrossRef]
- Wu, Z.; Sloan, D.B. Recombination and intraspecific polymorphism for the presence and absence of entire chromosomes in mitochondrial genomes. Heredity 2019, 122, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.M.; Salinas-Giegé, T.; Triant, D.A.; Taylor, D.R.; Drouard, L.; Sloan, D.B. Rapid shifts in mitochondrial tRNA import in a plant lineage with extensive mitochondrial tRNA gene loss. Mol. Biol. Evol. 2021, 38, 5735–5751. [Google Scholar] [CrossRef]
- Lei, B.; Li, S.; Liu, G.; Chen, Z.; Su, A.; Li, P.; Li, Z.; Hua, J. Evolution of mitochondrial gene content: Loss of genes, tRNAs and introns between Gossypium harknessii and other plants. Plant Syst. Evol. 2013, 299, 1889–1897. [Google Scholar] [CrossRef]
- Van de Paer, C.; Bouchez, O.; Besnard, G. Prospects on the evolutionary mitogenomics of plants: A case study on the olive family (Oleaceae). Mol. Ecol. Resour. 2018, 18, 407–423. [Google Scholar] [CrossRef]
- Li, Y.X.; Li, Z.H.; Schuiteman, A.; Chase, M.W.; Li, J.W.; Huang, W.C.; Hidayat, A.; Wu, S.S.; Jin, X.H. Phylogenomics of Orchidaceae based on plastid and mitochondrial genomes. Mol. Phylogenetics Evol. 2019, 139, 106540. [Google Scholar] [CrossRef]
- Pérez-Escobar, O.A.; Bogarín, D.; Przelomska, N.A.S.; Ackerman, J.D.; Balbuena, J.A.; Bellot, S.; Bühlmann, R.P.; Cabrera, B.; Cano, J.A.; Charitonidou, M.; et al. The origin and speciation of orchids. New Phytol. 2024, 242, 700–716. [Google Scholar] [CrossRef]
- Ke, S.J.; Liu, D.K.; Tu, X.D.; He, X.; Zhang, M.M.; Zhu, M.J.; Zhang, D.Y.; Zhang, C.L.; Lan, S.R.; Liu, Z.J. Apostasia mitochondrial genome analysis and monocot mitochondria phylogenomics. Int. J. Mol. Sci. 2023, 24, 7837. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, L.; Liu, D.K.; Zhao, X.W.; Zhang, D.; Ke, S.; Chen, G.Z.; Zheng, Q.; Liu, Z.J.; Lan, S. Apostasia fujianica (Apostasioideae, Orchidaceae), a new Chinese species: Evidence from morphological, genome size and molecular analyses. Phytotaxa 2023, 583, 277–284. [Google Scholar] [CrossRef]
- Zhang, G.Q.; Liu, K.W.; Li, Z.; Lohaus, R.; Hsiao, Y.Y.; Niu, S.C.; Wang, J.Y.; Lin, Y.C.; Xu, Q.; Chen, L.J. The Apostasia genome and the evolution of orchids. Nature 2017, 549, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Hu, Y.; Huang, M.Z.; Huang, W.C.; Liu, D.K.; Zhang, D.; Hu, H.; Downing, J.L.; Liu, Z.J.; Ma, H. Comprehensive phylogenetic analyses of Orchidaceae using nuclear genes and evolutionary insights into epiphytism. J. Integr. Plant Biol. 2023, 65, 1204–1225. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, H.N.; Rasmussen, F.N. Seedling mycorrhiza: A discussion of origin and evolution in Orchidaceae. Bot. J. Linn. Soc. 2014, 175, 313–327. [Google Scholar] [CrossRef]
- Carrie, C.; Murcha, M.W.; Giraud, E.; Ng, S.; Zhang, M.F.; Narsai, R.; Whelan, J. How do plants make mitochondria? Planta 2013, 237, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Wynn, E.L.; Christensen, A.C. Repeats of unusual size in plant mitochondrial genomes: Identification, incidence and evolution. G3 Genes Genomes Genet 2019, 9, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Jin, X.; Liu, J.; Zhao, X.; Zhou, J.; Wang, X.; Wang, D.; Lai, C.; Xu, W.; Huang, J. The Gastrodia elata genome provides insights into plant adaptation to heterotrophy. Nat. Commun. 2018, 9, 1615. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Li, Y.; Zhai, J.W.; Liu, Z.J.; Li, M.H. Organelle Genomes of Epipogium roseum Provide Insight into the Evolution of Mycoheterotrophic Orchids. Int. J. Mol. Sci. 2024, 25, 1578. [Google Scholar] [CrossRef]
- Chen, T.C.; Su, Y.Y.; Wu, C.H.; Liu, Y.C.; Huang, C.H.; Chang, C.C. Analysis of mitochondrial genomics and transcriptomics reveal abundant RNA edits and differential editing status in moth orchid, Phalaenopsis aphrodite subsp. formosana. Sci. Hortic. 2020, 267, 109304. [Google Scholar] [CrossRef]
- Yang, J.X.; Dierckxsens, N.; Bai, M.Z.; Guo, Y.Y. Multichromosomal mitochondrial genome of Paphiopedilum micranthum: Compact and fragmented genome, and rampant intracellular gene transfer. Int. J. Mol. Sci. 2023, 24, 3976. [Google Scholar] [CrossRef] [PubMed]
- Petersen, G.; Cuenca, A.; Moller, I.M.; Seberg, O. Massive gene loss in mistletoe (Viscum, Viscaceae) mitochondria. Sci. Rep. 2015, 5, 17588. [Google Scholar] [CrossRef] [PubMed]
- Skippington, E.; Barkman, T.J.; Rice, D.W.; Palmer, J.D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. USA 2015, 112, E3515–E3524. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Cuthbert, J.M.; Taylor, D.R.; Sloan, D.B. The massive mitochondrial genome of the angiosperm Silene noctiflora is evolving by gain or loss of entire chromosomes. Proc. Natl. Acad. Sci. USA 2015, 112, 10185–10191. [Google Scholar] [CrossRef]
- Feng, Y.; Xiang, X.; Akhter, D.; Pan, R.; Fu, Z.; Jin, X. Mitochondrial phylogenomics of fagales provides insights into plant mitogenome mosaic evolution. Front. Plant Sci. 2021, 12, 762195. [Google Scholar] [CrossRef]
- Sloan, D.B.; Alverson, A.J.; Štorchová, H.; Palmer, J.D.; Taylor, D.R. Extensive loss of translational genes in the structurally dynamic mitochondrial genome of the angiosperm Silene latifolia. BMC Evol. Biol. 2010, 10, 274. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Grewe, F.; Fan, W.; Young, G.J.; Knoop, V.; Palmer, J.D.; Mower, J.P. Ginkgo and Welwitschia mitogenomes reveal extreme contrasts in gymnosperm mitochondrial evolution. Mol. Biol. Evol. 2016, 33, 1448–1460. [Google Scholar] [CrossRef]
- Choi, K.S.; Park, S. Complete plastid and mitochondrial genomes of Aeginetia indica reveal intracellular gene transfer (IGT), horizontal gene transfer (HGT), and cytoplasmic male sterility (CMS). Int. J. Mol. Sci. 2021, 22, 6143. [Google Scholar] [CrossRef] [PubMed]
- Spencer, D.F.; Schnare, M.N.; Coulthart, M.B.; Gray, M.W. Sequence and organization of a 7.2 kb region of wheat mitochondrial DNA containing the large subunit (26S) rRNA gene. Plant Mol. Biol. 1992, 20, 347–352. [Google Scholar] [CrossRef]
- Wang, M.; Yu, W.; Yang, J.; Hou, Z.; Li, C.; Niu, Z.; Zhang, B.; Xue, Q.; Liu, W.; Ding, X. Mitochondrial genome comparison and phylogenetic analysis of Dendrobium (Orchidaceae) based on whole mitogenomes. BMC Plant Biol. 2023, 23, 586. [Google Scholar] [CrossRef]
- Adams, K.L.; Qiu, Y.L.; Stoutemyer, M.; Palmer, J.D. Punctuated evolution of mitochondrial gene content: High and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc. Natl. Acad. Sci. USA 2002, 99, 9905–9912. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.S.; Schwarz, E.N.; Ruhlman, T.A.; Khiyami, M.A.; Sabir, J.S.M.; Hajarah, N.H.; Sabir, M.J.; Rabah, S.O.; Jansen, R.K. Fluctuations in Fabaceae mitochondrial genome size and content are both ancient and recent. BMC Plant Biol. 2019, 19, 448. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Grewe, F.; Zhu, A.; Ruhlman, T.A.; Sabir, J.; Mower, J.P.; Jansen, R.K. Dynamic evolution of Geranium mitochondrial genomes through multiple horizontal and intracellular gene transfers. New Phytol. 2015, 208, 570–583. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, A.; Small, I.; Cosset, A.; Weil, J.H.; Maréchal-Drouard, L. Editing and import: Strategies for providing plant mitochondria with a complete set of functional transfer RNAs. Biochimie 1996, 78, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Knoop, V.; Volkmar, U.; Hecht, J.; Grewe, F. Mitochondrial genome evolution in the plant lineage. In Plant Mitochondria; Springer: Berlin/Heidelberg, Germany, 2011; pp. 3–29. [Google Scholar]
- Qiu, Y.L.; Li, L.; Wang, B.; Xue, J.Y.; Hendry, T.A.; Li, R.Q.; Brown, J.W.; Liu, Y.; Hudson, G.T.; Chen, Z.D. Angiosperm phylogeny inferred from sequences of four mitochondrial genes. J. Syst. Evol. 2010, 48, 391–425. [Google Scholar] [CrossRef]
- Asaf, S.; Khan, A.L.; Khan, A.R.; Waqas, M.; Kang, S.M.; Khan, M.A.; Shahzad, R.; Seo, C.W.; Shin, J.H.; Lee, I.J. Mitochondrial genome analysis of wild rice (Oryza minuta) and its comparison with other related species. PLoS ONE 2016, 11, e0152937. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B.; Langley, C.H.; Stephan, W. The evolution of restricted recombination and the accumulation of repeated DNA sequences. Genetics 1986, 112, 947–962. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Wang, Y.; Wang, Q.; Li, A.; Hou, F.; Zhang, L. Evolution analysis of simple sequence repeats in plant genome. PLoS ONE 2015, 10, e0144108. [Google Scholar] [CrossRef]
- Li, J.; Chen, Y.; Liu, Y.; Wang, C.; Li, L.; Chao, Y. Complete mitochondrial genome of Agrostis stolonifera: Insights into structure, Codon usage, repeats, and RNA editing. BMC Genom. 2023, 24, 466. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Drummond, A. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3. 1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Wu, Y.; Ke, S.J.; Liu, D.K.; Liu, Z.J. Characteristics and Comparative Analysis of the Complete Plastomes of Apostasia fujianica and Neuwiedia malipoensis (Apostasioideae). Horticulturae 2024, 10, 383. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic. Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contig | Length (bp) | GC Content (%) | Genes |
|---|---|---|---|
| mt1 | 120 | 37.5 | / |
| mt2 | 22,467 | 42.9 | nad6 |
| mt3 | 3110 | 49.8 | / |
| mt4 | 116 | 44.8 | nad3 * |
| mt5 | 382 | 43.5 | nad3 * |
| mt6 | 188 | 47.3 | / |
| mt7 | 12,321 | 42.0 | cob, rps14, rpl15 |
| mt8 | 20,753 | 44.4 | trnQ-UUG, rps12 |
| mt9 | 38,670 | 45.4 | trnK-UUU, ccmC |
| mt10 | 23,805 | 44.9 | / |
| mt11 | 7824 | 43.9 | / |
| mt12 | 37,110 | 44.1 | atp9, cox3, rrn5, rrn18 |
| mt13 | 38,175 | 46.2 | nad7, trnE-UUC |
| mt14 | 164,718 | 44.3 | nad5, trnC-GCA, cox1, ccmFc |
| mt15 | 37,436 | 44.7 | matR, tRNA-Cys |
| mt16 | 6018 | 47.7 | trnN-GUU |
| mt17 | 3434 | 44.5 | / |
| mt18 | 34,527 | 45.0 | rps10 |
| mt19 | 25,307 | 45.8 | nad4L, atp4, trnW-CCA, trnP-UGG, psaJ, rpl16, rps3, rps19, rpl2 |
| mt20 | 10,861 | 41.8 | tRNA-Met, ccmFn, atp1 |
| mt21 | 25,659 | 44.5 | rps11, trnQ-UUG, rps13 |
| mt22 | 16,559 | 43.0 | trnD-GUC, atp8 |
| mt23 | 5887 | 44.9 | nad9, trnY-GUA |
| mt24 | 10,780 | 43.6 | rps4 |
| mt25 | 27,385 | 44.1 | rps7 |
| Feature | A. fujianica | A. shenzhenica | D. henanense | D. wilsonii | G. elata | G. pubilabiata | P. micranthum | P. aphrodite |
|---|---|---|---|---|---|---|---|---|
| Genome size (bp) | 573,612 | 672,872 | 807,551 | 763,005 | 1,339,825 | 867,349 | 447,368 | 576,203 |
| GC content (%) | 44.5 | 44.4 | 43.1 | 43.7 | 44.6 | 42.1 | 44.4 | 44.3 |
| Number of PCGs | 30 | 36 | 38 | 38 | 37 | 38 | 39 | 38 |
| Number of tRNA | 12 | 16 | 40 | 33 | 20 | 9 | 16 | 9 |
| Number of rRNA | 2 | 2 | 3 | 3 | 3 | 3 | 3 | 3 |
| Total genes | 44 | 54 | 81 | 74 | 60 | 50 | 58 | 50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Q.; Luo, X.; Huang, Y.; Ke, S.-J.; Liu, Z.-J. The Complete Mitogenome of Apostasia fujianica Y.Li & S.Lan and Comparative Analysis of Mitogenomes across Orchidaceae. Int. J. Mol. Sci. 2024, 25, 8151. https://doi.org/10.3390/ijms25158151
Zheng Q, Luo X, Huang Y, Ke S-J, Liu Z-J. The Complete Mitogenome of Apostasia fujianica Y.Li & S.Lan and Comparative Analysis of Mitogenomes across Orchidaceae. International Journal of Molecular Sciences. 2024; 25(15):8151. https://doi.org/10.3390/ijms25158151
Chicago/Turabian StyleZheng, Qinyao, Xiaoting Luo, Ye Huang, Shi-Jie Ke, and Zhong-Jian Liu. 2024. "The Complete Mitogenome of Apostasia fujianica Y.Li & S.Lan and Comparative Analysis of Mitogenomes across Orchidaceae" International Journal of Molecular Sciences 25, no. 15: 8151. https://doi.org/10.3390/ijms25158151
APA StyleZheng, Q., Luo, X., Huang, Y., Ke, S.-J., & Liu, Z.-J. (2024). The Complete Mitogenome of Apostasia fujianica Y.Li & S.Lan and Comparative Analysis of Mitogenomes across Orchidaceae. International Journal of Molecular Sciences, 25(15), 8151. https://doi.org/10.3390/ijms25158151