Abstract
APC is a tumor suppressor gene that exerts its effect through the regulation of the Wnt signaling pathway. Loss of function mutations of the gene are associated with familial adenomatous polyposis (FAP). Early diagnosis in FAP patients is essential to prevent the development of colorectal cancer. Extraintestinal manifestations often precede the formation of the polyposis; therefore, these manifestations may serve as a clinical indicator for the condition. The aim of this study was to assess genotype–phenotype associations between the location of APC mutations and various extraintestinal features, mainly focusing on osseous and dental anomalies. Analyses of our cases and the mutations available in the literature with these manifestations revealed that mutations in the N-terminal region (amino acids 1–~1000) of the protein are more frequently associated with only osseous anomalies, whereas dental manifestations are more prevalent in mutations in the middle region (amino acids 1000–~2100). In addition, supernumerary teeth were found to be the most common dental feature. Since dental abnormalities often precede intestinal polyposis, dentists have a crucial role in the early identification of patients at risk.
1. Introduction
The human adenomatous polyposis coli (APC) gene located on chromosome 5q21-q22 encodes a 300 kDa, ubiquitously expressed tumor suppressor protein [1]. The gene consist of 15 exons and plays a crucial role in preserving the colonic epithelial structure [2]. In addition to its main function as an antagonist of the Wnt pathway, APC is essential, as it interacts with a number of different proteins. Through its complex function, the APC protein is involved in cell adhesion and migration, spindle assembly, neuronal differentiation, chromosome segregation and cell cycle control [1,3,4,5].
Since the identification and characterization of the gene [6], a lot of different research has been carried out. It has been revealed that APC is involved in the maturation process of the epithelial cells of the colon, and its expression increases from the base of crypt to the top of the villi, extending into the lumen [2,6]. Mutations in the APC gene can induce the development of numerous adenomatous polyps, therefore contributing to the development of colorectal cancers (CRC) [6]. According to Hankey et al. [7], biallelic mutations in the APC gene are responsible for 45–80% of CRC, which is the second leading cause of cancer deaths.
The loss of APC function has been associated with multiple disorders [8,9]. The most well-known among these is the classic familial adenomatous polyposis (FAP). The estimated prevalence for FAP is approximately 1 in 8000 to 1 in 18,000, with a worldwide distribution [10]. It is estimated that in about 25% of FAP patients, de novo mutations develop the disorder [10]. In addition, large deletions may account for approximately 15% of the cases [11]. It follows an autosomal dominant inheritance pattern and is characterized by hundreds to thousands of adenomatous polyps in the colon and rectum. They usually appear during childhood or adolescence [12,13]. According to previous studies, the development of adenomas requires inactivating mutations in both APC alleles, which typically result from an inherited and a somatic mutation [14]. The vast number of early-onset adenomas carry an increased risk of CRC. The number of polyps is generally related to the risk of developing cancer [15]. The mean age of the development of polyps is 16 years, and generally, patients are diagnosed between 20 and 40 years of age. In addition, extraintestinal manifestations may also occur in 70% of FAP patients [16], such as odontomas, osteomas, congenital hypertrophy of the retinal pigment epithelium (CHRPE), papillary carcinoma of the thyroid, hepatoblastoma, medulloblastoma, desmoid, soft tissue, gastrointestinal, and benign tumors, and various dental findings [17,18]. Although almost complete penetrance of colonic polyposis can be observed, the extracolonic manifestations display variable penetrance and severity [10].
If fewer than 100 adenomatous polyps appear, a phenotypic variant of FAP known as attenuated FAP (AFAP) can be differentiated [19]. Compared to the classic FAP, adenomas appear later and are more proximally distributed in the colon. Nevertheless, cancer is diagnosed at a later age and, predominantly, the risk of colorectal cancer is slightly lower [20]. Compared to FAP patients, generally fewer extraintestinal manifestations occur.
In addition to classic and attenuated FAP, other APC-associated polyposis conditions are also known, such as gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS), Turcot syndrome (associates FAP with central nervous system tumors), and Gardner syndrome (GS, association of FAP with osteomas and soft tissue tumors). Moreover, APC mutations have been associated with various neurological disorders and intellectual disabilities, as well [21,22,23].
Gardner syndrome [24] is a variant of FAP that is mainly characterized by a triad of multiple gastrointestinal polyps, cutaneous and subcutaneous soft tissue tumors, and osteomas. The prevalence of GS is estimated to range from 1 in 7000 to 1 in 30,000 live births, with variable expressivity, affecting women and men equally [25]. Familial accumulation has been observed in most cases, but approximately 20–30% of cases occur de novo [25,26]. Osteomas are one of the most frequent bone alterations, which most commonly affect the maxilla, mandible, and/or the frontal bone [25,27,28]. In comparison, orbital osteomas are extremely rare [25]. The gastrointestinal symptoms, such as diarrhea, anemia, lower gastrointestinal bleeding, abdominal pain, etc., occur around the age of 30 [26]. More than 70% of patients will develop extracolonic features, such as neoplastic lesions, CHRPE, fibromas, thyroid cancer, lipomas, gastric fundic gland polyps, juvenile nasopharyngeal angiofibromas, dental abnormalities, epidermoid cysts, or desmoid, solid organ, and/or brain tumors [16,25,26]. Desmoid tumors, which may cause life-threatening complications, develop in approximately 3.5–5.7% of GS patients, usually at the retroperitoneum or the abdominal cavity [29]. CHRPE is present in about 90% of the patients [29]. Approximately 30–70% of GS patients present with dental abnormalities, such as odontomas, impacted/obstructed/unerupted teeth, osteomas of the jaw, missing or supernumerary teeth, or other abnormal tooth morphology [30,31]. The presence of both dental abnormalities and osteomas is suggestive of underlying GS [32,33,34,35].
Certain extraintestinal manifestations in GS, such as dento-osseous features often precede the intestinal polyposis; therefore, they can potentially serve as an early marker of this syndrome. Previous research has stated that bone and cutaneous abnormalities develop approximately 10 years before the onset of polyposis [36,37]. Altogether, the specific extraintestinal manifestations may help in the early diagnosis and proper treatment of GS [26].
According to the ClinVar database, approximately 12,500 mutations have been observed within the APC gene, of which roughly 2500 are pathogenic or likely pathogenic variants. The majority of the mutations are intragenic, small-scale variants (<50 base pairs) manifesting as nonsense, frameshifts, deletions, or insertions that could lead to a premature stop codon, thus potentially resulting in either a truncated protein or nonsense-mediated decay. A wide variety of mutations have been observed throughout the APC gene, but the majority (~75%) of both germline and somatic mutations occur in the 15th exon of the APC gene [38]. A mutational cluster region (MCR) containing about 60% of all identified mutation sites has been defined [39], which localizes between amino acids 1000 and 1600 [7]. In addition to MCR, other mutational hotspots can be distinguished, and several specific genotype–phenotype associations have been demonstrated between the location of a mutation and the presence, frequency, and/or severity of particular manifestations [40,41]. These investigations mainly focused on colonic manifestations. Regarding dental anomalies, only a few studies have discussed the genotype related to the dental phenotype [40,42].
In our study, we aimed to study the association between APC mutations and dental or osseous manifestations. For this purpose, we collected APC mutations accompanying dental and/or osseous manifestations published in the literature and added two of our cases. In order to find a link between the dental or osseous symptoms and the APC mutations, we have analyzed the localizations of various mutations, their possible role in the protein function, and the observed manifestations.
2. Results
Altogether, 49 different cases (with 30 various mutations), including our two cases, have been found with various dental and/or osseous abnormalities in which the presence of an APC mutation was approved (Table 1). Based on the location of the mutation (N-terminal or middle region of the protein), the individuals with dental and/or osseous anomalies were classified into two groups. Almost half of the patients (n = 23) carried a mutation in the N-terminal region; among these, 15 (~65%) cases presented only osseous abnormalities, 4 (~17%) individuals showed only dental abnormalities, and 4 (~17%) patients had both dental and osseous anomalies. In 24 patients, the mutation was located in the middle region, of which 7 (~29%) demonstrated only osseous abnormalities, 9 (~38%) cases displayed only dental anomalies, and 8 (~33%) patients had both dental and osseous anomalies. Two patients with mutations localized in the C-terminal region presented only dental anomalies; they were not involved in the statistical analyses.

Table 1.
APC germline mutations with dento-osseous manifestations and FAP phenotype.
In order to ascertain a potential influence of mutation localization on specific dental or osseous anomalies, a comparative analysis between patients with mutations in the N-terminal or middle region was conducted. The following manifestations were involved in osseous anomalies: osteomas (OS); dense bone island (DBI); and hazy sclerosis (HS). In the case of dental anomalies, mesiodens; supernumerary teeth (ST); impacted teeth (IT); odontomas (OD); and other dental abnormalities were included.
Mutations in the N-terminal region are more frequently associated with only osseous anomalies (15 out of 23) compared to mutations in the middle region (7 out of 24). A statistical analysis further confirmed this association with a significant difference (p = 0.014). Notably, osteoma was the predominant symptom in osseous anomalies and was observed across both regions in 20 out of 22 cases (~91%).
Dental manifestations appeared to be more prevalent among cases with mutations in the middle region (17 cases out of 24) compared to those in the N-terminal region (8 cases out of 23). A significant difference (p = 0.014) was observed in the frequencies. Furthermore, an examination of specific dental manifestations has been carried out. Mesiodens are a type of supernumerary teeth; therefore, in our calculations, they were treated together. A significant difference was observed in the frequencies of the supernumerary teeth (p = 0.030) and the impacted teeth (p = 0.042) presented in the patients with a mutation located in the middle region compared to the N-terminal.
Although twice as many cases (8 out of 24 vs. 4 out of 23) presented dental abnormalities with osseous anomalies, and nearly twice as many (7 out of 24 vs. 4 out of 23) demonstrated only dental anomalies, the statistical analysis could not determine a significant difference in either case (p = 0.180, p = 0.272, respectively).
Among dental cases, supernumerary teeth were present in 10 cases out of 17 (59%) in the middle domain and 3 out of 8 cases (37.5%) in the N-terminal domain; however, no significant difference was established (p = 0.278).
In conclusion, while mutations in the N-terminal domain appear to be predominantly associated with osseous anomalies, mutations in the middle domain may contribute to a higher incidence of dental anomalies. These findings underscore the potential role of mutation localization in determining the phenotypic expression of dental and osseous abnormalities.
Regarding the polyposis phenotype among the presented cases, the same number of patients were diagnosed with classic FAP and Gardner syndrome. The majority of patients with classic FAP carry mutations in the N-terminal region, whereas patients with Gardner syndrome harbor mutations localized in the middle region of the APC protein.
3. Discussion
The early diagnosis of individuals affected by FAP, a rare genetically determined disorder, is crucial to prevent the development of colorectal cancer. Moreover, presymptomatic diagnosis is essential, as two-thirds of symptomatic patients will already have developed carcinoma and consequently have a worse prognosis [58].
Since the disorder is inherited in an autosomal dominant manner, family members have a 50% risk of inheriting the disease. Therefore, genetic testing in individuals at risk has an important role in the identification of the disease-causing variants in the APC gene. Due to the state-of-the-art NGS-based technology, it is feasible nowadays; however, in the general population, it is not applicable because of ethical and other considerations. Furthermore, genetic analysis plays an important role in the early diagnosis of patients with atypical manifestations as was also the case in our patients. Our proband had initial atypical skeletal symptoms and dental anomalies only, and the diagnosis was established with the help of whole exome sequencing [57].
Extraintestinal manifestations, such as various dental and osseous abnormalities, accompanying FAP have a potential role as a clinical indicator in early diagnosis. It is well-known from previous studies [59] that these manifestations often precede the development of polyposis. Thakker et al. [60] developed a weighted dental panoramic radiograph score (DPRS) system as a diagnostic tool for individuals with high risk of FAP. DPRS took into consideration the nature, extent, and site of osseous and dental changes on dental panoramic radiographs in FAP patients and was shown to have a sensitivity of up to 69% and specificity of 100%. Using this tool, differences in the frequency of dental anomalies and osseous jaw lesions between FAP and unaffected groups were observed. Bone changes and dental anomalies were found in 81% and 37% of the FAP patients, respectively. Aggarwal and colleagues evaluated the validity of this DPRS tool on an independent patient group, and they found a 100% specificity. Within their patient cohort, osseous jaw lesions were seen in 69% of FAP patients, and dental anomalies were seen in 35% [58]. Bone alterations (osteomas, islands of bone condensation, and diffuse sclerosis) are more frequent than dental anomalies in FAP patients [43,47,58,60]. Almeida et al. performed a meta-analysis investigating the oral manifestations of FAP and their frequency in affected individuals. It was found that the frequency of osseous jaw lesions ranged from 21% to 95%, and the frequency of dental anomalies ranged from 9.3% to 56% in FAP patients [61]. The frequency of some dental and osseous anomalies was found to be higher in FAP patients than in the general population (Table 2).
Table 2.
Frequency of dental and osseous manifestations in patients with FAP and in the general population.
In several studies, a genotype–phenotype association was performed to reveal an association between the location of APC mutations and extraintestinal manifestations in FAP patients. In their study, Septer and colleagues investigated pediatric patients with FAP [42]. They found that in patients carrying a mutation in or upstream of codon 1309, a higher frequency of osteomas (77.8%) and jawbone sclerosis (44.4%) was observed, and 77% of these patients had at least one dental anomaly. Moreover, they observed that osteomas were present in the jaw in 42.9% of genetic variants in the 5′-codon 516 region, 66.7% in variants between codons 849 and 1309, and 77.8% of genetic variants situated between codons 1310 and 3′. Previously, Bertario et al. found that mutations beyond codon 1444 were significantly associated with osteomas [66]. Another study by Bertario [67] revealed that dental abnormalities were strongly associated with osteomas, and both conditions were associated with mutations between codons 1256 and 1303. Davies et al. [40] observed an association of significantly more abnormalities on dental panoramic radiographs with mutations distal to codon 1444 compared to mutations at codons 1–1444. In a study by Bisgaard and colleagues, osteomas were only identified in patients with mutations between codons 767 and 1513 [68].
In this study, a mutation found in our patients was analyzed along with 29 previously published APC mutations in 47 patients. Our patients, who were carrying a c.4700C>G (p.Ser1567*) mutation presented with impacted teeth, odontomas, and osteomas. The detailed clinical description was published previously [57]. This mutation has been reported previously [69], but no dento-osseous anomalies were associated with it. A detailed clinical evaluation of dental and osseous anomalies of patients with known APC mutations revealed that osseous manifestations were more frequent than dental abnormalities (34 out of 46 vs. 24 out of 46, respectively), similar to previous findings [43,47,58,60]. Osteoma was the most frequent bone lesion, and supernumerary teeth were more prevalent among the dental anomalies. The investigation of genotype–phenotype associations in APC mutations and these extraintestinal manifestations revealed that mutations localized in the N-terminal region (1–1000 aa’s) of the APC protein are more frequently associated with only osseous anomalies compared to mutations localized in the middle region (1000–2100 aa’s) (65% vs. 32%). Moreover, dental manifestations were more frequently associated with mutations in the middle region than those in the N-terminal region (68% vs. 35%). Supernumerary teeth were found to be the most prevalent dental anomaly, and they were also found to be associated more frequently with mutations in the middle region compared to those in the N-terminal region.
APC is a multifunctional protein containing multiple functional regions, including specific repeats, definite motifs, nuclear localization, and export signals (Figure 1). The protein can be divided into three major sections known as the N-terminal (APC-N), middle (APC-M), and C-terminal (APC-C) regions. The N-terminal region contains amino acids 1–~1000, the middle region covers amino acids ~1000–2100 and the C-terminal region spans over amino acids 2130–2843 [70]. The structure of the protein provides binding sites for one or more different partner proteins, such as β-catenin, Axin, microtubule, human DISCS large protein (hDLG), and APC-stimulated guanine nucleotide exchange factor (Asef).
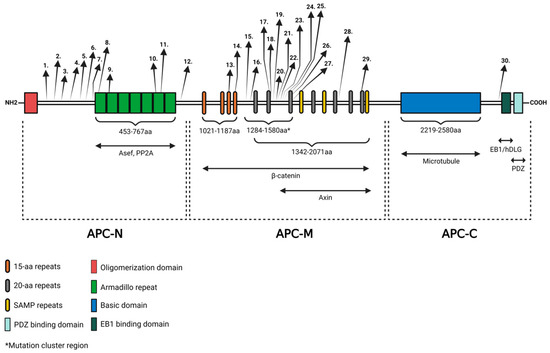
Figure 1.
The structure of the APC protein and the locations of known APC mutations. APC contains regions required for oligomerization, armadillo repeats, β-catenin-binding domain, AXIN-binding domain, basic domain, EB1-binding domain, and HDLG-binding domain. Three major region names and the interacting partners of the protein are shown underneath the APC diagram. APC-N is the N-terminal region, APC-M is the middle region, and APC-C is the C-terminal region. Locations of the mutations are indicated by arrows. Numbers are in correspondence with the mutations listed in Table 1. The figure was created with BioRender.com (accessed on 29 March 2024).
The N-terminal region contains an oligomerization domain and seven repeats, known as the armadillo region, which has been shown to bind to PP2A and Asef [17]. The middle region encompasses various repeats, such as 15- and 20-amino acid signature repeats and characteristic SAMP amino acid sequences. They provide binding sites for the Wnt signaling complex (β-catenin, Axin, and KIF3a). The C-terminal region contains a basic domain, which provides a microtubule-binding site, a PDZ-binding motif, and domains for the interaction between end-binding protein 1 (EB1) and hDLG. The protein interacts with multiple cytoskeletal proteins, thereby influencing various cellular processes, with special respect to the Wnt signaling pathway [1,17].
The role of Wnt in odontogenesis and bone development has been addressed previously [71]. Previous experiments on mouse models established that balanced Wnt/β-catenin activity is essential in tooth development. Disruption of the Wnt pathway leads to either impaired tooth formation or supernumerary teeth [72,73]. This was further demonstrated in research by Panyarat et al. [50], in which they hypothesized that as a result of mutations in the APC gene, the β-catenin levels will increase. They suggested that the reduction in CTBP interaction and disruption of axin or microtubule interaction will affect the WNT/β-catenin signaling.
Normally β-catenin would be phosphorylated and broken down by a destruction complex [74]. The so-called β-catenin destruction complex consists of axin, β-catenin, and APC and phosphorylates β-catenin with the help of CK1α and GSK-3β [75]. Previous studies [17,74,76,77] have stated that the accumulation of β-catenin in the cellular cytoplasm and nucleus can occur in various cases, including the direct mutation of β-catenin as a result of the Wnt signal or the inactivation of APC. Truncation of the APC is assumed to disrupt the interaction of the destruction complex, thereby disrupting the degradation of β-catenin [75]. As a result of elevated β-catenin, it will form a complex with TCF/LEF in the nucleus and activate the gene expression of target genes [17] (Figure 2).
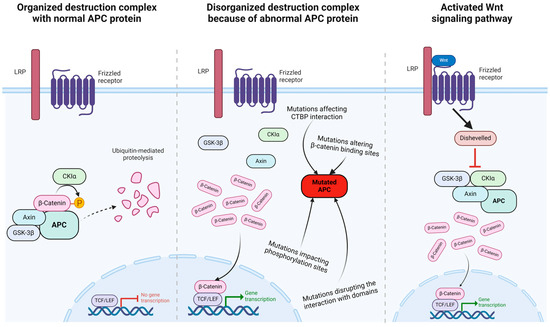
Figure 2.
Mutations in APC protein mimic the effects of Wnt signaling pathway. The normal APC protein inactivates the transcriptional role of β-catenin through phosphorylation leading to ubiquitinylation and degradation (left), whereas mutated APC protein allows β-catenin to enter the nucleus (middle), similar to the result of an active Wnt signal transduction pathway (right). The figure was adapted from “Wnt Signaling Pathway Activation and Inhibition”, by BioRender.com (accessed on 29 March 2024). Retrieved from https://app.biorender.com/biorender-templates.
Our statistical analysis proved that patients with dental abnormalities were more pronounced in cases with a mutation in the middle region. Moreover, supernumerary teeth were more frequently associated with mutations in the middle region. The majority of the mutations in this region are frameshift or nonsense mutations resulting in the termination of protein translation that could lead to a non-functional APC protein or the nonsense-mediated decay of the transcript [47]. There are a few missense mutations that may cause the modification of the APC protein structure, which probably has an effect on its binding properties [50]. Numerous binding sites for the axin and β-catenin interaction are present in the middle region of the APC protein. Therefore, the mutations, especially in the middle domain, are expected to result in a defective APC protein formation, which can affect the assembly of the protein complex, thus contributing to an increased β-catenin level. The elevated β-catenin leads to increased gene expression, which may contribute to supernumerary tooth formation [1,17,72,73].
The limitations of our study are that we found only a limited number of genetic mutations in the APC gene with clear clinical information. Although several cases of Gardner syndrome have been published in the literature, their exact genetic variants are not available.
4. Materials and Methods
4.1. Literature Search and Keyword Specification
A relevant literature search was applied in the PubMed database using the following keywords and phrases: “dental anomalies” or “dental abnormalities” or “teeth abnormality” or teeth anomalies” or “osseous abnormalities” or “osseous anomalies” and “Gardner’s syndrome” or “APC gene mutation” or “familial adenomatous polyposis”. After a thorough analysis of potential publications with genotype–phenotype data, patients with dental and/or osseous abnormalities were collected from the literature. Dental anomalies included supernumerary teeth, ST; impacted teeth, IT; odontomas, OD; mesiodens; and other dental abnormalities. Osseous anomalies included osteomas, OS; dense bone island, DBI; and hazy sclerosis, HS.
4.2. Statistical Analysis
A statistical analysis was carried out to see the relationship between the location of mutations and specific manifestations of dental and/or osseous anomalies. A descriptive statistical analysis, including a chi-square test, was applied using SPSS version 27.0 (IBM Corp. Released 2020. IBM SPSS Statistics for Windows, Version 27.0. Armonk, NY, USA: IBM Corp.). The expected values for each cell were five or higher, and p < 0.05 was considered the level of significance.
5. Conclusions
Germline mutations of the APC gene are responsible for the development of FAP, a tumor predisposition syndrome. The early diagnosis of FAP is essential to prevent the development of colorectal cancer and improve the prognosis of the disease. Extraintestinal manifestations of FAP, such as osseous or dental anomalies, often precede the development of polyposis and may serve as a clinical marker for the presence of this condition. Dentists should be familiar with the typical dento-osseous features of this disorder and refer the patients to genetic counseling. In our study, we demonstrated that mutations in the N-terminal region of the protein are associated with osseous lesions more frequently, whereas mutations in the middle region are linked to dental anomalies. However, further studies are needed to better characterize the mutational spectrum of this gene and the associated extraintestinal features.
Author Contributions
Conceptualization, J.B.; writing—original draft preparation, G.B. and G.A.; writing—review and editing, J.B.; visualization G.B.; supervision, J.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
This study was conducted in accordance with the Declaration of Helsinki of 1975 and with the Hungarian legal requirements of genetic examination, research, and biobanking. The studies involving human participants were reviewed and approved by the Ethics Committee of the University of Pécs (Protocol 8770-PTE 2021).
Informed Consent Statement
Informed consent was obtained from all subjects involved in this study.
Data Availability Statement
The data presented in this study are available upon request from the corresponding author.
Acknowledgments
We would like to thank the patients who participated in this study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hanson, C.A.; Miller, J.R. Non-traditional roles for the Adenomatous Polyposis Coli (APC) tumor suppressor protein. Gene 2005, 361, 1–12. [Google Scholar] [CrossRef]
- Smith, K.J.; Johnson, K.A.; Bryan, T.M.; Hill, D.E.; Markowitz, S.; Willson, J.K.; Paraskeva, C.; Petersen, G.M.; Hamilton, S.R.; Vogelstein, B.; et al. The APC gene product in normal and tumor cells. Proc. Natl. Acad. Sci. USA 1993, 90, 2846–2850. [Google Scholar] [CrossRef]
- Faux, M.C.; Ross, J.L.; Meeker, C.; Johns, T.; Ji, H.; Simpson, R.J.; Layton, M.J.; Burgess, A.W. Restoration of full-length adenomatous polyposis coli (APC) protein in a colon cancer cell line enhances cell adhesion. J. Cell Sci. 2004, 117, 427–439. [Google Scholar] [CrossRef]
- Pollack, A.L.; Barth, A.I.; Altschuler, Y.; Nelson, W.J.; Mostov, K.E. Dynamics of beta-catenin interactions with APC protein regulate epithelial tubulogenesis. J. Cell Biol. 1997, 137, 1651–1662. [Google Scholar] [CrossRef]
- Wen, Y.; Eng, C.H.; Schmoranzer, J.; Cabrera-Poch, N.; Morris, E.J.; Chen, M.; Wallar, B.J.; Alberts, A.S.; Gundersen, G.G. EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration. Nat. Cell Biol. 2004, 6, 820–830. [Google Scholar] [CrossRef]
- Groden, J.; Thliveris, A.; Samowitz, W.; Carlson, M.; Gelbert, L.; Albertsen, H.; Joslyn, G.; Stevens, J.; Spirio, L.; Robertson, M.; et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991, 66, 589–600. [Google Scholar] [CrossRef]
- Hankey, W.; Frankel, W.L.; Groden, J. Functions of the APC tumor suppressor protein dependent and independent of canonical WNT signaling: Implications for therapeutic targeting. Cancer Metastasis Rev. 2018, 37, 159–172. [Google Scholar] [CrossRef]
- Zhang, L.; Shay, J.W. Multiple Roles of APC and its Therapeutic Implications in Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109, djw332. [Google Scholar] [CrossRef]
- Al-Tameemi, H.K.; Al-Husseini, R.M.; Al-Mudhafer, R.H.; Abid, H.A.; Al-Gazali, H.R.; Abdullah, D.A.A.; Albaldawy, M.T. Molecular and immunohistochemical study of APC exon 16 and its possible role in colorectal carcinoma development. Heliyon 2024, 10, e23443. [Google Scholar] [CrossRef]
- Bisgaard, M.L.; Fenger, K.; Bulow, S.; Niebuhr, E.; Mohr, J. Familial adenomatous polyposis (FAP): Frequency, penetrance, and mutation rate. Hum. Mutat. 1994, 3, 121–125. [Google Scholar] [CrossRef]
- Aretz, S.; Stienen, D.; Uhlhaas, S.; Pagenstecher, C.; Mangold, E.; Caspari, R.; Propping, P.; Friedl, W. Large submicroscopic genomic APC deletions are a common cause of typical familial adenomatous polyposis. J. Med. Genet. 2005, 42, 185–192. [Google Scholar] [CrossRef]
- Fearnhead, N.S.; Wilding, J.L.; Bodmer, W.F. Genetics of colorectal cancer: Hereditary aspects and overview of colorectal tumorigenesis. Br. Med. Bull. 2002, 64, 27–43. [Google Scholar] [CrossRef]
- Bronner, M.P. Gastrointestinal inherited polyposis syndromes. Mod. Pathol. 2003, 16, 359–365. [Google Scholar] [CrossRef]
- Lamlum, H.; Papadopoulou, A.; Ilyas, M.; Rowan, A.; Gillet, C.; Hanby, A.; Talbot, I.; Bodmer, W.; Tomlinson, I. APC mutations are sufficient for the growth of early colorectal adenomas. Proc. Natl. Acad. Sci. USA 2000, 97, 2225–2228. [Google Scholar] [CrossRef]
- Debinski, H.S.; Love, S.; Spigelman, A.D.; Phillips, R.K. Colorectal polyp counts and cancer risk in familial adenomatous polyposis. Gastroenterology 1996, 110, 1028–1030. [Google Scholar] [CrossRef]
- Antohi, C.; Haba, D.; Caba, L.; Ciofu, M.L.; Drug, V.L.; Barboi, O.B.; Dobrovat, B.I.; Panzaru, M.C.; Gorduza, N.C.; Lupu, V.V.; et al. Novel Mutation in APC Gene Associated with Multiple Osteomas in a Family and Review of Genotype-Phenotype Correlations of Extracolonic Manifestations in Gardner Syndrome. Diagnostics 2021, 11, 1560. [Google Scholar] [CrossRef]
- Fearnhead, N.S.; Britton, M.P.; Bodmer, W.F. The ABC of APC. Hum. Mol. Genet. 2001, 10, 721–733. [Google Scholar] [CrossRef]
- Galiatsatos, P.; Foulkes, W.D. Familial adenomatous polyposis. Am. J. Gastroenterol. 2006, 101, 385–398. [Google Scholar] [CrossRef]
- Spirio, L.; Olschwang, S.; Groden, J.; Robertson, M.; Samowitz, W.; Joslyn, G.; Gelbert, L.; Thliveris, A.; Carlson, M.; Otterud, B.; et al. Alleles of the APC gene: An attenuated form of familial polyposis. Cell 1993, 75, 951–957. [Google Scholar] [CrossRef]
- Hernegger, G.S.; Moore, H.G.; Guillem, J.G. Attenuated familial adenomatous polyposis: An evolving and poorly understood entity. Dis. Colon. Rectum 2002, 45, 127–134, discussion 134–126. [Google Scholar] [CrossRef]
- Gonzalez, L.; Alvarez, J.; Weinstein, E.; Korenis, P. Familial adenomatous polyposis in an adolescent with coexisting schizophrenia: Treatment strategies and implications. Mol. Genet. Genom. Med. 2015, 3, 391–395. [Google Scholar] [CrossRef][Green Version]
- Mohn, J.L.; Alexander, J.; Pirone, A.; Palka, C.D.; Lee, S.Y.; Mebane, L.; Haydon, P.G.; Jacob, M.H. Adenomatous polyposis coli protein deletion leads to cognitive and autism-like disabilities. Mol. Psychiatry 2014, 19, 1133–1142. [Google Scholar] [CrossRef]
- Onouchi, T.; Kobayashi, K.; Sakai, K.; Shimomura, A.; Smits, R.; Sumi-Ichinose, C.; Kurosumi, M.; Takao, K.; Nomura, R.; Iizuka-Kogo, A.; et al. Targeted deletion of the C-terminus of the mouse adenomatous polyposis coli tumor suppressor results in neurologic phenotypes related to schizophrenia. Mol. Brain 2014, 7, 21. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gardner, E.J. A genetic and clinical study of intestinal polyposis, a predisposing factor for carcinoma of the colon and rectum. Am. J. Hum. Genet. 1951, 3, 167–176. [Google Scholar]
- Avila, S.A.; Nguyen, G.; Wojno, T.; Kim, H.J. Orbital osteomas associated with Gardner’s syndrome: A case presentation and review of literature. Orbit 2024, 43, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Yao, X.; Pan, Y.; Gu, H.; Xiong, F.; Yin, X.; Wu, B.; Chen, T. A novel APC mutation associated with Gardner syndrome in a Chinese family. Gene 2024, 896, 148051. [Google Scholar] [CrossRef] [PubMed]
- Dawlatly, E.D.; Al-Qurain, A.A.; Salih-Mahmud, M. Gardner’s syndrome presenting with a giant osteoma. Ann. Saudi Med. 1997, 17, 542–544. [Google Scholar] [CrossRef]
- Madani, M.; Madani, F. Gardner’s syndrome presenting with dental complaints. Arch. Iran. Med. 2007, 10, 535–539. [Google Scholar]
- Fotiadis, C.; Tsekouras, D.K.; Antonakis, P.; Sfiniadakis, J.; Genetzakis, M.; Zografos, G.C. Gardner’s syndrome: A case report and review of the literature. World J. Gastroenterol. 2005, 11, 5408–5411. [Google Scholar] [CrossRef]
- Dinarvand, P.; Davaro, E.P.; Doan, J.V.; Ising, M.E.; Evans, N.R.; Phillips, N.J.; Lai, J.; Guzman, M.A. Familial Adenomatous Polyposis Syndrome: An Update and Review of Extraintestinal Manifestations. Arch. Pathol. Lab. Med. 2019, 143, 1382–1398. [Google Scholar] [CrossRef]
- Davies, A.S. Gardner’s syndrome--a case report. Br. J. Oral Surg. 1970, 8, 51–57. [Google Scholar] [CrossRef]
- Gardner, E.J.; Richards, R.C. Multiple cutaneous and subcutaneous lesions occurring simultaneously with hereditary polyposis and osteomatosis. Am. J. Hum. Genet. 1953, 5, 139–147. [Google Scholar]
- Yu, D.; Ng Cw, B.; Zhu, H.; Liu, J.; Lin, Y. Bone and dental abnormalities as first signs of familial Gardner’s syndrome in a Chinese family: A literature review and a case report. Med. Sci. (Paris) 2018, 34, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Cristofaro, M.G.; Giudice, A.; Amantea, M.; Riccelli, U.; Giudice, M. Gardner’s syndrome: A clinical and genetic study of a family. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2013, 115, e1-6. [Google Scholar] [CrossRef] [PubMed]
- Ida, M.; Nakamura, T.; Utsunomiya, J. Osteomatous changes and tooth abnormalities found in the jaw of patients with adenomatosis coli. Oral Surg. Oral Med. Oral Pathol. 1981, 52, 2–11. [Google Scholar] [CrossRef]
- Nandakumar, G.; Morgan, J.A.; Silverberg, D.; Steinhagen, R.M. Familial polyposis coli: Clinical manifestations, evaluation, management and treatment. Mt. Sinai J. Med. 2004, 71, 384–391. [Google Scholar] [PubMed]
- Newman, C.A.; Reuther, W.L., 3rd; Wakabayashi, M.N.; Payette, M.M.; Plavsic, B.M. Gastrointestinal case of the day. Gardner syndrome. Radiographics 1999, 19, 546–548. [Google Scholar] [CrossRef][Green Version]
- Beroud, C.; Soussi, T. APC gene: Database of germline and somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1996, 24, 121–124. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Nagase, H.; Ando, H.; Horii, A.; Ichii, S.; Nakatsuru, S.; Aoki, T.; Miki, Y.; Mori, T.; Nakamura, Y. Somatic mutations of the APC gene in colorectal tumors: Mutation cluster region in the APC gene. Hum. Mol. Genet. 1992, 1, 229–233. [Google Scholar] [CrossRef]
- Davies, D.R.; Armstrong, J.G.; Thakker, N.; Horner, K.; Guy, S.P.; Clancy, T.; Sloan, P.; Blair, V.; Dodd, C.; Warnes, T.W.; et al. Severe Gardner syndrome in families with mutations restricted to a specific region of the APC gene. Am. J. Hum. Genet. 1995, 57, 1151–1158. [Google Scholar]
- Newton, K.F.; Mallinson, E.K.; Bowen, J.; Lalloo, F.; Clancy, T.; Hill, J.; Evans, D.G. Genotype-phenotype correlation in colorectal polyposis. Clin. Genet. 2012, 81, 521–531. [Google Scholar] [CrossRef]
- Septer, S.; Bohaty, B.; Onikul, R.; Kumar, V.; Williams, K.B.; Attard, T.M.; Friesen, C.A.; Friesen, L.R. Dental anomalies in pediatric patients with familial adenomatous polyposis. Fam. Cancer 2018, 17, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Almeida, F.T.; Leite, A.F.; de Souza Figueiredo, P.T.; Dos Santos, P.A.C.; Rosa, E.; Mazzeu, J.F.; Sousa, J.B.; Pogue, R.; Acevedo, A.C.; Guerra, E.N.S. Dento-osseous anomalies in patients with familial adenomatous polyposis: A follow-up study. Clin. Oral Investig. 2020, 24, 3501–3511. [Google Scholar] [CrossRef] [PubMed]
- Seehra, J.; Patel, S.; Bryant, C. Gardner’s Syndrome revisited: A clinical case and overview of the literature. J. Orthod. 2016, 43, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Kerr, S.E.; Thomas, C.B.; Thibodeau, S.N.; Ferber, M.J.; Halling, K.C. APC germline mutations in individuals being evaluated for familial adenomatous polyposis: A review of the Mayo Clinic experience with 1591 consecutive tests. J Mol Diagn 2013, 15, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Nishisho, I.; Nakamura, Y.; Miyoshi, Y.; Miki, Y.; Ando, H.; Horii, A.; Koyama, K.; Utsunomiya, J.; Baba, S.; Hedge, P. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991, 253, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Arruda, K.A.R.; Normando, A.G.C.; Pacheco-Pereira, C.; Amorim Dos Santos, J.; Yamaguti, P.M.; Mazzeu, J.F.; Almeida, F.T.; Acevedo, A.C.; Guerra, E.N.S. Phenotypic dento-osseous characterization of a Brazilian family with Familial Adenomatous Polyposis. Arch. Oral Biol. 2021, 129, 105206. [Google Scholar] [CrossRef] [PubMed]
- Eccles, D.M.; Lunt, P.W.; Wallis, Y.; Griffiths, M.; Sandhu, B.; McKay, S.; Morton, D.; Shea-Simonds, J.; Macdonald, F. An unusually severe phenotype for familial adenomatous polyposis. Arch. Dis. Child. 1997, 77, 431–435. [Google Scholar] [CrossRef]
- Soravia, C.; Sugg, S.L.; Berk, T.; Mitri, A.; Cheng, H.; Gallinger, S.; Cohen, Z.; Asa, S.L.; Bapat, B.V. Familial adenomatous polyposis-associated thyroid cancer: A clinical, pathological, and molecular genetics study. Am. J. Pathol. 1999, 154, 127–135. [Google Scholar] [CrossRef]
- Panyarat, C.; Nakornchai, S.; Chintakanon, K.; Leelaadisorn, N.; Intachai, W.; Olsen, B.; Tongsima, S.; Adisornkanj, P.; Ngamphiw, C.; Cox, T.C.; et al. Rare Genetic Variants in Human APC Are Implicated in Mesiodens and Isolated Supernumerary Teeth. Int. J. Mol. Sci. 2023, 24, 4255. [Google Scholar] [CrossRef]
- Pereira, D.L.; Carvalho, P.A.; Achatz, M.I.; Rocha, A.; TardinTorrezan, G.; Alves, F.A. Oral and maxillofacial considerations in Gardner’s syndrome: A report of two cases. Ecancermedicalscience 2016, 10, 623. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Cai, W.; Jiang, B.; Xu, L.; Liu, S.; Zhao, S. A novel mutation of adenomatous polyposis coli (APC) gene results in the formation of supernumerary teeth. J. Cell Mol. Med. 2018, 22, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Martin-Denavit, T.; Duthel, S.; Giraud, S.; Olschwang, S.; Saurin, J.C.; Plauchu, H. Phenotype variability of two FAP families with an identical APC germline mutation at codon 1465: A potential modifier effect? Clin. Genet. 2001, 60, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; He, F.; Xu, X.; Xiong, F.; Zhang, L. APC c.4621C>T variant causing Gardner’s syndrome in a Han Chinese family may be inherited through maternal mosaicism. Exp. Ther. Med. 2021, 21, 488. [Google Scholar] [CrossRef] [PubMed]
- Nilbert, M.; Fernebro, J.; Kristoffersson, U. Novel germline APC mutations in Swedish patients with familial adenomatous polyposis and Gardner syndrome. Scand. J. Gastroenterol. 2000, 35, 1200–1203. [Google Scholar] [CrossRef] [PubMed]
- Oku, T.; Takayama, T.; Sato, Y.; Sato, Y.; Takada, K.; Hayashi, T.; Takahashi, M.; Kuroda, M.; Kato, J.; Niitsu, Y. A case of Gardner syndrome with a mutation at codon 1556 of APC: A suggested case of genotype-phenotype correlation in dental abnormality. Eur. J. Gastroenterol. Hepatol. 2004, 16, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Antal, G.; Zsigmond, A.; Till, A.; Orsi, E.; Szanto, I.; Buki, G.; Kereskai, L.; Herbert, Z.; Hadzsiev, K.; Bene, J. Case report: Initial atypical skeletal symptoms and dental anomalies as first signs of Gardner syndrome: The importance of genetic analysis in the early diagnosis. Pathol. Oncol. Res. 2024, 30, 1611768. [Google Scholar] [CrossRef]
- Aggarwal, V.R.; Sloan, P.; Horner, K.; Macfarlane, T.V.; Clancy, T.; Evans, G.; Thakker, N. Dento-osseous changes as diagnostic markers in familial adenomatous polyposis families. Oral Dis. 2003, 9, 29–33. [Google Scholar] [CrossRef]
- D’Agostino, S.; Dell’Olio, F.; Tempesta, A.; Cervinara, F.; D’Amati, A.; Dolci, M.; Favia, G.; Capodiferro, S.; Limongelli, L. Osteoma of the Jaw as First Clinical Sign of Gardner’s Syndrome: The Experience of Two Italian Centers and Review. J. Clin. Med. 2023, 12, 1496. [Google Scholar] [CrossRef]
- Thakker, N.; Davies, R.; Horner, K.; Armstrong, J.; Clancy, T.; Guy, S.; Harris, R.; Sloan, P.; Evans, G. The dental phenotype in familial adenomatous polyposis: Diagnostic application of a weighted scoring system for changes on dental panoramic radiographs. J. Med. Genet. 1995, 32, 458–464. [Google Scholar] [CrossRef][Green Version]
- Almeida, F.T.; Pacheco-Pereira, C.; Porporatti, A.L.; Flores-Mir, C.; Leite, A.F.; De Luca Canto, G.; Guerra, E.N. Oral manifestations in patients with familial adenomatous polyposis: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Bulow, S.; Sondergaard, J.O.; Witt, I.; Larsen, E.; Tetens, G. Mandibular osteomas in familial polyposis coli. Dis. Colon Rectum 1984, 27, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Katou, F.; Motegi, K.; Baba, S. Mandibular lesions in patients with adenomatosis coli. J. Cranio-Maxillo-Facial Surg. Off. Publ. Eur. Assoc. Cranio-Maxillo-Facial Surg. 1989, 17, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Wijn, M.A.; Keller, J.J.; Giardiello, F.M.; Brand, H.S. Oral and maxillofacial manifestations of familial adenomatous polyposis. Oral Dis. 2007, 13, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Budzynska, A. Dentistry anomalies in patients with Lynch syndrome and familial adenomatous polyposis. Ann. Acad. Medicae Stetin. 2008, 54, 106–111. [Google Scholar]
- Bertario, L.; Russo, A.; Sala, P.; Eboli, M.; Giarola, M.; D’Amico, F.; Gismondi, V.; Varesco, L.; Pierotti, M.A.; Radice, P.; et al. Genotype and phenotype factors as determinants of desmoid tumors in patients with familial adenomatous polyposis. Int. J. Cancer 2001, 95, 102–107. [Google Scholar] [CrossRef]
- Bertario, L.; Russo, A.; Sala, P.; Varesco, L.; Giarola, M.; Mondini, P.; Pierotti, M.; Spinelli, P.; Radice, P.; Hereditary Colorectal Tumor, R. Multiple approach to the exploration of genotype-phenotype correlations in familial adenomatous polyposis. J. Clin. Oncol. 2003, 21, 1698–1707. [Google Scholar] [CrossRef] [PubMed]
- Bisgaard, M.L.; Bulow, S. Familial adenomatous polyposis (FAP): Genotype correlation to FAP phenotype with osteomas and sebaceous cysts. Am. J. Med. Genet. A 2006, 140, 200–204. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Ando, H.; Nagase, H.; Nishisho, I.; Horii, A.; Miki, Y.; Mori, T.; Utsunomiya, J.; Baba, S.; Petersen, G.; et al. Germ-line mutations of the APC gene in 53 familial adenomatous polyposis patients. Proc. Natl. Acad. Sci. USA 1992, 89, 4452–4456. [Google Scholar] [CrossRef]
- Fang, X.; Svitkina, T.M. Adenomatous Polyposis Coli (APC) in cell migration. Eur. J. Cell Biol. 2022, 101, 151228. [Google Scholar] [CrossRef]
- Wang, X.P.; O’Connell, D.J.; Lund, J.J.; Saadi, I.; Kuraguchi, M.; Turbe-Doan, A.; Cavallesco, R.; Kim, H.; Park, P.J.; Harada, H.; et al. Apc inhibition of Wnt signaling regulates supernumerary tooth formation during embryogenesis and throughout adulthood. Development 2009, 136, 1939–1949. [Google Scholar] [CrossRef] [PubMed]
- Hermans, F.; Hemeryck, L.; Lambrichts, I.; Bronckaers, A.; Vankelecom, H. Intertwined Signaling Pathways Governing Tooth Development: A Give-and-Take Between Canonical Wnt and Shh. Front. Cell Dev. Biol. 2021, 9, 758203. [Google Scholar] [CrossRef] [PubMed]
- Thesleff, I.; Sharpe, P. Signalling networks regulating dental development. Mech. Dev. 1997, 67, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Honma, T.; Matsuda, Y.; Suzuki, Y.; Narisawa, R.; Ajioka, Y.; Asakura, H. Nuclear translocation of beta-catenin in colorectal cancer. Br. J. Cancer 2000, 82, 1689–1693. [Google Scholar] [CrossRef] [PubMed]
- Barua, D.; Hlavacek, W.S. Modeling the effect of APC truncation on destruction complex function in colorectal cancer cells. PLoS Comput. Biol. 2013, 9, e1003217. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.; Hocking, A.M.; Brown, J.D.; Moon, R.T. Mechanism and function of signal transduction by the Wnt/beta-catenin and Wnt/Ca2+ pathways. Oncogene 1999, 18, 7860–7872. [Google Scholar] [CrossRef]
- Akiyama, T. Wnt/beta-catenin signaling. Cytokine Growth Factor. Rev. 2000, 11, 273–282. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).