
Metabolome and Transcriptome Unveil the Correlated Metabolites and Transcripts with 2-acetyl-1-pyrroline in Fragrant Rice

Abstract
:1. Introduction
2. Results
2.1. Rice Fragrance Confirmation through Sensory and Genetic Analysis
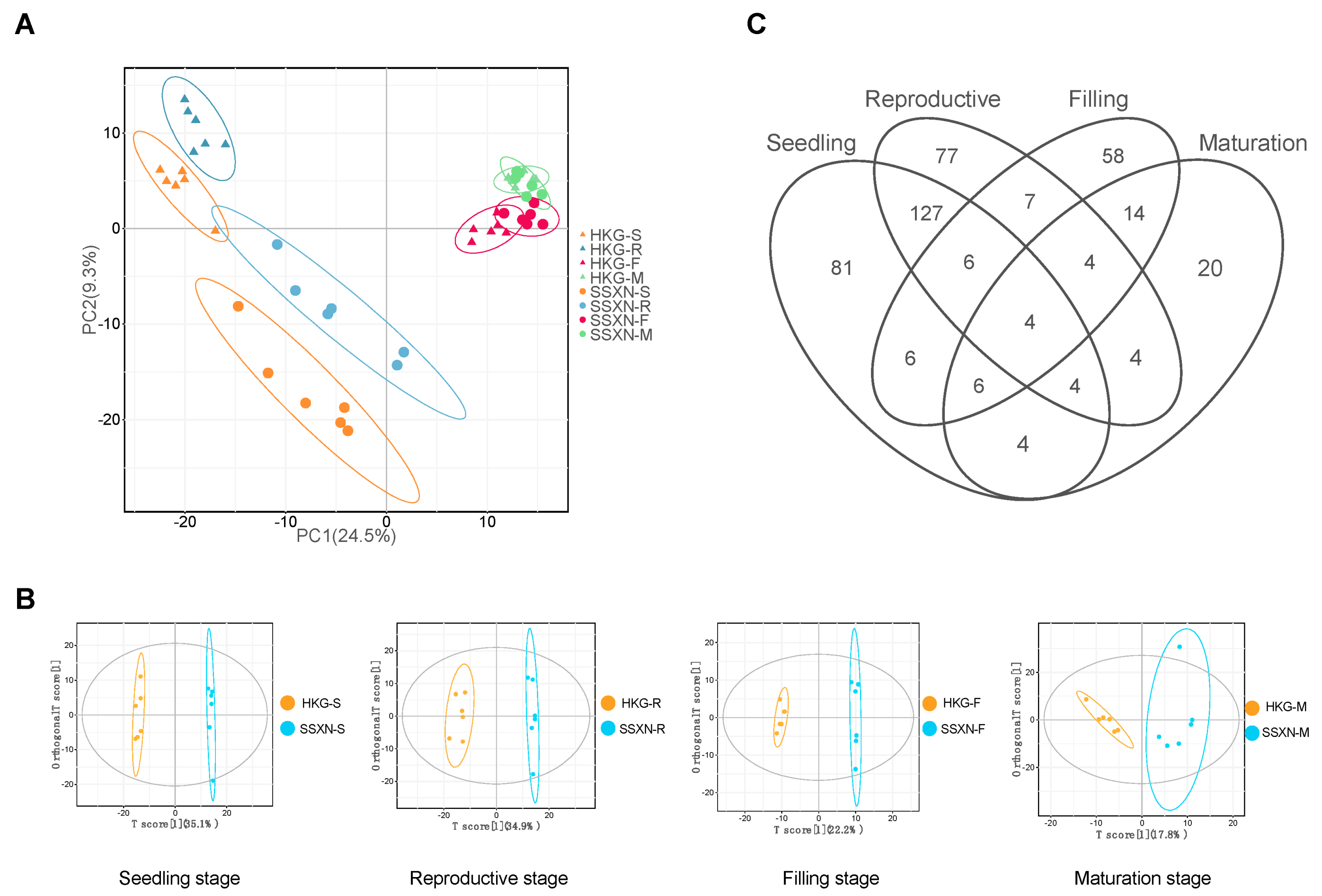
2.2. Comparative Volatile Profiling in Fragrant and Non-Fragrant Rice Varieties
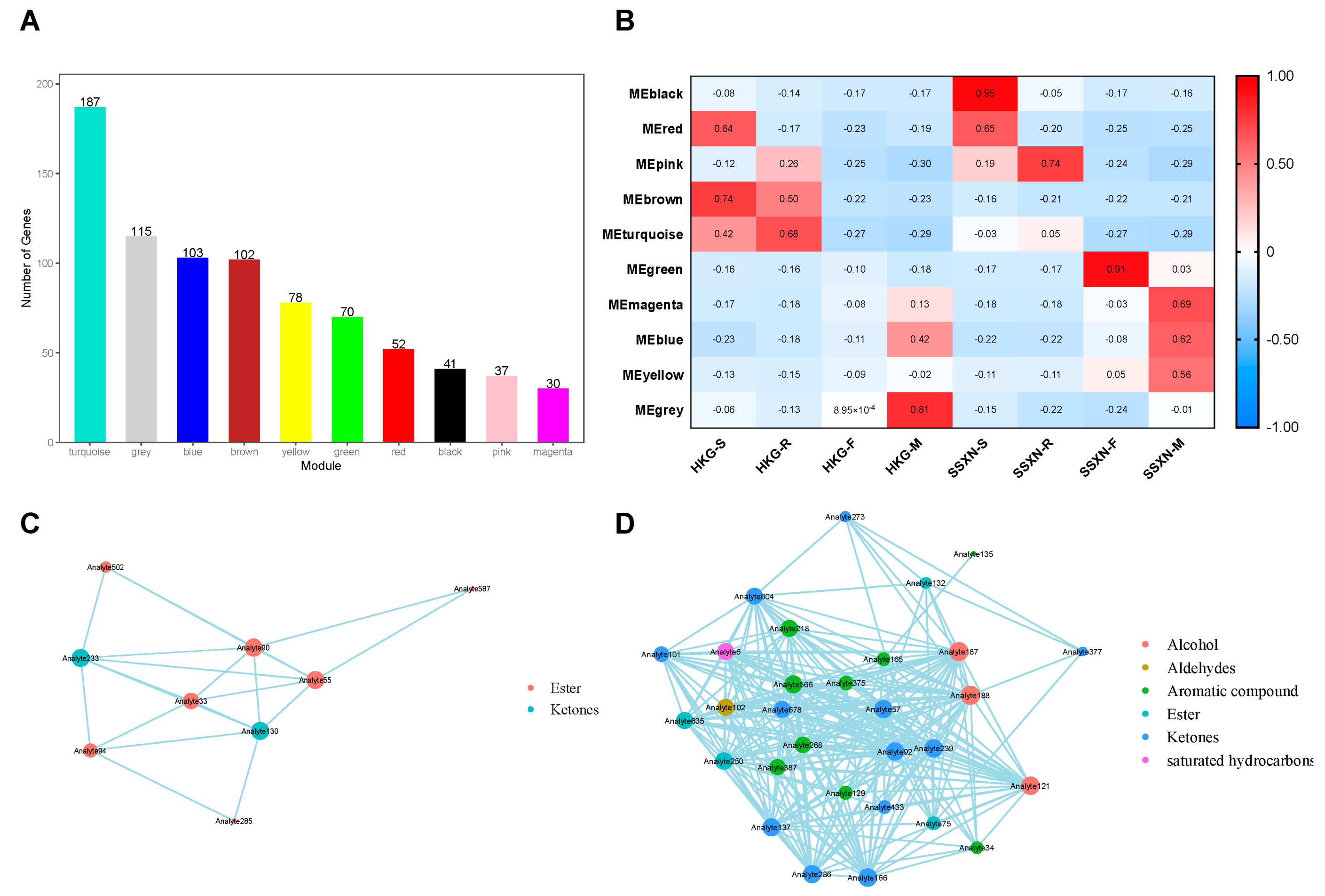
2.3. The Correlation Analysis of 2AP and Metabolome Co-Expression Analysis
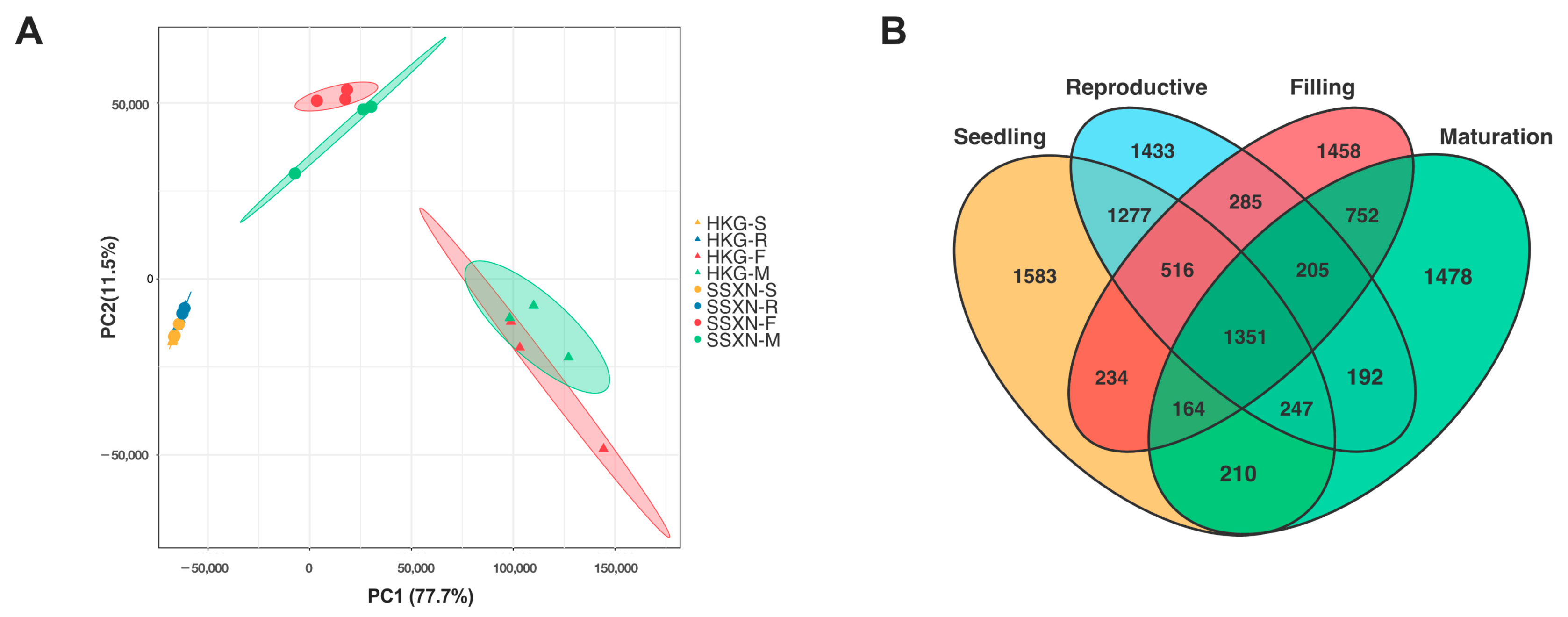
2.4. Comparative Transcriptome Profiling in Fragrant and Non-Fragrant Rice Varieties
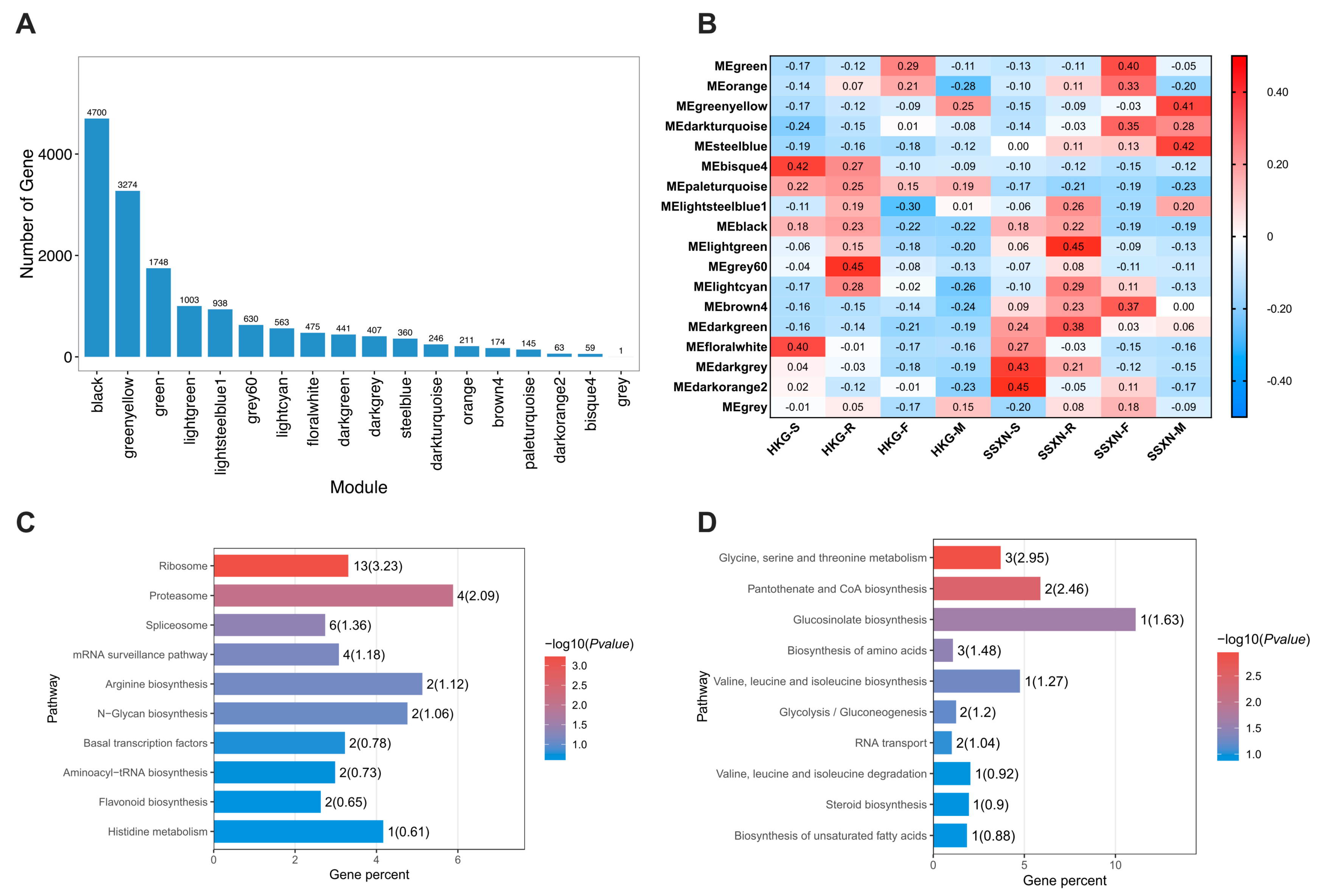
2.5. Comparative Transcriptome Profiling in Fragrant and Non-Fragrant Rice Varieties
2.6. The Correlation between Metabolome and Transcriptome
3. Discussion
4. Materials and Methods
4.1. Material and Planting
4.2. Fragrance Determination
4.3. Sequence Analysis of Fragrant Gene
4.4. Gas Chromatography–Time-of-Flight Mass Spectrometry Analysis and Data Annotation
4.5. Ribosomal Nuclear Acids (RNA) Extraction, Library Construction and Sequencing
4.6. Quantitative Real-Time Polymerase Chain Raction (PCR) Validation
4.7. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bairagi, S.; Demont, M.; Custodio, M.C.; Ynion, J. What drives consumer demand for rice fragrance? Evidence from South and Southeast Asia. Br. Food J. 2020, 122, 3473–3498. [Google Scholar] [CrossRef]
- Sakthivel, K.; Sundaram, R.M.; Shobha Rani, N.; Balachandran, S.M.; Neeraja, C.N. Genetic and molecular basis of fragrance in rice. Biotechnol. Adv. 2009, 27, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.K.; Srivastav, P.P. A paradigm of volatile aroma compounds in rice and their product with extraction and identification methods: A comprehensive review. Food Res. Int. 2020, 130, 108924. [Google Scholar] [CrossRef] [PubMed]
- Bergman, C.J.; Delgado, J.T.; Bryant, R.; Grimm, C.; Cadwallader, K.R.; Webb, B.D. Rapid Gas Chromatographic Technique for Quantifying 2-Acetyl-1-Pyrroline and Hexanal in Rice (Oryza sativa, L.). Cereal Chem. 2000, 77, 454–458. [Google Scholar] [CrossRef]
- Yoshihashi, T.; Huong, N.T.T.; Inatomi, H. Precursors of 2-Acetyl-1-pyrroline, a Potent Flavor Compound of an Aromatic Rice Variety. J. Agric. Food Chem. 2002, 50, 2001–2004. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Li, Y.; Nie, J.; He, L.; Pan, S.; Duan, M.; Tian, H.; Xiao, L.; Zhong, K.; Tang, X. Nitrogen application and different water regimes at booting stage improved yield and 2-acetyl-1-pyrroline (2AP) formation in fragrant rice. Rice 2019, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Poonlaphdecha, J.; Gantet, P.; Maraval, I.; Sauvage, F.-X.; Menut, C.; Morère, A.; Boulanger, R.; Wüst, M.; Gunata, Z. Biosynthesis of 2-acetyl-1-pyrroline in rice calli cultures: Demonstration of 1-pyrroline as a limiting substrate. Food Chem. 2016, 197, 965–971. [Google Scholar] [CrossRef]
- Kaikavoosi, K.; Kad, T.D.; Zanan, R.L.; Nadaf, A.B. 2-Acetyl-1-Pyrroline Augmentation in Scented indica Rice (Oryza sativa L.) Varieties Through Δ1-Pyrroline-5-Carboxylate Synthetase (P5CS) Gene Transformation. Appl. Biochem. Biotechnol. 2015, 177, 1466–1479. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Waters, D.; Cordeiro, G.M.; Henry, R.J.; Reinke, R.F. A single nucleotide polymorphism (SNP) marker linked to the fragrance gene in rice (Oryza sativa L.). Plant Sci. 2003, 165, 359–364. [Google Scholar] [CrossRef]
- Bradbury, L.M.; Fitzgerald, T.L.; Henry, R.J.; Jin, Q.; Waters, D.L. The gene for fragrance in rice. Plant Biotechnol. J. 2005, 3, 363–370. [Google Scholar] [CrossRef]
- Chen, S.; Wu, J.; Yang, Y.; Shi, W.; Xu, M. The fgr gene responsible for rice fragrance was restricted within 69kb. Plant Sci. 2006, 171, 505–514. [Google Scholar] [CrossRef]
- Chen, S.; Yang, Y.; Shi, W.; Ji, Q.; He, F.; Zhang, Z.; Cheng, Z.; Liu, X.; Xu, M. Badh2, Encoding Betaine Aldehyde Dehydrogenase, Inhibits the Biosynthesis of 2-Acetyl-1-Pyrroline, a Major Component in Rice Fragrance. Plant Cell 2008, 20, 1850–1861. [Google Scholar] [CrossRef] [PubMed]
- Hinge, V.R.; Patil, H.B.; Nadaf, A.B. Aroma volatile analyses and 2AP characterization at various developmental stages in Basmati and Non-Basmati scented rice (Oryza sativa L.) cultivars. Rice 2016, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.-Z. The conservation of Chinese rice biodiversity: Genetic erosion, ethnobotany and prospects. Genet. Resour. Crop Evol. 2003, 50, 17–32. [Google Scholar] [CrossRef]
- Lei, Q.-Y.; Zhou, J.-J.; Xiong, Y.; Zhang, W.-H.; Luo, J.; Long, C.-L. Genetic Diversity Evaluation and Conservation of Kam Fragrant Glutinous Rice (Oryza sativa L.) Germplasm in Southeast Guizhou, China. Plants 2021, 10, 1898. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Zhang, D.; Zhang, H.; Wang, M.; Sun, J.; Wei, X.; Qiu, Z.; Tang, S.; Cao, Y.; Wang, X. Genetic diversity of rice cultivars (Oryza sativa L.) in China and the temporal trends in recent fifty years. Chin. Sci. Bull. 2006, 51, 681–688. [Google Scholar] [CrossRef]
- Han, L.; Cao, G. Status of collection, conservation and propagation of rice germplasm in China. J. Plant Genet. Resour. 2005, 6, 359–364. [Google Scholar]
- Wang, Y.; Wang, Y.; Sun, X.; Caiji, Z.; Yang, J.; Cui, D.; Cao, G.; Ma, X.; Han, B.; Xue, D.; et al. Influence of ethnic traditional cultures on genetic diversity of rice landraces under on-farm conservation in southwest China. J. Ethnobiol. Ethnomed. 2016, 12, 51. [Google Scholar] [CrossRef]
- Zeng, Y.; Xia, X.; Nong, B.; Zhang, Z.; Yang, X.; Deng, G. Identification of fragrance and genotype of local varieties of Guangxi characteristic aromatic rice. J. South. Agric. 2017, 48, 18–23. [Google Scholar]
- Liu, C.; Cui, D.; Jiao, A.; Ma, X.; Li, X.; Han, B.; Chen, H.; Ruan, R.; Wang, Y.; Han, L. Kam Sweet Rice (Oryza sativa L.) Is a Special Ecotypic Rice in Southeast Guizhou, China as Revealed by Genetic Diversity Analysis. Front. Plant Sci. 2022, 13, 830556. [Google Scholar] [CrossRef]
- Chan-in, P.; Jamjod, S.; Yimyam, N.; Rerkasem, B.; Pusadee, T. Grain Quality and Allelic Variation of the Badh2 Gene in Thai Fragrant Rice Landraces. Agronomy 2020, 10, 779. [Google Scholar] [CrossRef]
- Daygon, V.D.; Prakash, S.; Calingacion, M.; Riedel, A.; Ovenden, B.; Snell, P.; Mitchell, J.; Fitzgerald, M. Understanding the Jasmine phenotype of rice through metabolite profiling and sensory evaluation. Metabolomics 2016, 12, 63. [Google Scholar] [CrossRef]
- Phitaktansakul, R.; Kim, K.-W.; Aung, K.M.; Maung, T.Z.; Min, M.-H.; Somsri, A.; Lee, W.; Lee, S.-B.; Nam, J.; Kim, S.-H.; et al. Multi-omics analysis reveals the genetic basis of rice fragrance mediated by betaine aldehyde dehydrogenase 2. J. Adv. Res. 2022, 42, 303–314. [Google Scholar] [CrossRef]
- Setyaningsih, W.; Majchrzak, T.; Dymerski, T.; Namieśnik, J.; Palma, M. Key-Marker Volatile Compounds in Aromatic Rice (Oryza sativa) Grains: An HS-SPME Extraction Method Combined with GC×GC-TOFMS. Molecules 2019, 24, 4180. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Xue, Y.; Shen, Q. Changes in the major aroma-active compounds and taste components of Jasmine rice during storage. Food Res. Int. 2020, 133, 109160. [Google Scholar] [CrossRef] [PubMed]
- Jendoubi, T. Approaches to Integrating Metabolomics and Multi-Omics Data: A Primer. Metabolites 2021, 11, 184. [Google Scholar] [CrossRef] [PubMed]
- Xin, W.; Zhang, L.; Zhang, W.; Gao, J.; Yi, J.; Zhen, X.; Li, Z.; Zhao, Y.; Peng, C.; Zhao, C. An Integrated Analysis of the Rice Transcriptome and Metabolome Reveals Differential Regulation of Carbon and Nitrogen Metabolism in Response to Nitrogen Availability. Int. J. Mol. Sci. 2019, 20, 2349. [Google Scholar] [CrossRef] [PubMed]
- Zaghum, M.J.; Ali, K.; Teng, S. Integrated Genetic and Omics Approaches for the Regulation of Nutritional Activities in Rice (Oryza sativa L.). Agriculture 2022, 12, 1757. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, G. Transcriptome and metabolome profiling reveal the regulatory mechanism of protein accumulation in inferior grains of indica-japonica rice hybrids. Curr. Plant Biol. 2021, 28, 100226. [Google Scholar] [CrossRef]
- Shao, G.N.; Tang, A.; Tang, S.Q.; Luo, J.; Jiao, G.A.; Wu, J.L.; Hu, P.S. A new deletion mutation of fragrant gene and the development of three molecular markers for fragrance in rice. Plant Breed. 2011, 130, 172–176. [Google Scholar] [CrossRef]
- Cheng, S.; Fang, Z.; Wang, C.; Cheng, X.; Huang, F.; Yan, C.; Zhou, L.; Wu, X.; Li, Z.; Ren, Y. Modulation of 2-Acetyl-1-pyrroline (2-AP) Accumulation, Yield Formation and Antioxidant Attributes in Fragrant Rice by Exogenous Methylglyoxal (MG) Application. J. Plant Growth Regul. 2023, 42, 1444–1456. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhang, C.; Liu, K.; Liu, Q. Volatile Organic Compounds, Evaluation Methods and Processing Properties for Cooked Rice Flavor. Rice 2022, 15, 53. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Shafiq, S.; Ashraf, U.; Qi, J.; Mo, Z.; Tang, X. Biosynthesis of 2-Acetyl-1-pyrroline in Fragrant Rice: Recent Insights into Agro-management, Environmental Factors, and Functional Genomics. J. Agric. Food Chem. 2023, 71, 4201–4215. [Google Scholar] [CrossRef] [PubMed]
- Daygon, V.D.; Calingacion, M.; Forster, L.C.; Voss, J.J.D.; Schwartz, B.D.; Ovenden, B.; Alonso, D.E.; McCouch, S.R.; Garson, M.J.; Fitzgerald, M.A. Metabolomics and genomics combine to unravel the pathway for the presence of fragrance in rice. Sci. Rep. 2017, 7, 8767. [Google Scholar] [CrossRef] [PubMed]
- Jie, Y.; Shi, T.; Zhang, Z.; Yan, Q. Identification of Key Volatiles Differentiating Aromatic Rice Cultivars Using an Untargeted Metabolomics Approach. Metabolites 2021, 11, 528. [Google Scholar] [CrossRef]
- Maga, J.A. Pyrroles in foods. J. Agric. Food Chem. 1981, 29, 691–694. [Google Scholar] [CrossRef]
- Buttery, R.G.; Ling, L.C.; Juliano, B.O.; Turnbaugh, J.G. Cooked rice aroma and 2-acetyl-1-pyrroline. J. Agric. Food Chem. 1983, 31, 823–826. [Google Scholar] [CrossRef]
- Ma, R.; Tian, Y.; Zhang, H.; Cai, C.; Chen, L.; Jin, Z. Interactions between rice amylose and aroma compounds and their effect on rice fragrance release. Food Chem. 2019, 289, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Hu, Z.; Fang, C.; Hu, X. Characteristic Flavor Compounds and Functional Components of Fragrant Rice with Different Flavor Types. Foods 2023, 12, 2185. [Google Scholar] [CrossRef]
- Chen, C.L.; Li, C.C.; Sung, J.M. Carbohydrate metabolism enzymes in CO2-enriched developing rice grains of cultivars varying in grain size. Physiol. Plant. 1994, 90, 79–85. [Google Scholar] [CrossRef]
- Yamakawa, H.; Hakata, M. Atlas of rice grain filling-related metabolism under high temperature: Joint analysis of metabolome and transcriptome demonstrated inhibition of starch accumulation and induction of amino acid accumulation. Plant Cell Physiol. 2010, 51, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liu, M.; Chu, N.; Chen, G.; Wang, P.; Mo, J.; Guo, H.; Xu, J.; Zhou, H. Combined transcriptome and metabolome reveal glutathione metabolism plays a critical role in resistance to salinity in rice landraces HD961. Front. Plant Sci. 2022, 13, 952595. [Google Scholar] [CrossRef]
- Fitzgerald, T.L.; Waters, D.L.E.; Brooks, L.O.; Henry, R.J. Fragrance in rice (Oryza sativa) is associated with reduced yield under salt treatment. Environ. Exp. Bot. 2010, 68, 292–300. [Google Scholar] [CrossRef]
- Prodhan, Z.H.; Islam, S.A.; Alam, M.S.; Li, S.; Jiang, M.; Tan, Y.; Shu, Q. Impact of OsBadh2 Mutations on Salt Stress Response in Rice. Plants 2022, 11, 2829. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wu, G. Roles of dietary glycine, proline, and hydroxyproline in collagen synthesis and animal growth. Amino Acids 2018, 50, 29–38. [Google Scholar] [CrossRef]
- Poonlaphdecha, J.; Maraval, I.; Roques, S.; Audebert, A.; Boulanger, R.; Bry, X.; Gunata, Z. Effect of Timing and Duration of Salt Treatment during Growth of a Fragrant Rice Variety on Yield and 2-Acetyl-1-pyrroline, Proline, and GABA Levels. J. Agric. Food Chem. 2012, 60, 3824–3830. [Google Scholar] [CrossRef]
- Luo, H.; Zhang, T.; Zheng, A.; He, L.; Lai, R.; Liu, J.; Xing, P.; Tang, X. Exogenous proline induces regulation in 2-acetyl-1-pyrroline (2-AP) biosynthesis and quality characters in fragrant rice (Oryza sativa L.). Sci. Rep. 2020, 10, 13971. [Google Scholar] [CrossRef]
- Sood, B.C.; Siddiq, E.A. A rapid technique for scent determination in rice. Indian J. Genet. Plant Breed. 1978, 38, 268–275. [Google Scholar]
- Shi, W.; Yang, Y.; Chen, S.; Xu, M. Discovery of a new fragrance allele and the development of functional markers for the breeding of fragrant rice varieties. Mol. Breed. 2008, 22, 185–192. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolites | Seedling Stage + | Reproductive Stage | Filling Stage | Maturation Stage | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Log2FC | p Value | VIP * | Log2FC | p Value | VIP | Log2FC | p Value | VIP | Log2FC | p Value | VIP | |
| 2-Acetyl-1-pyrroline | 23.858 | 0.000 | 1.618 | 23.627 | 0.000 | 1.551 | 22.953 | 0.002 | 1.729 | 24.424 | 0.000 | 2.019 |
| (+)-epi-Bicyclosesquiphellandrene | −2.133 | 0.007 | 1.243 | −1.837 | 0.005 | 1.277 | 2.767 | 0.000 | 1.897 | 16.211 | 0.003 | 1.914 |
| Ethanone, 1-(1H-pyrrol-2-yl)- | 19.915 | 0.000 | 1.614 | 19.851 | 0.000 | 1.587 | 19.097 | 0.003 | 1.681 | 20.720 | 0.001 | 1.911 |
| Phenol, 3-amino- | 19.218 | 0.001 | 1.394 | 19.441 | 0.001 | 1.375 | 18.384 | 0.002 | 1.715 | 19.444 | 0.001 | 1.966 |
| Metabolite ID | Description | Flavor | Class |
|---|---|---|---|
| Analyte84 | 2-Butanol, (R)- | alcoholic, sweet, fruity | alcohol |
| Analyte428 | 2-(3-Methyl-but-1-ynyl)-cyclohexene-1-carboxaldehyde | jasmine | aldehydes |
| Analyte90 | Butanoic acid, ethyl ester | pineapple-like | ester |
| Analyte55 | Propanoic acid, 2-methyl-, ethyl ester | fruity | ester |
| Analyte33 | Ethyl Acetate | fruity, sweet, pear-like | ester |
| Analyte94 | Carbonic acid, ethyl-, methyl ester | sweet, ether-like | ester |
| Analyte502 | Benzoic acid, ethyl ester | sweet, fruity, cherry, grape | ester |
| Analyte587 | Benzeneacetic acid, ethyl ester | sweet, floral, fruity | ester |
| Analyte285 | Hepten-2-yl tiglate, 6-methyl-5- | fruity | ester |
| Analyte828 | Linoleic acid ethyl ester | faintly fruity | ester |
| Analyte431 | gamma-Nonalactone | coconut, creamy, fruity | ester |
| Analyte233 | 2-Octen-4-one, 2-methoxy- | sweet, mushroom-like, earthy | ketones |
| Analyte130 | 4-Methylheptane-3,5-dione | buttery, creamy | ketones |
| Metabolite ID | Description | Flavor | Class |
|---|---|---|---|
| Analyte188 | 1-butanol, 3-methyl- | blue cheese | alcohol |
| Analyte187 | 1-butanol, 2-methyl- | blue cheese | alcohol |
| Analyte121 | 1-propanol, 2-methyl- | sweet, musty | alcohol |
| Analyte102 | butanal, 2-ethyl-3-methyl- | cocoa | aldehydes |
| Analyte566 | naphthalene, 1,2,4a,5,8,8a-hexahydro-4,7-dimethyl-1-(1-methylethyl)-, [1S-(1,4a,8a)]- | woody | aromatic compound |
| Analyte218 | styrene | sweet odor | aromatic compound |
| Analyte387 | pyrrole | nutty odor | aromatic compound |
| Analyte268 | 2-acetyl-1-pyrroline | popcorn and cracker-like | aromatic compound |
| Analyte375 | .alfa.-copaene | balsamic, woody, sweet | aromatic compound |
| Analyte129 | benzene, 1,3-dimethyl- | aromatic | aromatic compound |
| Analyte165 | o-xylene | faint sweet | aromatic compound |
| Analyte34 | furan, 3-methyl- | chocolate | aromatic compound |
| Analyte135 | p-xylene | faint sweet | aromatic compound |
| Analyte250 | hexanoic acid, 3-hexenyl ester, (Z)- | sweet, fatty, grassy | ester |
| Analyte635 | 2,2,4-trimethyl-1,3-pentanediol diisobutyrate | mild and sweet | ester |
| Analyte75 | isobutyl acetate | sweet, banana-like | ester |
| Analyte132 | 1-butanol, 3-methyl-, acetate | sweet, banana-like | ester |
| Analyte166 | 2-heptanone | sweet, fruity, and camphor-like. | ketones |
| Analyte92 | 3-pentanone, 2,4-dimethyl- | sweet | ketones |
| Analyte57 | 2,3-butanedione | buttery | ketones |
| Analyte286 | 2-hydroxy-3-pentanone | Sweet, fruity | ketones |
| Analyte137 | 2,3-hexanedione | butter, caramel, creamy, | ketones |
| Analyte678 | ethanone, 1-(1H-pyrrol-2-yl)- | popcorn-like | ketones |
| Analyte604 | 2-tridecanone | milky, coconut, nutty | ketones |
| Analyte101 | 2,3-pentanedione | butter | ketones |
| Analyte433 | bicyclo[3.1.1]heptan-2-one, 6,6-dimethyl-, (1R)- | woody | ketones |
| Analyte273 | 2-hydroxy-3-hexanone | sweet, fruity | ketones |
| Analyte377 | 2-decanone | floral, fatty, and peach-like | ketones |
| Analyte269 | 5-hepten-2-one, 6-methyl- | fruity and apple-like | ketones |
| Analyte239 | 2-octanone | fruity, cheese-like | ketones |
| Analyte6 | pentane, 3-methyl- | sweet odor | saturated hydrocarbons |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, Y.; Nong, B.; Xia, X.; Zhang, Z.; Wang, Y.; Xu, Y.; Feng, R.; Guo, H.; Liang, Y.; Chen, C.; et al. Metabolome and Transcriptome Unveil the Correlated Metabolites and Transcripts with 2-acetyl-1-pyrroline in Fragrant Rice. Int. J. Mol. Sci. 2024, 25, 8207. https://doi.org/10.3390/ijms25158207
Zeng Y, Nong B, Xia X, Zhang Z, Wang Y, Xu Y, Feng R, Guo H, Liang Y, Chen C, et al. Metabolome and Transcriptome Unveil the Correlated Metabolites and Transcripts with 2-acetyl-1-pyrroline in Fragrant Rice. International Journal of Molecular Sciences. 2024; 25(15):8207. https://doi.org/10.3390/ijms25158207
Chicago/Turabian StyleZeng, Yu, Baoxuan Nong, Xiuzhong Xia, Zongqiong Zhang, Yuhao Wang, Yong Xu, Rui Feng, Hui Guo, Yuntao Liang, Can Chen, and et al. 2024. "Metabolome and Transcriptome Unveil the Correlated Metabolites and Transcripts with 2-acetyl-1-pyrroline in Fragrant Rice" International Journal of Molecular Sciences 25, no. 15: 8207. https://doi.org/10.3390/ijms25158207
APA StyleZeng, Y., Nong, B., Xia, X., Zhang, Z., Wang, Y., Xu, Y., Feng, R., Guo, H., Liang, Y., Chen, C., Liang, S., Jiang, X., Yang, X., & Li, D. (2024). Metabolome and Transcriptome Unveil the Correlated Metabolites and Transcripts with 2-acetyl-1-pyrroline in Fragrant Rice. International Journal of Molecular Sciences, 25(15), 8207. https://doi.org/10.3390/ijms25158207