
Bioenergetic and Inflammatory Alterations in Regressed and Non-Regressed Patients with Autism Spectrum Disorder

Abstract
:1. Introduction
2. Results
2.1. Clinical Data
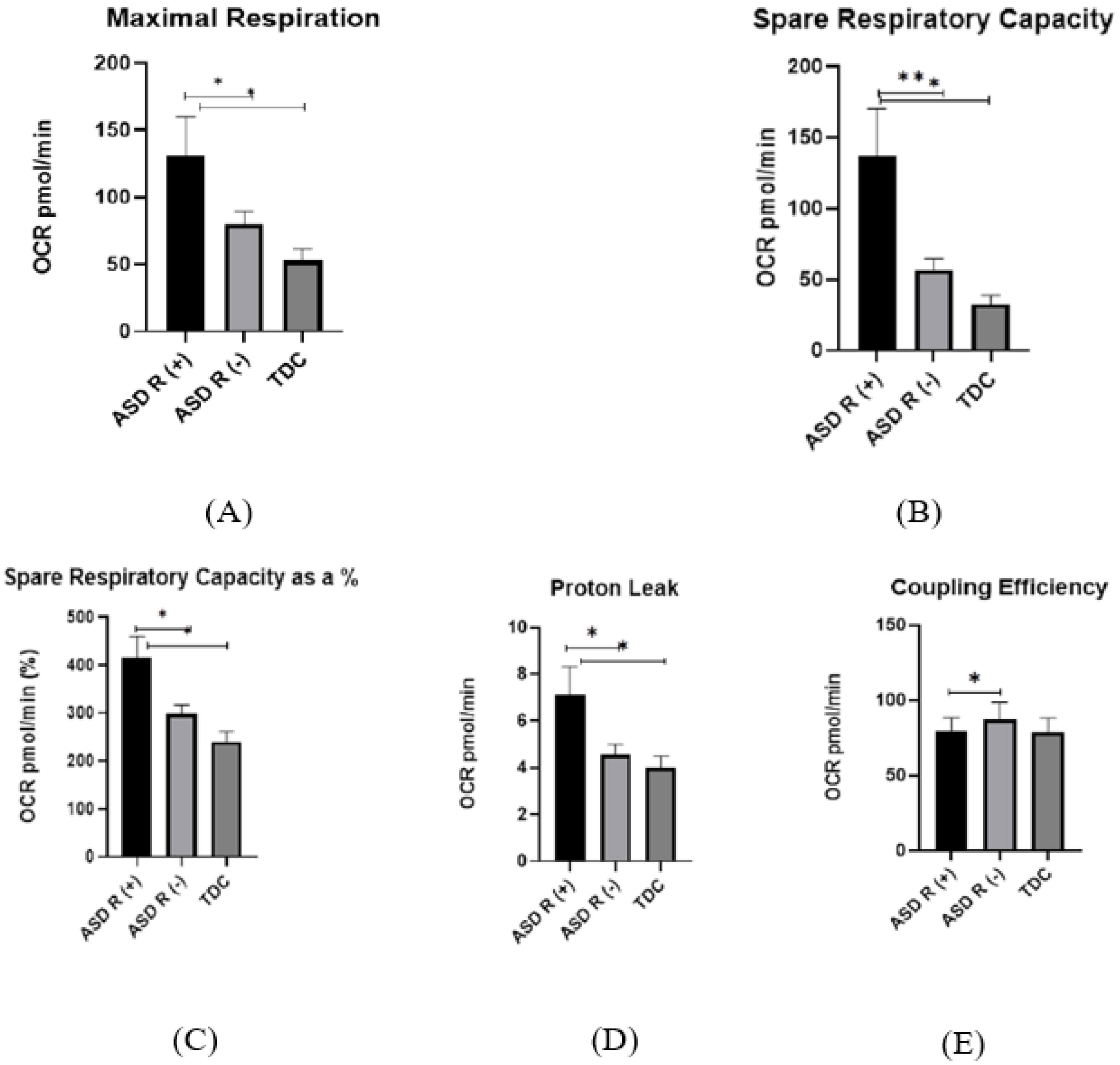
2.2. Mitochondrial Activity
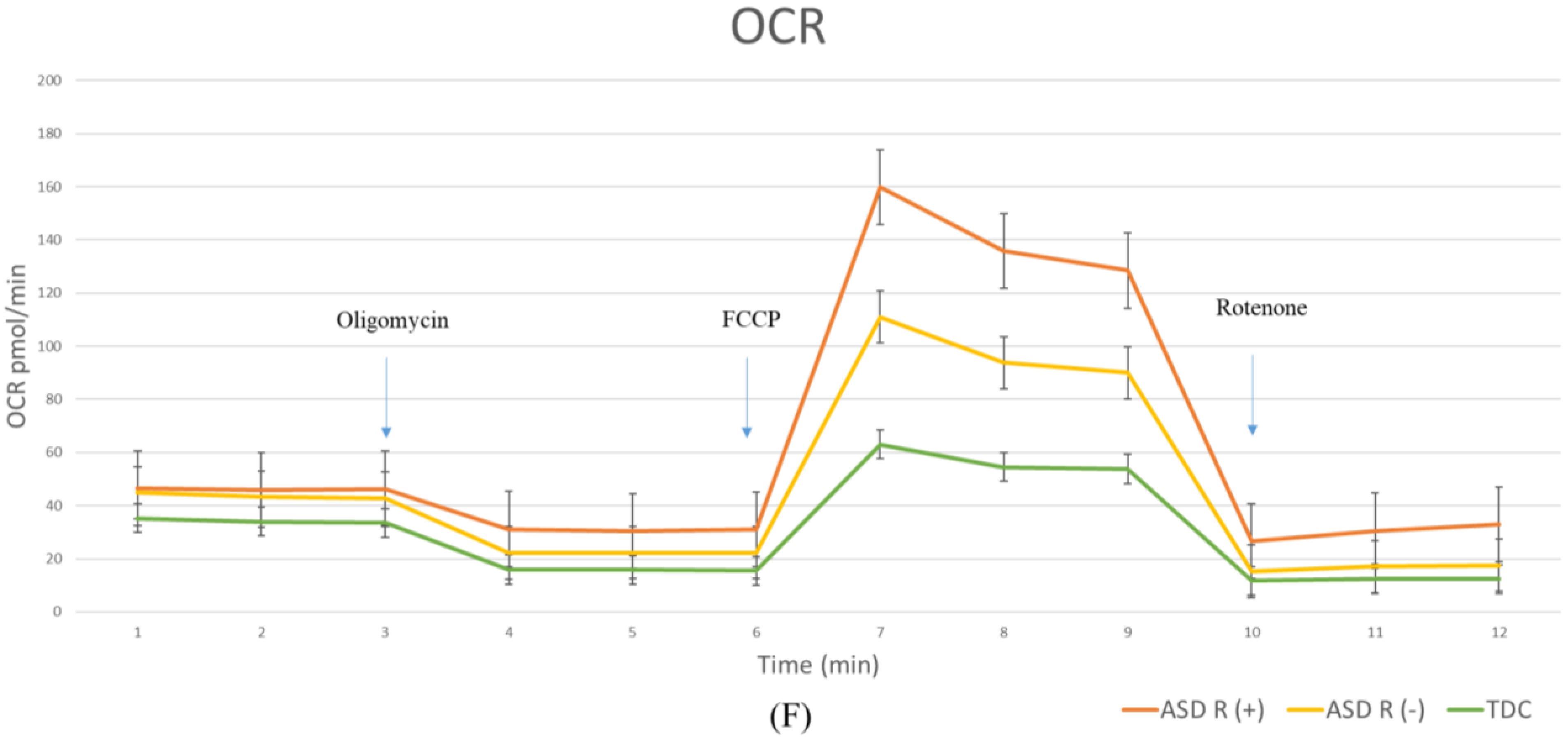
2.2.1. Oxygen Consumption Rate (OCR) in Children with Regressive/Non-Regressive ASD and TDC
- Basel Respiration
- Maximal Respiration
- Spare Respiratory Capacity
- Proton Leak
- Non-Mitochondrial Respiration
- Coupling efficiency
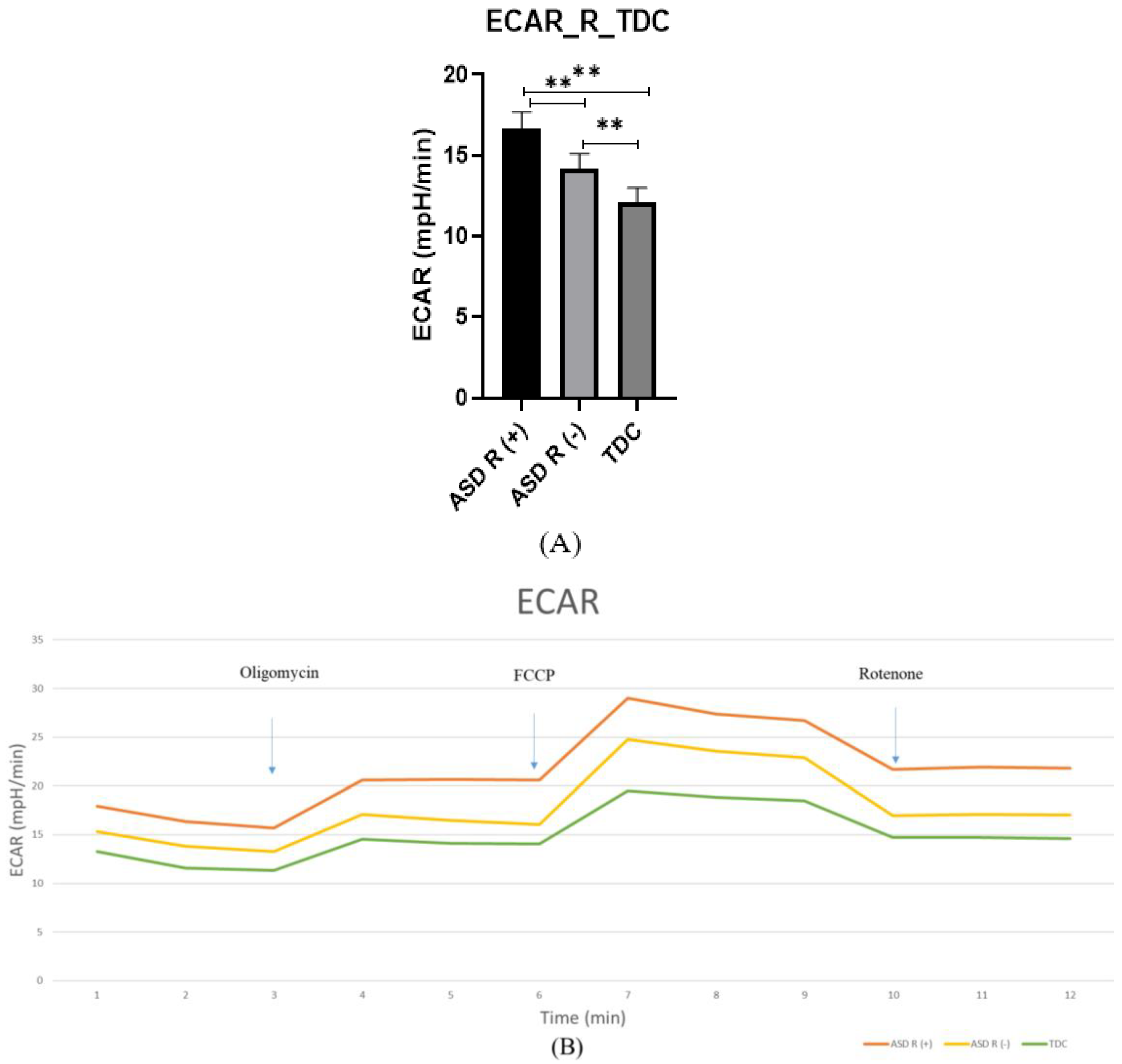
2.2.2. Extracellular Acidification Rate (ECAR)
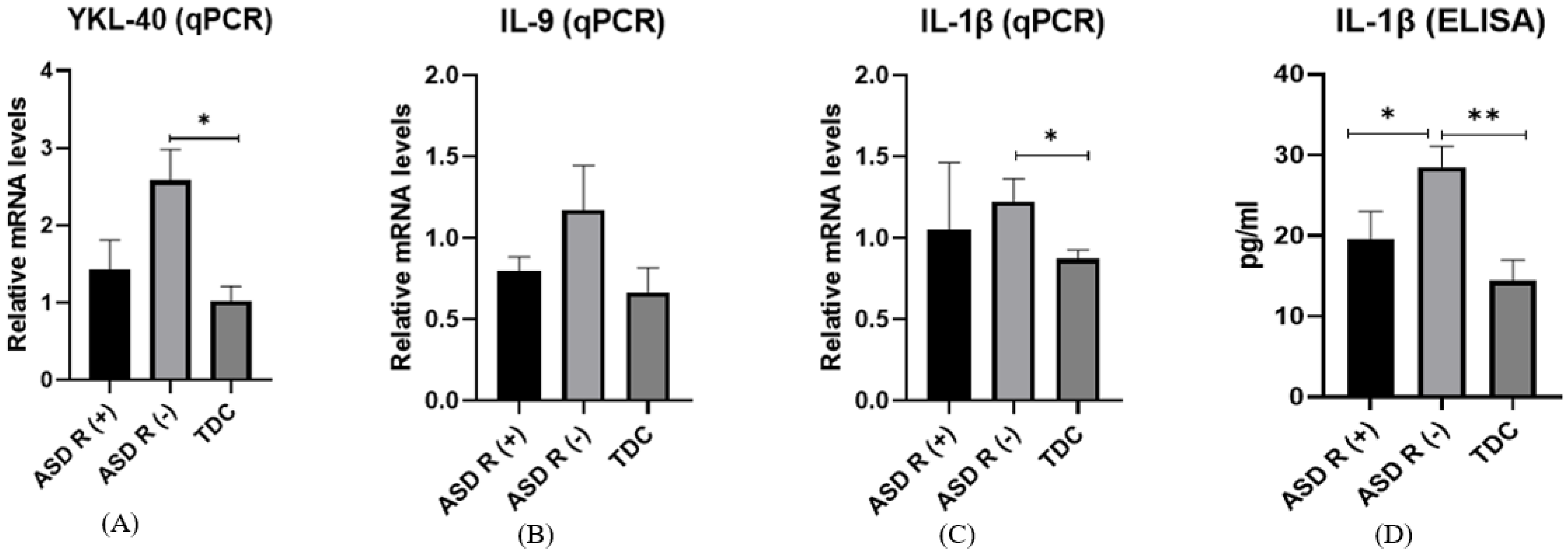
2.3. Gene and Protein Expression of Proinflammatory Markers
- Gene expression of COX-2
- Protein expression of COX-2
- Gene expression of YKL-40
- Protein expression of YKL-40
- Gene expression of IL-9
- Protein expression of IL-9
- Gene expression of IL-1β
- Protein expression of IL-1β
2.4. Correlation Analysis
3. Discussion
3.1. Novelty
3.2. PBMCs as a Model
3.3. Mitochondrial Function in Children with Regressive/Non-Regressive ASD and TDC
3.4. Inflammation
3.5. COX-2
3.6. YKL-40
3.7. Il-1 and Il-9
3.8. Correlation between Metabolic State and Regression in ASD
3.9. Limitations of This Study
4. Materials and Methods
4.1. Subjects
- Age 1.9–11 years.
- No previous intake of high-dose vitamins, mineral supplements, or immunomodulators in the past 3 months.
- No acute illness, epileptic seizures, or medication in the last month.
- No accompanying comorbidities.
- Completed informed consent from the parents for participation in the study in accordance with the instructions of the University Ethics Committee.
- If according to clinical criteria, the patient corresponds to probable syndromic/complex/ASD (performed by a team of pediatricians—child neurologists) or probable syndromic/complex/ASD proven through genetic or imaging methods.
- Chronic diseases such as infections, bronchial asthma, diabetes mellitus, etc.
- Healthy/not covering ASD according to DSM-5/children with normal intellectual development (good school performance), without chronic diseases such as asthma, diabetes, gastrointestinal disorders, etc.
- No acute illness and intake of medicines, or nutritional supplements such as vitamins, minerals, or immunostimulants one month before the examination.
- Completed informed consent from the parents for participation in the study in accordance with the instructions of the University Ethics Committee.
4.2. Mitochondrial Activity
4.2.1. Isolation of Peripheral Blood Mononuclear Cells (PBMCs)
4.2.2. Mito Stress Test
4.3. Gene Expression of COX-2, YKL-40, IL-1β, and IL-9
4.3.1. Isolation of Total RNA from White Blood Cells
4.3.2. Reverse Transcription and qPCR
4.4. Detection of COX-2, YKL-40, IL-1β and IL-9 in Plasma by ELISA
4.5. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Maenner, M.J.; Warren, Z.; Williams, A.R.; Amoakohene, E.; Bakian, A.V.; Bilder, D.A.; Durkin, M.S.; Fitzgerald, R.T.; Furnier, S.M.; Hughes, M.M.; et al. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2020. Surveill. Summ. 2023, 72, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Loomes, R.; Hull, L.; Mandy, W.P.L. What Is the Male-to-Female Ratio in Autism Spectrum Disorder? A Systematic Review and Meta-Analysis. J. Am. Acad. Child. Adolesc. Psychiatry 2017, 56, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Gevezova, M.; Sbirkov, Y.; Sarafian, V.; Plaimas, K.; Suratanee, A.; Maes, M. Autistic spectrum disorder (ASD)—Gene, molecular and pathway signatures linking systemic inflammation, mitochondrial dysfunction, transsynaptic signalling, and neurodevelopment. Brain Behav. Immun. Health 2023, 30, 100646. [Google Scholar] [CrossRef] [PubMed]
- Gevezova, M.; Minchev, D.; Pacheva, I.; Sbirkov, Y.; Yordanova, R.; Timova, E.; Kotetarov, V.; Ivanov, I.; Sarafian, V. Cellular Bioenergetic and Metabolic Changes in Patients with Autism Spectrum Disorder. Curr. Top. Med. Chem. 2021, 21, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Gevezova, M.; Sarafian, V.; Anderson, G.; Maes, M. Inflammation and Mitochondrial Dysfunction in Autism Spectrum Disorder. CNS Neurol. Disord. Drug Targets 2020, 19, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Al Dera, H. Cellular and molecular mechanisms underlying autism spectrum disorders and associated comorbidities: A pathophysiological review. Biomed. Pharmacother. 2022, 148, 112688. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lin, J.; Zhang, H.; Khan, N.U.; Zhang, J.; Tang, X.; Cao, X.; Shen, L. Oxidative Stress in Autism Spectrum Disorder-Current Progress of Mechanisms and Biomarkers. Front. Psychiatry 2022, 13, 813304. [Google Scholar] [CrossRef] [PubMed]
- De Sales-Millan, A.; Aguirre-Garrido, J.F.; Gonzalez-Cervantes, R.M.; Velazquez-Aragon, J.A. Microbiome-Gut-Mucosal-Immune-Brain Axis and Autism Spectrum Disorder (ASD): A Novel Proposal of the Role of the Gut Microbiome in ASD Aetiology. Behav. Sci. 2023, 13, 548. [Google Scholar] [CrossRef]
- Rose, S.; Bennuri, S.C.; Davis, J.E.; Wynne, R.; Slattery, J.C.; Tippett, M.; Delhey, L.; Melnyk, S.; Kahler, S.G.; MacFabe, D.F.; et al. Butyrate enhances mitochondrial function during oxidative stress in cell lines from boys with autism. Transl. Psychiatry 2018, 8, 42. [Google Scholar] [CrossRef]
- Rose, S.; Niyazov, D.M.; Rossignol, D.A.; Goldenthal, M.; Kahler, S.G.; Frye, R.E. Clinical and Molecular Characteristics of Mitochondrial Dysfunction in Autism Spectrum Disorder. Mol. Diagn. Ther. 2018, 22, 571–593. [Google Scholar] [CrossRef]
- Giulivi, C.; Zhang, Y.F.; Omanska-Klusek, A.; Ross-Inta, C.; Wong, S.; Hertz-Picciotto, I.; Tassone, F.; Pessah, I.N. Mitochondrial dysfunction in autism. JAMA 2010, 304, 2389–2396. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Wong, S.; Hertz-Picciotto, I.; Giulivi, C. Deficits in bioenergetics and impaired immune response in granulocytes from children with autism. Pediatrics 2014, 133, e1405–e1410. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, L.; Papaleo, V.; Porcelli, V.; Scarcia, P.; Gaita, L.; Sacco, R.; Hager, J.; Rousseau, F.; Curatolo, P.; Manzi, B.; et al. Altered calcium homeostasis in autism-spectrum disorders: Evidence from biochemical and genetic studies of the mitochondrial aspartate/glutamate carrier AGC1. Mol. Psychiatry 2010, 15, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Frye, R.E.; Slattery, J.; Wynne, R.; Tippett, M.; Melnyk, S.; James, S.J. Oxidative stress induces mitochondrial dysfunction in a subset of autistic lymphoblastoid cell lines. Transl. Psychiatry 2014, 4, e377. [Google Scholar] [CrossRef]
- Rose, S.; Frye, R.E.; Slattery, J.; Wynne, R.; Tippett, M.; Pavliv, O.; Melnyk, S.; James, S.J. Oxidative stress induces mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines in a well-matched case control cohort. PLoS ONE 2014, 9, e85436. [Google Scholar] [CrossRef] [PubMed]
- Barger, B.D.; Campbell, J.M.; McDonough, J.D. Prevalence and onset of regression within autism spectrum disorders: A meta-analytic review. J. Autism Dev. Disord. 2013, 43, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, W.A.; Osann, K.; Filipek, P.A.; Laulhere, T.; Jarvis, K.; Modahl, C.; Flodman, P.; Spence, M.A. Language and other regression: Assessment and timing. J. Autism Dev. Disord. 2003, 33, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Shulman, C.; DiLavore, P. Regression and word loss in autistic spectrum disorders. J. Child. Psychol. Psychiatry 2004, 45, 936–955. [Google Scholar] [CrossRef]
- Pickles, A.; Simonoff, E.; Conti-Ramsden, G.; Falcaro, M.; Simkin, Z.; Charman, T.; Chandler, S.; Loucas, T.; Baird, G. Loss of language in early development of autism and specific language impairment. J. Child. Psychol. Psychiatry 2009, 50, 843–852. [Google Scholar] [CrossRef]
- Thurm, A.; Powell, E.M.; Neul, J.L.; Wagner, A.; Zwaigenbaum, L. Loss of skills and onset patterns in neurodevelopmental disorders: Understanding the neurobiological mechanisms. Autism Res. 2018, 11, 212–222. [Google Scholar] [CrossRef]
- Singh, K.; Singh, I.N.; Diggins, E.; Connors, S.L.; Karim, M.A.; Lee, D.; Zimmerman, A.W.; Frye, R.E. Developmental regression and mitochondrial function in children with autism. Ann. Clin. Transl. Neurol. 2020, 7, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.F.; Elwell, C.; Johnson, M.H. Mitochondrial Dysfunction in Autism Spectrum Disorders. Autism Open Access 2016, 6, 1000190. [Google Scholar] [CrossRef] [PubMed]
- Gevezova, M.; Minchev, D.; Pacheva, I.; Todorova, T.; Yordanova, R.; Timova, E.; Ivanov, I.; Sarafian, V. Association of NGF and Mitochondrial Respiration with Autism Spectrum Disorder. Int. J. Mol. Sci. 2022, 23, 11917. [Google Scholar] [CrossRef] [PubMed]
- Shoffner, J.; Hyams, L.; Langley, G.N.; Cossette, S.; Mylacraine, L.; Dale, J.; Ollis, L.; Kuoch, S.; Bennett, K.; Aliberti, A.; et al. Fever plus mitochondrial disease could be risk factors for autistic regression. J. Child. Neurol. 2010, 25, 429–434. [Google Scholar] [CrossRef]
- Poling, J.S.; Frye, R.E.; Shoffner, J.; Zimmerman, A.W. Developmental regression and mitochondrial dysfunction in a child with autism. J. Child. Neurol. 2006, 21, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.K.; Geier, D.A.; Sykes, L.K.; Geier, M.R. Relevance of Neuroinflammation and Encephalitis in Autism. Front. Cell Neurosci. 2015, 9, 519. [Google Scholar] [CrossRef] [PubMed]
- Saad, K.; Abdallah, A.M.; Abdel-Rahman, A.A.; Al-Atram, A.A.; Abdel-Raheem, Y.F.; Gad, E.F.; Abo-Elela, M.G.M.; Elserogy, Y.M.; Elhoufey, A.; Nigm, D.A.; et al. Polymorphism of interleukin-1beta and interleukin-1 receptor antagonist genes in children with autism spectrum disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 103, 109999. [Google Scholar] [CrossRef]
- Molloy, C.A.; Morrow, A.L.; Meinzen-Derr, J.; Schleifer, K.; Dienger, K.; Manning-Courtney, P.; Altaye, M.; Wills-Karp, M. Elevated cytokine levels in children with autism spectrum disorder. J. Neuroimmunol. 2006, 172, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Angelidou, A.; Asadi, S.; Alysandratos, K.D.; Karagkouni, A.; Kourembanas, S.; Theoharides, T.C. Perinatal stress, brain inflammation and risk of autism-review and proposal. BMC Pediatr. 2012, 12, 89. [Google Scholar] [CrossRef]
- Conrady, C.D.; Drevets, D.A.; Carr, D.J. Herpes simplex type I (HSV-1) infection of the nervous system: Is an immune response a good thing? J. Neuroimmunol. 2010, 220, 1–9. [Google Scholar] [CrossRef]
- Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Pessah, I.N.; Van de Water, J. Associations of impaired behaviors with elevated plasma chemokines in autism spectrum disorders. J. Neuroimmunol. 2011, 232, 196–199. [Google Scholar] [CrossRef]
- Gomez-Fernandez, A.; de la Torre-Aguilar, M.J.; Gil-Campos, M.; Flores-Rojas, K.; Cruz-Rico, M.D.; Martin-Borreguero, P.; Perez-Navero, J.L. Children with Autism Spectrum Disorder with Regression Exhibit a Different Profile in Plasma Cytokines and Adhesion Molecules Compared to Children without Such Regression. Front. Pediatr. 2018, 6, 264. [Google Scholar] [CrossRef]
- Masi, A.; Glozier, N.; Dale, R.; Guastella, A.J. The Immune System, Cytokines, and Biomarkers in Autism Spectrum Disorder. Neurosci. Bull. 2017, 33, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Mead, J.; Ashwood, P. Evidence supporting an altered immune response in ASD. Immunol. Lett. 2015, 163, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.N.; Miao, H.; Yu, X.Y.; Guo, Y.; Su, W.; Liu, F.; Cao, G.; Zhao, Y.Y. Oxidative stress and inflammation are mediated via aryl hydrocarbon receptor signalling in idiopathic membranous nephropathy. Free Radic. Biol. Med. 2023, 207, 89–106. [Google Scholar] [CrossRef]
- Luo, X.; Xiong, H.; Jiang, Y.; Fan, Y.; Zuo, C.; Chen, D.; Chen, L.; Lin, H.; Gao, J. Macrophage Reprogramming via Targeted ROS Scavenging and COX-2 Downregulation for Alleviating Inflammation. Bioconjug. Chem. 2023, 34, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.J.; Lee, N.; Choi, S.E.; Jeon, J.Y.; Han, S.J.; Kim, D.J.; Kang, Y.; Lee, K.W.; Kim, H.J. Amphiregulin Induces iNOS and COX-2 Expression through NF-kappaB and MAPK Signaling in Hepatic Inflammation. Mediat. Inflamm. 2023, 2023, 2364121. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.F.; Chen, G.W.; Chen, Y.C.; Shen, C.K.; Lu, D.Y.; Yang, L.Y.; Chen, J.H.; Yeh, W.L. Regulatory Effects of Quercetin on M1/M2 Macrophage Polarization and Oxidative/Antioxidative Balance. Nutrients 2021, 14, 67. [Google Scholar] [CrossRef]
- Yao, C.; Narumiya, S. Prostaglandin-cytokine crosstalk in chronic inflammation. Br. J. Pharmacol. 2019, 176, 337–354. [Google Scholar] [CrossRef]
- Kondo, M.; Shibata, T.; Kumagai, T.; Osawa, T.; Shibata, N.; Kobayashi, M.; Sasaki, S.; Iwata, M.; Noguchi, N.; Uchida, K. 15-Deoxy-Delta(12,14)-prostaglandin J(2): The endogenous electrophile that induces neuronal apoptosis. Proc. Natl. Acad. Sci. USA 2002, 99, 7367–7372. [Google Scholar] [CrossRef]
- Chen, J.; Deng, J.C.; Zemans, R.L.; Bahmed, K.; Kosmider, B.; Zhang, M.; Peters-Golden, M.; Goldstein, D.R. Age-induced prostaglandin E(2) impairs mitochondrial fitness and increases mortality to influenza infection. Nat. Commun. 2022, 13, 6759. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.R.; Kwon, C.H.; Park, J.Y.; Kim, Y.K.; Woo, J.S. 15-Deoxy-Delta(12,14)-prostaglandin J(2) induces mitochondrial-dependent apoptosis through inhibition of PKA/NF-kappaB in renal proximal epithelial cells. Toxicology 2009, 258, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, M.; Sanchez-Rodriguez, J.; Santos, A.; Perez-Castillo, A. 15-deoxy-Delta-12,14-prostaglandin J2 induces programmed cell death of breast cancer cells by a pleiotropic mechanism. Carcinogenesis 2005, 26, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Kalpage, H.A.; Bazylianska, V.; Recanati, M.A.; Fite, A.; Liu, J.; Wan, J.; Mantena, N.; Malek, M.H.; Podgorski, I.; Heath, E.I.; et al. Tissue-specific regulation of cytochrome c by post-translational modifications: Respiration, the mitochondrial membrane potential, ROS, and apoptosis. FASEB J. 2019, 33, 1540–1553. [Google Scholar] [CrossRef] [PubMed]
- Villa, M.; Sanin, D.E.; Apostolova, P.; Corrado, M.; Kabat, A.M.; Cristinzio, C.; Regina, A.; Carrizo, G.E.; Rana, N.; Stanczak, M.A.; et al. Prostaglandin E (2) controls the metabolic adaptation of T cells to the intestinal microenvironment. bioRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- Plewes, M.R.; Przygrodzka, E.; Monaco, C.F.; Snider, A.P.; Keane, J.A.; Burns, P.D.; Wood, J.R.; Cupp, A.S.; Davis, J.S. Prostaglandin F2alpha regulates mitochondrial dynamics and mitophagy in the bovine corpus luteum. Life Sci. Alliance 2023, 6, e202301968. [Google Scholar] [CrossRef]
- Bonneh-Barkay, D.; Wang, G.; Starkey, A.; Hamilton, R.L.; Wiley, C.A. In vivo CHI3L1 (YKL-40) expression in astrocytes in acute and chronic neurological diseases. J. Neuroinflamm. 2010, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Ferretti, M.T. Function and Dysfunction of Microglia during Brain Development: Consequences for Synapses and Neural Circuits. Front. Synaptic Neurosci. 2017, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Purves-Tyson, T.D.; Robinson, K.; Brown, A.M.; Boerrigter, D.; Cai, H.Q.; Weissleder, C.; Owens, S.J.; Rothmond, D.A.; Shannon Weickert, C. Increased Macrophages and C1qA, C3, C4 Transcripts in the Midbrain of People with Schizophrenia. Front. Immunol. 2020, 11, 2002. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, C.; Zhang, Y.; Sun, R.; Wang, H.; Li, G.; Zhang, J. Elevated CHI3L1 and OPN levels in patients with anti-N-methyl-d-aspartate receptor encephalitis. J. Neuroimmunol. 2019, 334, 577005. [Google Scholar] [CrossRef]
- Lee, D.H.; Han, J.H.; Lee, Y.S.; Jung, Y.S.; Roh, Y.S.; Yun, J.S.; Han, S.B.; Hong, J.T. Chitinase-3-like-1 deficiency attenuates ethanol-induced liver injury by inhibition of sterol regulatory element binding protein 1-dependent triglyceride synthesis. Metabolism 2019, 95, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, E.; Sadanaga, T.; Nanni, L.; Wang, S.; Mizoguchi, A. Recently updated role of Chitinase 3-like 1 on various cell types as a major infuencer of chronic inflammation. Cells 2024, 13, 678. [Google Scholar] [CrossRef] [PubMed]
- Kovacheva, E.; Gevezova, M.; Maes, M.; Sarafian, V. The mast cells—Cytokines axis in Autism Spectrum Disorder. Neuropharmacology 2024, 249, 109890. [Google Scholar] [CrossRef] [PubMed]
- Vancolen, S.; Ayash, T.; Allard, M.J.; Sebire, G. Sex-Specific Dysconnective Brain Injuries and Neuropsychiatric Conditions such as Autism Spectrum Disorder Caused by Group B Streptococcus-Induced Chorioamnionitis. Int. J. Mol. Sci. 2023, 24, 14090. [Google Scholar] [CrossRef] [PubMed]
- Borreca, A.; Mantovani, C.; Desiato, G.; Corradini, I.; Filipello, F.; Elia, C.A.; D’Autilia, F.; Santamaria, G.; Garlanda, C.; Morini, R.; et al. Loss of interleukin 1 signaling causes impairment of microglia- mediated synapse elimination and autistic-like behaviour in mice. Brain Behav. Immun. 2024, 117, 493–509. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.F.; Nadeem, A.; Ansari, M.A.; Bakheet, S.A.; Al-Ayadhi, L.Y.; Attia, S.M. Upregulation of IL-9 and JAK-STAT signaling pathway in children with autism. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79 Pt B, 472–480. [Google Scholar] [CrossRef]
- Tomar, S.; Ganesan, V.; Sharma, A.; Zeng, C.; Waggoner, L.; Smith, A.; Kim, C.H.; Licona-Limon, P.; Reinhardt, R.L.; Flavell, R.A.; et al. IL-4-BATF signaling directly modulates IL-9 producing mucosal mast cell (MMC9) function in experimental food allergy. J. Allergy Clin. Immunol. 2021, 147, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Nagura, N.; Uchida, K.; Kenmoku, T.; Inoue, G.; Nakawaki, M.; Miyagi, M.; Takaso, M. IL-1beta mediates NGF and COX-2 expression through transforming growth factor-activating kinase 1 in subacromial bursa cells derived from rotator cuff tear patients. J. Orthop. Sci. 2019, 24, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.C.; Shen, H.H.; Wang, C.J.; Zhang, X.Y.; Wu, J.N.; Lu, H.C.; Qiu, X.M.; Ding, J.Y.; Tan, X.F.; Liu, L.B.; et al. A positive COX-2/IL-1beta loop promotes decidualization by upregulating CD82. Reproduction 2021, 162, 227–236. [Google Scholar] [CrossRef]
- Nemeth, D.P.; Quan, N. Modulation of Neural Networks by Interleukin-1. Brain Plast. 2021, 7, 17–32. [Google Scholar] [CrossRef]
- Liao, Y.C.; Lim, Y.S.; Chu, P.W.; Chen, S.K. Inflammatory Milieu Induces Mitochondrial Alterations and Neuronal Activations in Hypothalamic POMC Neurons in a Time-Dependent Manner. Mol. Neurobiol. 2023, 60, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Jyonouchi, H.; Geng, L.; Rose, S.; Bennuri, S.C.; Frye, R.E. Variations in Mitochondrial Respiration Differ in IL-1ss/IL-10 Ratio Based Subgroups in Autism Spectrum Disorders. Front. Psychiatry 2019, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; McCarty, P.J.; Werner, B.A.; Rose, S.; Scheck, A.C. Bioenergetic signatures of neurodevelopmental regression. Front. Physiol. 2024, 15, 1306038. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Li, H.; Li, J.; Luo, X.; Hao, Y. Microglia: Synaptic modulator in autism spectrum disorder. Front. Psychiatry 2022, 13, 958661. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wang, Z. The Impact of Microglia on Neurodevelopment and Brain Function in Autism. Biomedicines 2024, 12, 210. [Google Scholar] [CrossRef] [PubMed]
- Memis, I.; Mittal, R.; Furar, E.; White, I.; Eshraghi, R.S.; Mittal, J.; Eshraghi, A.A. Altered Blood Brain Barrier Permeability and Oxidative Stress in Cntnap2 Knockout Rat Model. J. Clin. Med. 2022, 11, 2725. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.I.; Kern, J.K. Evidence of microglial activation in autism and its possible role in brain underconnectivity. Neuron Glia Biol. 2011, 7, 205–213. [Google Scholar] [CrossRef]
- Fattorusso, A.; Di Genova, L.; Dell’Isola, G.B.; Mencaroni, E.; Esposito, S. Autism Spectrum Disorders and the Gut Microbiota. Nutrients 2019, 11, 521. [Google Scholar] [CrossRef]
- Hossain, M.M.; Khan, N.; Sultana, A.; Ma, P.; McKyer, E.L.J.; Ahmed, H.U.; Purohit, N. Prevalence of comorbid psychiatric disorders among people with autism spectrum disorder: An umbrella review of systematic reviews and meta-analyses. Psychiatry Res. 2020, 287, 112922. [Google Scholar] [CrossRef]
- Al-Beltagi, M. Autism medical comorbidities. World J. Clin. Pediatr. 2021, 10, 15–28. [Google Scholar] [CrossRef]
- Schirinzi, T.; Salvatori, I.; Zenuni, H.; Grillo, P.; Valle, C.; Martella, G.; Mercuri, N.B.; Ferri, A. Pattern of Mitochondrial Respiration in Peripheral Blood Cells of Patients with Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 10863. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Nankova, B.; Bhattacharyya, S.; Rose, S.; Bennuri, S.C.; MacFabe, D.F. Modulation of Immunological Pathways in Autistic and Neurotypical Lymphoblastoid Cell Lines by the Enteric Microbiome Metabolite Propionic Acid. Front. Immunol. 2017, 8, 1670. [Google Scholar] [CrossRef] [PubMed]
- Voinsky, I.; Bennuri, S.C.; Svigals, J.; Frye, R.E.; Rose, S.; Gurwitz, D. Peripheral Blood Mononuclear Cell Oxytocin and Vasopressin Receptor Expression Positively Correlates with Social and Behavioral Function in Children with Autism. Sci. Rep. 2019, 9, 13443. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Bennuri, S.C.; Wynne, R.; Melnyk, S.; James, S.J.; Frye, R.E. Mitochondrial and redox abnormalities in autism lymphoblastoid cells: A sibling control study. FASEB J. 2017, 31, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Bennuri, S.C.; Murray, K.F.; Buie, T.; Winter, H.; Frye, R.E. Mitochondrial dysfunction in the gastrointestinal mucosa of children with autism: A blinded case-control study. PLoS ONE 2017, 12, e0186377. [Google Scholar] [CrossRef] [PubMed]
- Pfleger, J.; He, M.; Abdellatif, M. Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell Death Dis. 2015, 6, e1835. [Google Scholar] [CrossRef] [PubMed]
- Desler, C.; Hansen, T.L.; Frederiksen, J.B.; Marcker, M.L.; Singh, K.K.; Juel Rasmussen, L. Is There a Link between Mitochondrial Reserve Respiratory Capacity and Aging? J. Aging Res. 2012, 2012, 192503. [Google Scholar] [CrossRef] [PubMed]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef]
- Mithal, D.S.; Chandel, N.S. Mitochondrial Dysfunction in Fragile-X Syndrome: Plugging the Leak May Save the Ship. Mol. Cell 2020, 80, 381–383. [Google Scholar] [CrossRef]
- Sriwichaiin, S.; Apaijai, N.; Phrommintikul, A.; Jaiwongkam, T.; Kerdphoo, S.; Pratchayasakul, W.; Thongmung, N.; Mahantassanapong, U.; Vathesatogkit, P.; Kitiyakara, C.; et al. Increased Efficiency of Mitochondrial Coupling with a Reduction in Other Mitochondrial Respiratory Parameters in Peripheral Blood Mononuclear Cells Is Observed in Older Adults. J. Gerontol. A Biol. Sci. Med. Sci. 2023, 78, 384–391. [Google Scholar] [CrossRef]
- Harper, C.; Gopalan, V.; Goh, J. Exercise rescues mitochondrial coupling in aged skeletal muscle: A comparison of different modalities in preventing sarcopenia. J. Transl. Med. 2021, 19, 71. [Google Scholar] [CrossRef] [PubMed]
- Soto-Heredero, G.; Gomez de Las Heras, M.M.; Gabande-Rodriguez, E.; Oller, J.; Mittelbrunn, M. Glycolysis—A key player in the inflammatory response. FEBS J. 2020, 287, 3350–3369. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, E.M.; Gonzalez-Cotto, M.; Baseler, W.A.; Davies, L.C.; Ghesquiere, B.; Maio, N.; Rice, C.M.; Rouault, T.A.; Cassel, T.; Higashi, R.M.; et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun. 2020, 11, 698. [Google Scholar] [CrossRef] [PubMed]
- Hughes, H.K.; Moreno, R.J.; Ashwood, P. Innate immune dysfunction and neuroinflammation in autism spectrum disorder (ASD). Brain Behav. Immun. 2023, 108, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, B.; Wu, C.; Wang, J.; Sun, M. Autism Spectrum Disorder: Neurodevelopmental Risk Factors, Biological Mechanism, and Precision Therapy. Int. J. Mol. Sci. 2023, 24, 1819. [Google Scholar] [CrossRef] [PubMed]
- Gzielo, K.; Nikiforuk, A. Astroglia in Autism Spectrum Disorder. Int. J. Mol. Sci. 2021, 22, 11544. [Google Scholar] [CrossRef]
- Kealy, J.; Greene, C.; Campbell, M. Blood-brain barrier regulation in psychiatric disorders. Neurosci. Lett. 2020, 726, 133664. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.T.; Bestard-Lorigados, I.; Crawford, D.A. Autism-related behaviors in the cyclooxygenase-2-deficient mouse model. Genes Brain Behav. 2019, 18, e12506. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.; Rozemuller, A.J.; Janssen, I.; De Groot, C.J.; Veerhuis, R.; Eikelenboom, P. Cyclooxygenase expression in microglia and neurons in Alzheimer’s disease and control brain. Acta Neuropathol. 2001, 101, 2–8. [Google Scholar] [CrossRef]
- Kordulewska, N.K.; Kostyra, E.; Chwala, B.; Moszynska, M.; Cieslinska, A.; Fiedorowicz, E.; Jarmolowska, B. A novel concept of immunological and allergy interactions in autism spectrum disorders: Molecular, anti-inflammatory effect of osthole. Int. Immunopharmacol. 2019, 72, 1–11. [Google Scholar] [CrossRef]
- Cillero-Pastor, B.; Carames, B.; Lires-Dean, M.; Vaamonde-Garcia, C.; Blanco, F.J.; Lopez-Armada, M.J. Mitochondrial dysfunction activates cyclooxygenase 2 expression in cultured normal human chondrocytes. Arthritis Rheum. 2008, 58, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, R.B.; Emery, J.G.; Connor, J.R.; Dodds, R.; Lysko, P.G.; Rosenberg, M. Induction and expression of human cartilage glycoprotein 39 in rheumatoid inflammatory and peripheral blood monocyte-derived macrophages. Exp. Cell Res. 1997, 237, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Muszynski, P.; Groblewska, M.; Kulczynska-Przybik, A.; Kulakowska, A.; Mroczko, B. YKL-40 as a Potential Biomarker and a Possible Target in Therapeutic Strategies of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Garbett, K.; Ebert, P.J.; Mitchell, A.; Lintas, C.; Manzi, B.; Mirnics, K.; Persico, A.M. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol. Dis. 2008, 30, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Samet Can Demirci, S.B. Ahmet Özaslan Investigating the Relationship of Serum Cd163, Ykl40 and Vilip-1 Levels with Autism Severity and Language-Cognitive Development in Preschool Children with Autism. Preprints 2023. [Google Scholar] [CrossRef]
- Bringer, M.A.; Billard, E.; Glasser, A.L.; Colombel, J.F.; Darfeuille-Michaud, A. Replication of Crohn’s disease-associated AIEC within macrophages is dependent on TNF-alpha secretion. Lab. Investig. 2012, 92, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Bojesen, S.E.; Johansen, J.S.; Nordestgaard, B.G. Plasma YKL-40 levels in healthy subjects from the general population. Clin. Chim. Acta 2011, 412, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Shao, R. YKL-40 acts as an angiogenic factor to promote tumor angiogenesis. Front. Physiol. 2013, 4, 122. [Google Scholar] [CrossRef] [PubMed]
- Dichev, V.; Mehterov, N.; Kazakova, M.; Karalilova, R.; Batalov, A.; Sarafian, V. The lncRNAs/miR-30e/CHI3L1 Axis Is Dysregulated in Systemic Sclerosis. Biomedicines 2022, 10, 496. [Google Scholar] [CrossRef]
- Jafari-Nakhjavani, M.R.; Ghorbanihaghjo, A.; Bagherzadeh-Nobari, B.; Malek-Mahdavi, A.; Rashtchizadeh, N. Serum YKL-40 levels and disease characteristics in patients with rheumatoid arthritis. Caspian J. Intern. Med. 2019, 10, 92–97. [Google Scholar] [CrossRef]
- Baldacci, F.; Lista, S.; Palermo, G.; Giorgi, F.S.; Vergallo, A.; Hampel, H. The neuroinflammatory biomarker YKL-40 for neurodegenerative diseases: Advances in development. Expert. Rev. Proteom. 2019, 16, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Than, U.T.T.; Nguyen, L.T.; Nguyen, P.H.; Nguyen, X.H.; Trinh, D.P.; Hoang, D.H.; Nguyen, P.A.T.; Dang, V.D. Inflammatory mediators drive neuroinflammation in autism spectrum disorder and cerebral palsy. Sci. Rep. 2023, 13, 22587. [Google Scholar] [CrossRef] [PubMed]
- Erbescu, A.; Papuc, S.M.; Budisteanu, M.; Arghir, A.; Neagu, M. Re-emerging concepts of immune dysregulation in autism spectrum disorders. Front. Psychiatry 2022, 13, 1006612. [Google Scholar] [CrossRef] [PubMed]
- Decker, M.L.; Grobusch, M.P.; Ritz, N. Influence of Age and Other Factors on Cytokine Expression Profiles in Healthy Children-A Systematic Review. Front. Pediatr. 2017, 5, 255. [Google Scholar] [CrossRef] [PubMed]
- Graf, W.D.; Marin-Garcia, J.; Gao, H.G.; Pizzo, S.; Naviaux, R.K.; Markusic, D.; Barshop, B.A.; Courchesne, E.; Haas, R.H. Autism associated with the mitochondrial DNA G8363A transfer RNA(Lys) mutation. J. Child. Neurol. 2000, 15, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Delhey, L.; Kilinc, E.N.; Yin, L.; Slattery, J.; Tippett, M.; Wynne, R.; Rose, S.; Kahler, S.; Damle, S.; Legido, A.; et al. Bioenergetic variation is related to autism symptomatology. Metab. Brain Dis. 2017, 32, 2021–2031. [Google Scholar] [CrossRef] [PubMed]
- Hyder, F.; Rothman, D.L.; Bennett, M.R. Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. USA 2013, 110, 3549–3554. [Google Scholar] [CrossRef] [PubMed]
- Curtin, P.; Austin, C.; Curtin, A.; Gennings, C.; Arora, M.; Tammimies, K.; Willfors, C.; Berggren, S.; Siper, P.; Rai, D.; et al. Dynamical features in fetal and postnatal zinc-copper metabolic cycles predict the emergence of autism spectrum disorder. Sci. Adv. 2018, 4, eaat1293. [Google Scholar] [CrossRef]
- Frye, R.E.; Cakir, J.; Rose, S.; Delhey, L.; Bennuri, S.C.; Tippett, M.; Palmer, R.F.; Austin, C.; Curtin, P.; Arora, M. Early life metal exposure dysregulates cellular bioenergetics in children with regressive autism spectrum disorder. Transl. Psychiatry 2020, 10, 223. [Google Scholar] [CrossRef]
- Murray, K.N.; Parry-Jones, A.R.; Allan, S.M. Interleukin-1 and acute brain injury. Front. Cell. Neurosci. 2015, 9, 18. [Google Scholar] [CrossRef]
- Lord, C.; Luyster, R.J.; Gotham, K.; Guthrie, W. Autism Diagnostic Observation Schedule, Second Edition (ADOS-2) Manual (Part II): Toddler Module; Western Psychological Services: Torrance, CA, USA, 2012. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | ASD R(+) | ASD R(-) | TDC |
|---|---|---|---|
| Number of patients | 21 | 35 | 12 |
| Age (mean) | 3.05 ± 1.4 | 4.46 ± 2.5 | 7.63 ± 3.28 |
| Sex M/F | 19/2 | 28/7 | 8/4 |
| Normal intelligence | IQ ≥ 70 | ||
| IQ/DQ | 50.06 ± 10.73 | 53.6 ± 14.05 | not measured. |
| ADOS total points | 27.53 ± 4.41 | 24.59 ± 5.59 | not measured |
| Communication pts (ADOS) | 6.23 ± 1.78 | 5.62 ± 1.88 | not measured |
| Social Interaction pts (ADOS) | 14.92 ± 3.45 | 13.48 ± 3.32 | not measured |
| Repetitive Behavior pts (ADOS) | 6.38 ± 1.27 | 5.74 ± 1.87 | not measured. |
| Parameters | r | |
|---|---|---|
| Regression | Spare respiratory capacity | 0.499 |
| Spare respiratory capacity (%) | 0.469 | |
| Coupling efficiency | −0.343 | |
| Proton leak | 0.535 | |
| IL-1β | −0.326 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gevezova, M.; Ivanov, Z.; Pacheva, I.; Timova, E.; Kazakova, M.; Kovacheva, E.; Ivanov, I.; Sarafian, V. Bioenergetic and Inflammatory Alterations in Regressed and Non-Regressed Patients with Autism Spectrum Disorder. Int. J. Mol. Sci. 2024, 25, 8211. https://doi.org/10.3390/ijms25158211
Gevezova M, Ivanov Z, Pacheva I, Timova E, Kazakova M, Kovacheva E, Ivanov I, Sarafian V. Bioenergetic and Inflammatory Alterations in Regressed and Non-Regressed Patients with Autism Spectrum Disorder. International Journal of Molecular Sciences. 2024; 25(15):8211. https://doi.org/10.3390/ijms25158211
Chicago/Turabian StyleGevezova, Maria, Zdravko Ivanov, Iliana Pacheva, Elena Timova, Maria Kazakova, Eleonora Kovacheva, Ivan Ivanov, and Victoria Sarafian. 2024. "Bioenergetic and Inflammatory Alterations in Regressed and Non-Regressed Patients with Autism Spectrum Disorder" International Journal of Molecular Sciences 25, no. 15: 8211. https://doi.org/10.3390/ijms25158211