
Empagliflozin Prevent High-Glucose Stimulation Inducing Apoptosis and Mitochondria Fragmentation in H9C2 Cells through the Calcium-Dependent Activation Extracellular Signal-Regulated Kinase 1/2 Pathway
 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Empagliflozin Ameliorates the HG-Caused Decrease in H9C2 Viability
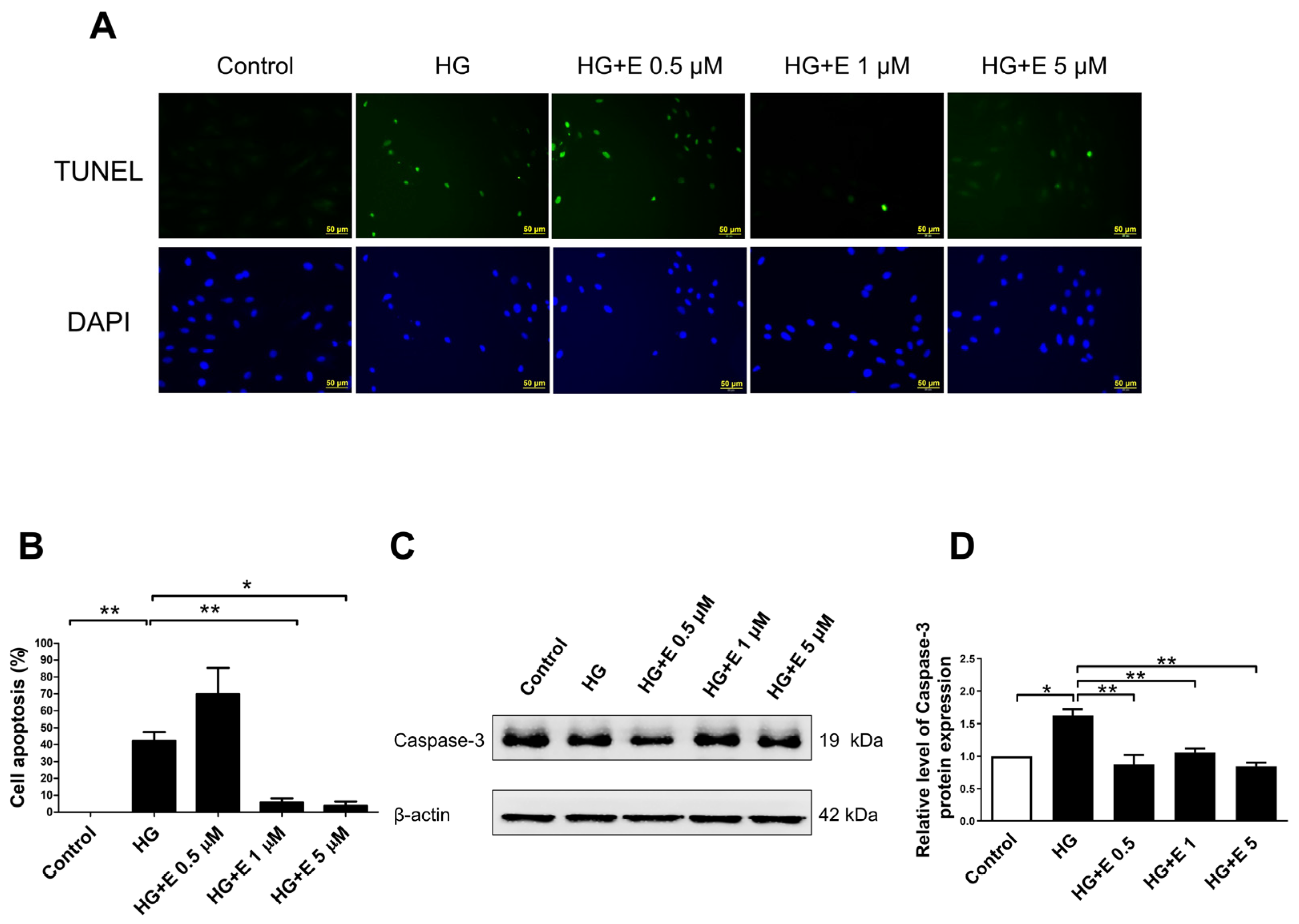
2.2. Empagliflozin Rescues HG-Induced Apoptosis in H9C2 Cells
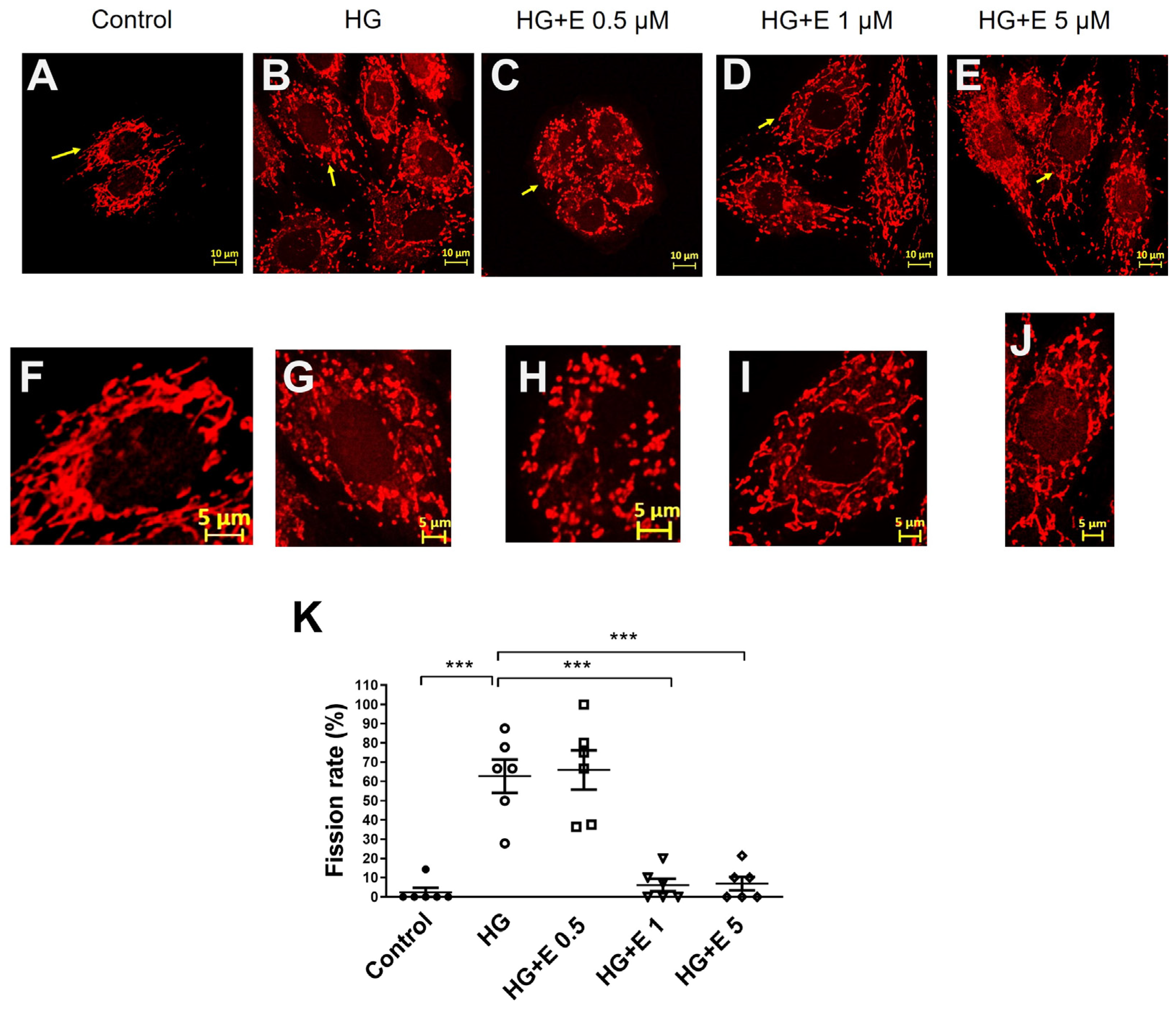
2.3. Empagliflozin Improves the Mitochondrial Fragmentation of HG-Treated H9C2 Cells
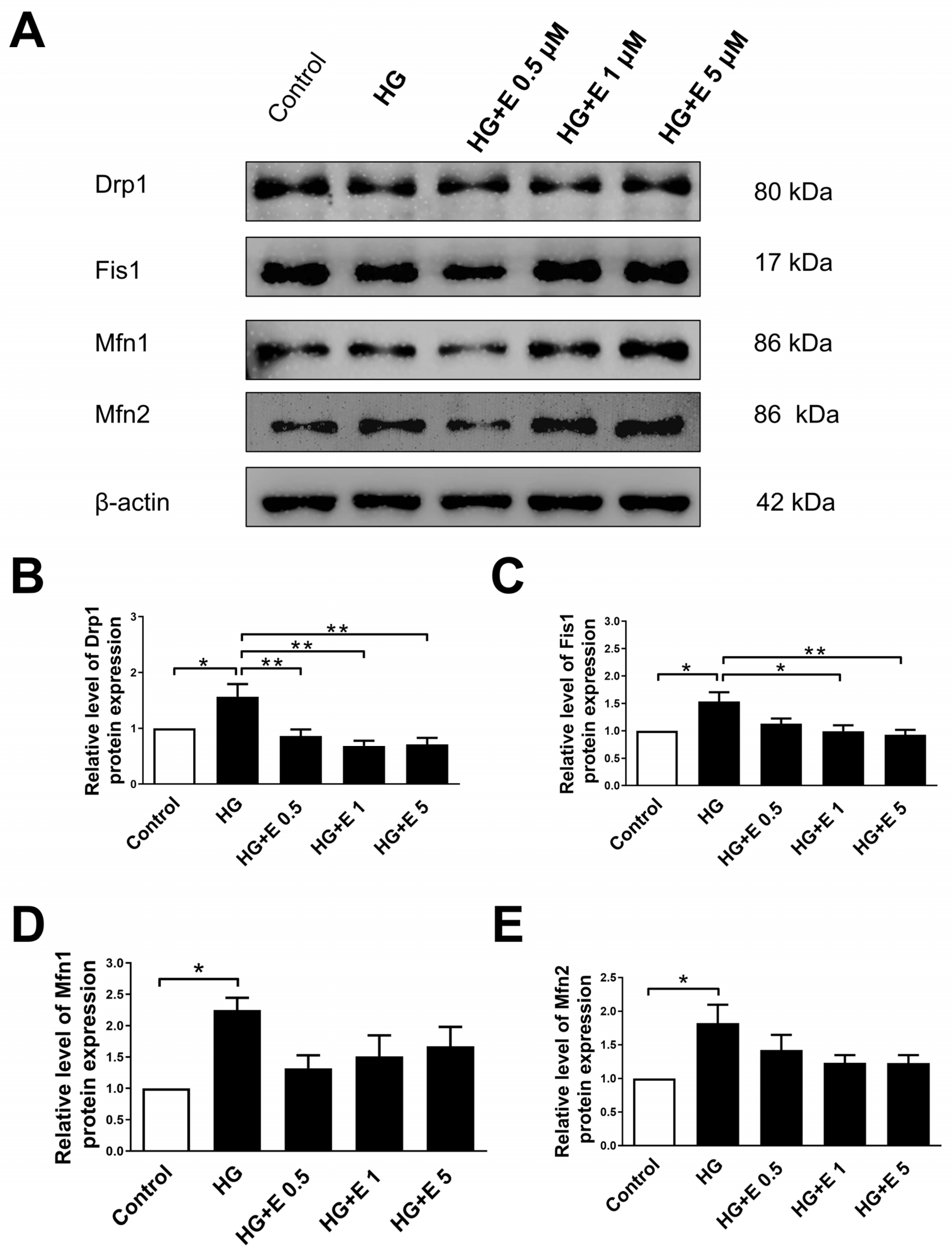
2.4. Empagliflozin Reduces the Expression Levels of Mitochondrial Fission Proteins of H9C2 Cells Incubated in HG Condition
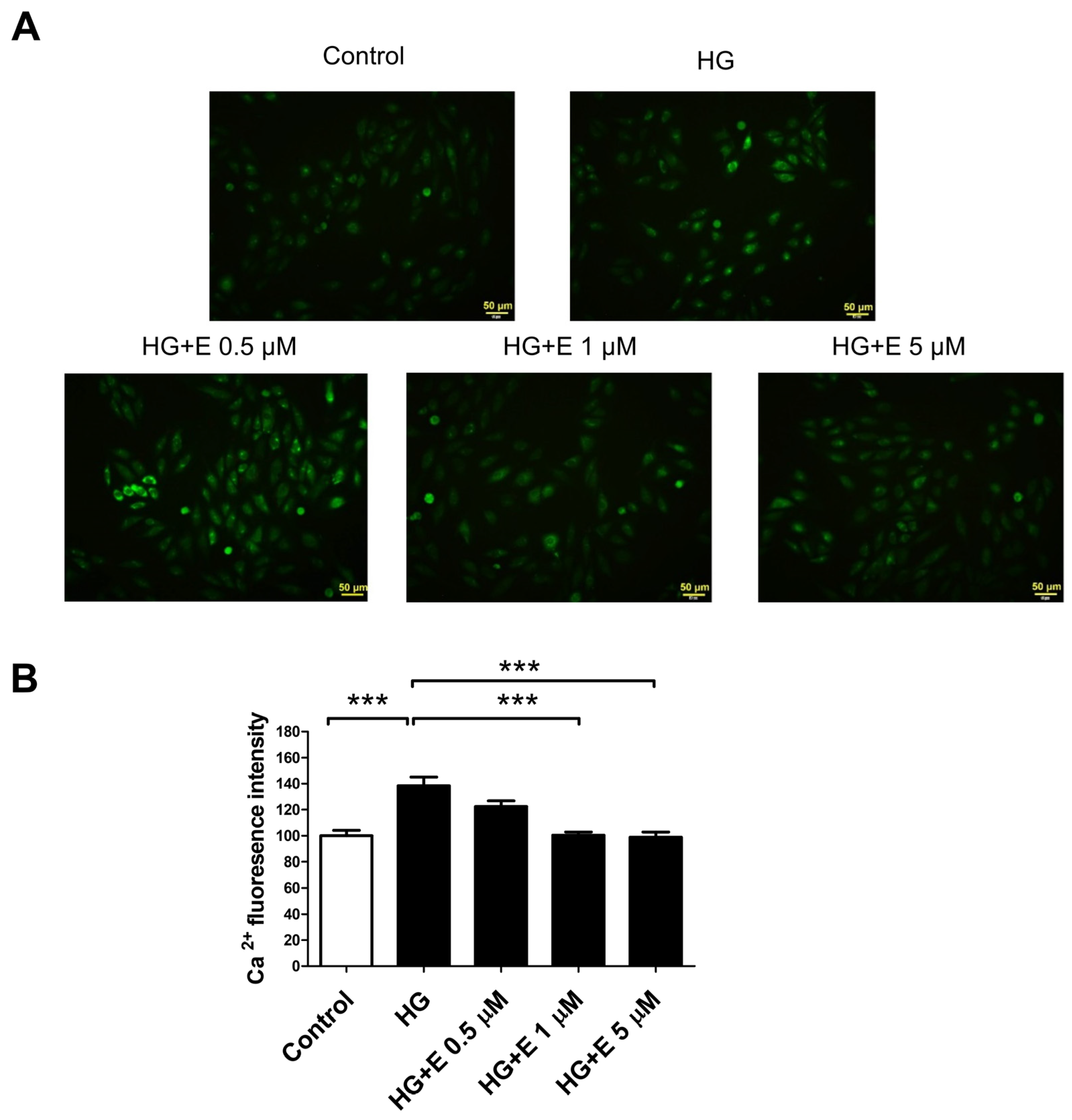
2.5. Empagliflozin Reduces HG-Induced Intracellular Calcium Overload Accumulation in H9C2 Cells
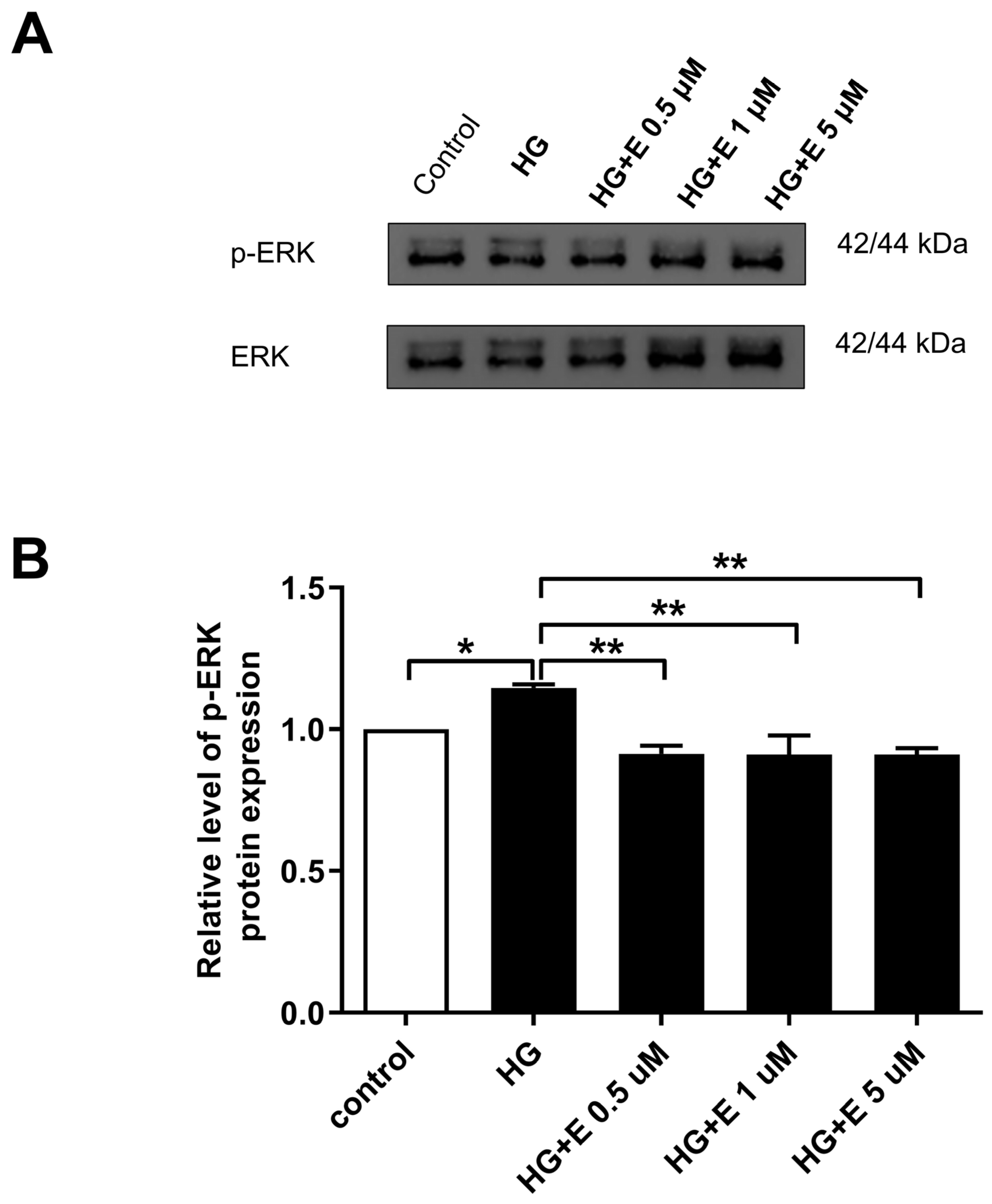
2.6. Impact of Empagliflozin on the ERK1/2 Activation, Which Leads to Mitochondrial Fragmentation in HG-Treated H9C2 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Chemicals
4.2. Cell Viability Assay
4.3. Western Blot Analysis
4.4. Mitochondrial Fragmentation Index
4.5. Cellular Apoptotic Assay
4.6. Calcium Live Cell Imaging
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Demir, S.; Nawroth, P.P.; Herzig, S.; Ekim Ustunel, B. Emerging Targets in Type 2 Diabetes and Diabetic Complications. Adv. Sci. 2021, 8, e2100275. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B.; et al. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Stefansson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Buse, J.B.; Wexler, D.J.; Tsapas, A.; Rossing, P.; Mingrone, G.; Mathieu, C.; D’Alessio, D.A.; Davies, M.J. 2019 update to: Management of hyperglycaemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2020, 63, 221–228. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes (KDIGO) Diabetes Work Group. KDIGO 2020 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int. 2020, 98, S1–S115. [Google Scholar] [CrossRef]
- Lee, W.C.; Chau, Y.Y.; Ng, H.Y.; Chen, C.H.; Wang, P.W.; Liou, C.W.; Lin, T.K.; Chen, J.B. Empagliflozin Protects HK-2 Cells from High Glucose-Mediated Injuries via a Mitochondrial Mechanism. Cells 2019, 8, 1085. [Google Scholar] [CrossRef]
- Liu, X.; Xu, C.; Xu, L.; Li, X.; Sun, H.; Xue, M.; Li, T.; Yu, X.; Sun, B.; Chen, L. Empagliflozin improves diabetic renal tubular injury by alleviating mitochondrial fission via AMPK/SP1/PGAM5 pathway. Metabolism 2020, 111, 154334. [Google Scholar] [CrossRef] [PubMed]
- Margonato, D.; Galati, G.; Mazzetti, S.; Cannistraci, R.; Perseghin, G.; Margonato, A.; Mortara, A. Renal protection: A leading mechanism for cardiovascular benefit in patients treated with SGLT2 inhibitors. Heart Fail. Rev. 2021, 26, 337–345. [Google Scholar] [CrossRef]
- Scheen, A.J. Effect of SGLT2 Inhibitors on the Sympathetic Nervous System and Blood Pressure. Curr. Cardiol. Rep. 2019, 21, 70. [Google Scholar] [CrossRef] [PubMed]
- Zelniker, T.A.; Braunwald, E. Mechanisms of Cardiorenal Effects of Sodium-Glucose Cotransporter 2 Inhibitors: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 422–434. [Google Scholar] [CrossRef]
- Cowie, M.R.; Fisher, M. SGLT2 inhibitors: Mechanisms of cardiovascular benefit beyond glycaemic control. Nat. Rev. Cardiol. 2020, 17, 761–772. [Google Scholar] [CrossRef]
- Verma, S.; McMurray, J.J.V. SGLT2 inhibitors and mechanisms of cardiovascular benefit: A state-of-the-art review. Diabetologia 2018, 61, 2108–2117. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Jhun, B.S.; Yoon, Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxid. Redox Signal. 2011, 14, 425–437. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, S.H.; Kang, J.M.; Heo, J.H.; Kim, D.J.; Park, S.H.; Sung, M.; Kim, J.; Oh, J.; Yang, D.H.; et al. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am. J. Physiol. Renal Physiol. 2019, 317, F767–F780. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, X.; Liu, H.; Chen, Y.; Li, P.; Liu, L.; Li, J.; Ren, Y.; Huang, J.; Xiong, E.; et al. Empagliflozin Ameliorates Diabetic Cardiomyopathy via Attenuating Oxidative Stress and Improving Mitochondrial Function. Oxid. Med. Cell Longev. 2022, 2022, 1122494. [Google Scholar] [CrossRef]
- Chung, C.C.; Lin, Y.K.; Chen, Y.C.; Kao, Y.H.; Yeh, Y.H.; Trang, N.N.; Chen, Y.J. Empagliflozin suppressed cardiac fibrogenesis through sodium-hydrogen exchanger inhibition and modulation of the calcium homeostasis. Cardiovasc. Diabetol. 2023, 22, 27. [Google Scholar] [CrossRef]
- Agell, N.; Bachs, O.; Rocamora, N.; Villalonga, P. Modulation of the Ras/Raf/MEK/ERK pathway by Ca2+, and calmodulin. Cell Signal. 2002, 14, 649–654. [Google Scholar] [CrossRef]
- Abdelhamid, A.M.; Saber, S.; Youssef, M.E.; Gaafar, A.G.A.; Eissa, H.; Abd-Eldayem, M.A.; Alqarni, M.; Batiha, G.E.; Obaidullah, A.J.; Shahien, M.A.; et al. Empagliflozin adjunct with metformin for the inhibition of hepatocellular carcinoma progression: Emerging approach for new application. Biomed. Pharmacother. 2022, 145, 112455. [Google Scholar] [CrossRef] [PubMed]
- Heimke, M.; Lenz, F.; Rickert, U.; Lucius, R.; Cossais, F. Anti-Inflammatory Properties of the SGLT2 Inhibitor Empagliflozin in Activated Primary Microglia. Cells 2022, 11, 3107. [Google Scholar] [CrossRef]
- Das, N.A.; Carpenter, A.J.; Belenchia, A.; Aroor, A.R.; Noda, M.; Siebenlist, U.; Chandrasekar, B.; DeMarco, V.G. Empagliflozin reduces high glucose-induced oxidative stress and miR-21-dependent TRAF3IP2 induction and RECK suppression, and inhibits human renal proximal tubular epithelial cell migration and epithelial-to-mesenchymal transition. Cell Signal. 2020, 68, 109506. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Flynn, E.R.; do Carmo, J.M.; Wang, Z.; da Silva, A.A.; Mouton, A.J.; Omoto, A.C.M.; Hall, M.E.; Hall, J.E. Direct Cardiac Actions of Sodium-Glucose Cotransporter 2 Inhibition Improve Mitochondrial Function and Attenuate Oxidative Stress in Pressure Overload-Induced Heart Failure. Front. Cardiovasc. Med. 2022, 9, 859253. [Google Scholar] [CrossRef]
- Koizumi, T.; Watanabe, M.; Yokota, T.; Tsuda, M.; Handa, H.; Koya, J.; Nishino, K.; Tatsuta, D.; Natsui, H.; Kadosaka, T.; et al. Empagliflozin suppresses mitochondrial reactive oxygen species generation and mitigates the inducibility of atrial fibrillation in diabetic rats. Front. Cardiovasc. Med. 2023, 10, 1005408. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Madreiter-Sokolowski, C.T.; Mangmool, S.; Parichatikanond, W. High Glucose-Induced Cardiomyocyte Damage Involves Interplay between Endothelin ET-1/ET(A)/ET(B) Receptor and mTOR Pathway. Int. J. Mol. Sci. 2022, 23, 13816. [Google Scholar] [CrossRef]
- Yang, M.; Lin, Y.; Wang, Y.; Wang, Y. High-glucose induces cardiac myocytes apoptosis through Foxo1/GRK2 signaling pathway. Biochem. Biophys. Res. Commun. 2019, 513, 154–158. [Google Scholar] [CrossRef]
- Kim, D.; Sankaramoorthy, A.; Roy, S. Downregulation of Drp1 and Fis1 Inhibits Mitochondrial Fission and Prevents High Glucose-Induced Apoptosis in Retinal Endothelial Cells. Cells 2020, 9, 1662. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Zou, R.; Shi, W.; Qiu, J.; Zhou, N.; Du, N.; Zhou, H.; Chen, X.; Ma, L. Empagliflozin attenuates cardiac microvascular ischemia/reperfusion injury through improving mitochondrial homeostasis. Cardiovasc. Diabetol. 2022, 21, 106. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, M.; Kuno, A.; Yano, T.; Miki, T.; Oshima, H.; Sato, T.; Nakata, K.; Kimura, Y.; Tanno, M.; Miura, T. Empagliflozin normalizes the size and number of mitochondria and prevents reduction in mitochondrial size after myocardial infarction in diabetic hearts. Physiol. Rep. 2018, 6, e13741. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xiang, H.; Zhao, S.; Sang, H.; Lv, F.; Chen, R.; Shu, Z.; Chen, A.F.; Chen, S.; Lu, H. Vildagliptin improves high glucose-induced endothelial mitochondrial dysfunction via inhibiting mitochondrial fission. J. Cell Mol. Med. 2019, 23, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Tahrir, F.G.; Langford, D.; Amini, S.; Mohseni Ahooyi, T.; Khalili, K. Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J. Cell Physiol. 2019, 234, 8122–8133. [Google Scholar] [CrossRef]
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial Quality Control as a Therapeutic Target. Pharmacol. Rev. 2016, 68, 20–48. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; He, Z.; Huang, H.; Zhuang, H.; Liu, H.; Liu, X.; Yang, S.; He, P.; Yang, H.; Feng, D. Mitochondrial Quality Control in Cardiomyocytes: A Critical Role in the Progression of Cardiovascular Diseases. Front. Physiol. 2020, 11, 252. [Google Scholar] [CrossRef] [PubMed]
- Smogorzewski, M.; Galfayan, V.; Massry, S.G. High glucose concentration causes a rise in [Ca2+]i of cardiac myocytes. Kidney Int. 1998, 53, 1237–1243. [Google Scholar] [CrossRef]
- Lee, T.I.; Chen, Y.C.; Lin, Y.K.; Chung, C.C.; Lu, Y.Y.; Kao, Y.H.; Chen, Y.J. Empagliflozin Attenuates Myocardial Sodium and Calcium Dysregulation and Reverses Cardiac Remodeling in Streptozotocin-Induced Diabetic Rats. Int. J. Mol. Sci. 2019, 20, 1680. [Google Scholar] [CrossRef]
- Radlinger, B.; Hornsteiner, F.; Folie, S.; Salvenmoser, W.; Haubner, B.J.; Schuetz, T.; Haas, S.; Ress, C.; Adolph, T.E.; Salzmann, K.; et al. Cardioprotective effects of short-term empagliflozin treatment in db/db mice. Sci. Rep. 2020, 10, 19686. [Google Scholar] [CrossRef]
- Szepes, M.; Janicsek, Z.; Benko, Z.; Cselenyak, A.; Kiss, L. Pretreatment of therapeutic cells with poly(ADP-ribose) polymerase inhibitor enhances their efficacy in an in vitro model of cell-based therapy in myocardial infarct. Int. J. Mol. Med. 2013, 31, 26–32. [Google Scholar] [CrossRef]
- Hwang, J.S.; Hur, J.; Lee, W.J.; Won, J.P.; Lee, H.G.; Lim, D.S.; Kim, E.; Seo, H.G. Catalase Mediates the Inhibitory Actions of PPARdelta against Angiotensin II-Triggered Hypertrophy in H9c2 Cardiomyocytes. Antioxidants 2021, 10, 1223. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-L.; Wang, H.-T.; Lee, W.-C.; Lin, P.-T.; Liu, W.-H.; Hsueh, S.-K. Empagliflozin Prevent High-Glucose Stimulation Inducing Apoptosis and Mitochondria Fragmentation in H9C2 Cells through the Calcium-Dependent Activation Extracellular Signal-Regulated Kinase 1/2 Pathway. Int. J. Mol. Sci. 2024, 25, 8235. https://doi.org/10.3390/ijms25158235
Chen Y-L, Wang H-T, Lee W-C, Lin P-T, Liu W-H, Hsueh S-K. Empagliflozin Prevent High-Glucose Stimulation Inducing Apoptosis and Mitochondria Fragmentation in H9C2 Cells through the Calcium-Dependent Activation Extracellular Signal-Regulated Kinase 1/2 Pathway. International Journal of Molecular Sciences. 2024; 25(15):8235. https://doi.org/10.3390/ijms25158235
Chicago/Turabian StyleChen, Yung-Lung, Hui-Ting Wang, Wen-Chin Lee, Pei-Ting Lin, Wen-Hao Liu, and Shu-Kai Hsueh. 2024. "Empagliflozin Prevent High-Glucose Stimulation Inducing Apoptosis and Mitochondria Fragmentation in H9C2 Cells through the Calcium-Dependent Activation Extracellular Signal-Regulated Kinase 1/2 Pathway" International Journal of Molecular Sciences 25, no. 15: 8235. https://doi.org/10.3390/ijms25158235