
Metabolic Insights into Neuropsychiatric Illnesses and Ketogenic Therapies: A Transcriptomic View
 ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. Final Pathways
2.2. Transcriptomic Results for Ketosis Datasets
2.3. Transcriptomic Results for Schizophrenia and Schizophrenia–Ketosis Comparison

{kind=link}
{kind=link}
| Metabolic Pathway | Significant Genes by Pathway in Schizophrenia (SCZ) Datasets (n = 35) | SCZ Average LFC Values | Significant Genes by Pathway in Ketogenic Intervention (KI) Datasets (n = 8) | KI Average LFC Values |
|---|---|---|---|---|
| Gluconeogenesis | Upregulated: PC, PCK1 | 1.08 | Upregulated: PC | 0.23 |
| Downregulated: G6PC3, PFKFB2 | −0.32 | Downregulated: PCK2 | −1.56 | |
| Glycolysis | Upregulated: ALDOA, ALDOB, ALDOC, GAPDHS, PGAM2, ENO1, ENO3, PKLR, PKM | 0.68 | ||
| Downregulated: GAPDH, PFKM, PFKP, PGK1, PGK2 | −0.58 | Downregulated: ENO1, GAPDH, PFKP | −0.56 | |
| Lactate Shuttle (Neuron–Astrocyte) | Upregulated: GLUL, LDHC, SLC16A3, SLC1A2, SLCIA3, SLC2A1 | 0.58 | Upregulated: SLC16A1 | 0.29 |
| Downregulated: GLS2, LDHA, SLC1A1, SLC2A11, SLC2A14 | −0.23 | |||
| Tricarboxylic Acid (TCA) Cycle | Upregulated: ACO1, ACO2, CS, DBT, IDH2, SDHA, SUCLG2 | 0.39 | ||
| Downregulated: DLD, IDH1, IDH3B, MDH2, SDHB, SDHD, SUCLA2 | −0.33 | Downregulated: CS, IDH3G, SDHA, SDHB, SUCLG2 | −0.67 | |
| Electron Transport Chain (ETC) | Upregulated: COX4I2, COX6B2, SDHA, UQCR10 | 0.56 | Upregulated: CYCS, COX4I2, SDHAF3 | 0.40 |
| Downregulated: ATP5F1A, ATP5F1B, ATP5F1C, ATP5F1E, COQ10B, COQ4, COQ7, COX5A, COX5B, COX6A1, COX6B1, COX6C, COX7A1, COX7A2, COX7B, COX7C, COX8A, CYC1, NDUFA12, NDUFA8, NDUFB5, NDUFC2, NDUFS4, NDUFS5, NDUFV1, NDUFV2, NDUFV3, SDHAF3, SDHB, SDHD, UQCR11, UQCRB, UQCRC1, UQCRFS1, UQCRQ | −0.30 | Downregulated: ATP5F1C, ATP5F1D, COX411, COX5B, COX6C, CYC1, SDHA, SDHB, NDUFV2, UQCRC1, UQCRFS1 | −0.62 | |
| Fatty Acid Synthesis | Upregulated: ACACB, DECR1, FASN, HADH | 0.50 | Upregulated: ACACB, HADH | 0.70 |
| Downregulated: ELOVL1, MCAT, OLAH, OXSM | −0.68 | Downregulated: DECR1, PPT1 | −0.69 | |
| Fatty Acid Oxidation | Upregulated: ACAA2, CPT1C, CPT2, HADHA, HSD17B10 | 0.56 | Upregulated: HADH | 0.41 |
| Downregulated: SCP2 | −0.48 | |||
| Ketogenesis | Upregulated: ACAA2, BDH1, BDH2, HMGCLL1 | 0.82 | ||
| Downregulated: ACAA1, HMGCS1 | −0.30 | |||
| Glycogenesis | Upregulated: HK2 | 0.33 | ||
| Downregulated: GBE1, GSK3A, GSK3B, GYS1 | −0.22 | Downregulated: PGM1 | −0.50 | |
| Glycogenolysis | Upregulated: PYGL | 0.54 | Upregulated: PYGL, PYGM | 0.56 |
| Downregulated: G6PC3 | −0.36 | Downregulated: PGM1 | −0.50 | |
| Urea Cycle | Upregulated: ACY1, AGMAT, CAD | 0.90 | ||
| Downregulated: ARG2, ASS1, CPS1, OAT | −0.32 | Downregulated: OAT | −0.56 | |
| Pentose Phosphate/ Glutathione Pathways | Upregulated: G6PD, GPI, HK2, RPE, TKT | 0.52 | ||
| Downregulated: GCLC, GSS, RPIA, SOD1, TALDO1 | −0.26 | Downregulated: G6PD, GSR, RPIA | −0.73 |
2.4. Transcriptomic Results for Bipolar Disorder and Bipolar Disorder–Ketosis Comparison
| Metabolic Pathway | Significant Genes by Pathway in Bipolar Disorder (BPD) Datasets (n = 55) | BPD Average LFC Values | Significant Genes by Pathway in Ketogenic Intervention (KI) Datasets (n = 8) | KI Average LFC Values |
|---|---|---|---|---|
| Gluconeogenesis | Upregulated: FBP2, PCK1 | 1.08 | Upregulated: PC | 0.23 |
| Downregulated: G6PC3 | −0.43 | Downregulated: PCK2 | −1.56 | |
| Glycolysis | Upregulated: BPGM, HK2, HK3, PGAM2 | 0.84 | ||
| Downregulated: ALDOA, ENO1, ENO2, GAPDH, GCK, GPI, PFKL, PFKM, PFKP, PGAM4, PGK1, PKLR | −0.58 | Downregulated: ENO1, GAPDH, PFKP | −0.56 | |
| Lactate Shuttle (Neuron–Astrocyte) | Upregulated: LDHC, SLC16A1, SLC2A1, SLC2A10, SLC2A12 | 1.06 | Upregulated: SLC16A1 | 0.29 |
| Downregulated: SLC1A2, SLC2A11, SLC2A3 | −0.84 | |||
| Tricarboxylic Acid (TCA) Cycle | Upregulated: ACO1, DBT, IDH1 | 0.38 | ||
| Downregulated: ACLY, ACO2, CS, DLAT, IDH2, IDH3B, IDH3G, MDH2, OGDHL, SDHAF4, SDHC, SUCLA2 | −0.51 | Downregulated: CS, IDH3G, SDHA, SDHB, SUCLG2 | −0.67 | |
| Electron Transport Chain (ETC) | Upregulated: COX4I2, COX6A2, COX6B2, COX8C, MT-ATP6, MT-CO1, MT-CO2, MT-CO3, MT-CYB, MT-ND1, MT-ND2, MT-ND3, MT-ND4, MT-ND4L, MT-ND5, MT-ND6, NDUFV2, UQCRC2, UQCRH | 0.65 | Upregulated: CYCS, COX4I2, SDHAF3 | 0.40 |
| Downregulated: ATP5F1A, ATP5F1B, ATP5F1D, ATP5F1E, COQ10A, COQ4, COQ7, COX5B, COX7B2, COX8A, CYC1, CYCS, NDUFA11, NDUFA12, NDUFB5, NDUFC2, NDUFS1, NDUFV1, SDHAF3, SDHAF4, SDHC, UQCR10, UQCR11, UQCRC1 | −0.57 | Downregulated: ATP5F1C, ATP5F1D, COX411, COX5B, COX6C, CYC1, SDHA, SDHB, NDUFV2, UQCRC1, UQCRFS1 | −0.62 | |
| Fatty Acid Synthesis | Upregulated: DECR1, ELOVL3, OLAH, OXSM | 0.90 | Upregulated: ACACB, HADH | 0.70 |
| Downregulated: ACACA, SLOVL6, FASN, MCAT, PPT1 | −0.51 | Downregulated: DECR1, PPT1 | −0.69 | |
| Fatty Acid Oxidation | Upregulated: CPT2, HADHB, SCP2 | 0.46 | Upregulated: HADH | 0.41 |
| Downregulated: ACSBG2, CPT1C, ECHS1 | −0.76 | |||
| Ketogenesis | Upregulated: HMGCL, HMGCS2 | 1.16 | ||
| Downregulated: ACAT1, HMGCLL1, HMGCS1 | −1.05 | |||
| Glycogenesis | Upregulated: HK2, HK3, UGP2 | 0.90 | ||
| Downregulated: PGM2 | −0.67 | Downregulated: PGM1 | −0.50 | |
| Glycogenolysis | Upregulated: PYGL | 0.49 | Upregulated: PYGL, PYGM | 0.56 |
| Downregulated: AGL, G6PC3, PGM2 | −0.58 | Downregulated: PGM1 | −0.50 | |
| Urea Cycle | Upregulated: ACY1, ARG1, OTC | 0.68 | ||
| Downregulated: ARG2, ASL, ASS1, CAD, OAT | −0.45 | Downregulated: OAT | −0.56 | |
| Pentose Phosphate / Glutathione Pathways | Upregulated: GCLC, HK2, HK3, RPIA | 0.87 | ||
| Downregulated: G6PD, GPI, PGD, TKT | −0.61 | Downregulated: G6PD, GSR, RPIA | −0.73 |
2.5. Transcriptomic Results for Major Depressive Disorder and Major Depressive Disorder–Ketosis Comparison
| Metabolic Pathway | Significant Genes by Pathway in Major Depressive Disorder (MDD) Datasets (n = 36) | MDD Average LFC Values | Significant Genes by Pathway in Ketogenic Intervention (KI) Datasets (n = 8) | KI Average LFC Values |
|---|---|---|---|---|
| Gluconeogenesis | Upregulated: G6PC2 | 0.3 | Upregulated: PC | 0.23 |
| Downregulated: G6PC3, PCK1, PFKFB2 | −0.68 | Downregulated: PCK2 | −1.56 | |
| Glycolysis | Upregulated: ALDOA, ENO2, GCK, GPI, HK1, HK3, PFKP, PGAM1, PGAM4, PGK1, PKM, TPI1 | 0.59 | ||
| Downregulated: ENO4, PGK2 | −1.19 | Downregulated: ENO1, GAPDH, PFKP | −0.56 | |
| Lactate Shuttle (Neuron–Astrocyte) | Upregulated: GLS, LDHA, SLC16A1, SLC1A6 | 0.39 | Upregulated: SLC16A1 | 0.29 |
| Downregulated: GLUL, LDHD, SLC1A2, SLC1A3, SLC2A10, SLC2A11, SLC2A12, SLC2A3 | −0.53 | |||
| Tricarboxylic Acid (TCA) Cycle | Upregulated: ACO2, CS, DLD, FH, IDH3A, IDH3B, IDH3G, OGDH, PDHA1, SDHA, SDHB, SDHC, SDHD, SUCLA2, SUCLG1 | 0.45 | ||
| Downregulated: ACLY, ACO1, ALDH5A1, DBT, IDH1, IDH2, IREB2, SUCLG2 | −0.33 | Downregulated: CS, IDH3G, SDHA, SDHB, SUCLG2 | −0.67 | |
| Electron Transport Chain (ETC) | Upregulated: ATP5F1A, ATP5F1B, ATP5F1E, COQ10A, COQ10B, COQ7, COX5B, COX6B1, COX6C, COX7A1, COX7A2, COX7B, COX7C, CYC1, CYCS, MT-CO2, MT-CO3, NDUFA12, NDUFB5, NDUFC2, NDUFS4, NDUFS5, NDUFV1, SDHA, SDHAF3, SDHB, SDHC, SDHD, UQCR11, UQCRB, UQCRC1, UQCRC2, UQCRFS1, UQCRH, UQCRQ | 0.57 | Upregulated: CYCS, COX4I2, SDHAF3 | 0.40 |
| Downregulated: COX7B2, MT-ATP6, MT-ATP8, MT-ND2, MT-ND5 | −0.51 | Downregulated: ATP5F1C, ATP5F1D, COX411, COX5B, COX6C, CYC1, SDHA, SDHB, NDUFV2, UQCRC1, UQCRFS1 | −0.62 | |
| Fatty Acid Synthesis | Upregulated: ELOVL3, MCAT, OLAH | 0.24 | Upregulated: ACACB, HADH | 0.70 |
| Downregulated: DECR1, ELOVL1, FASN | −0.35 | Downregulated: DECR1, PPT1 | −0.69 | |
| Fatty Acid Oxidation | Upregulated: ACSBG2, HSD17B10, SLC27A2 | 0.29 | Upregulated: HADH | 0.41 |
| Downregulated: ACAA2, ACSBG1, CPT2, ECI1 | −0.39 | |||
| Ketogenesis | Upregulated: ACAT1, HMGCLL1 | 0.62 | ||
| Downregulated: ACAA1, ACAA2, BDH1, BDH2, HMGCS1 | −0.35 | |||
| Glycogenesis | Upregulated: HK1, HK3, UGP2 | 0.41 | ||
| Downregulated: ADPGK, GYS1, PGM2 | −0.33 | Downregulated: PGM1 | −0.50 | |
| Glycogenolysis | Upregulated: G6PC2, PYGL | 0.66 | Upregulated: PYGL, PYGM | 0.56 |
| Downregulated: AGL, G6PC3, PGM2, PYGB, PYGM | −0.44 | Downregulated: PGM1 | −0.50 | |
| Urea Cycle | Upregulated: ARG2, ASS1, OTC | 0.33 | ||
| Downregulated: ACY1, ASL, CPS1 | −0.62 | Downregulated: OAT | −0.56 | |
| Pentose Phosphate/Glutathione Pathways | Upregulated: GCLC, GPI, GSR, HK1, HK3, PGD, RPIA, SOD1, TALDO1 | 0.34 | ||
| Downregulated: G6PD, GSR, RPIA | −0.73 |
2.6. Transcriptomic Results for Medication Analysis
| Metabolic Pathway | Significantly Altered Genes in Chronic Antipsychotic-Treated vs. Vehicle-Treated Datasets (n = 24) | Average LFC Values | Significantly Altered Genes in Chronic Mood Stabilizer-Treated vs. Vehicle-Treated Datasets (n = 7) | Average LFC Values |
|---|---|---|---|---|
| Gluconeogenesis | Upregulated: PFKFB2, PCK2, G6PC1 | 0.39 | ||
| Downregulated: PFKFB2, PCK1, PCK2 | −0.23 | |||
| Glycolysis | Upregulated: PKLR, PGK1, PFKM, HK2, ALDOC, ENO2 | 0.36 | Upregulated: ENO3 | 0.37 |
| Downregulated: PFKP, GAPDH, ALDOB, GPI | −0.45 | Downregulated: PFKP, PKLR | −0.37 | |
| Lactate Shuttle (Neuron–Astrocyte) | Upregulated: SLC1A3, SLC1A2, SLC16A3, LDHD | 0.37 | Upregulated: GLUL, SLC1A3 | 0.37 |
| Downregulated: SLC2A3, SLC2A1, GLS, SLC1A6, GLUL | −0.31 | Downregulated: GLS2, GLS | −0.36 | |
| Tricarboxylic Acid (TCA) Cycle | Upregulated: SUCLG1, SDHB, IDH2, IDH1, ACO1 | 0.26 | Upregulated: SDHB, IDH2, ACO1, SDHAF4 | 0.35 |
| Downregulated: SDHAF4, PDHA2, IDH3A, ALDH5A1, ACLY | −0.47 | Downregulated: SUCLA2, ALDH5A1, DBT | −0.22 | |
| Electron Transport Chain (ETC) | Upregulated: UQCRH, SDHB, NDUFV2, NDUFC1, NDUFB5, COX7C, COX6B2, COX5B, COQ4, COQ10A, SDHAF2 | 0.24 | Upregulated: COQ7, COX7A2, COX6C, SDHB, SDHAF4 | 0.32 |
| Downregulated: SDHAF4, COX8A, COX7A1, COX6C, COX5A, COQ10B, MT-ND3 | −0.23 | Downregulated: NDUFS1, SDAHF2 | −0.26 | |
| Fatty Acid Synthesis | Upregulated: OXSM, HADH, ELOVL1, DECR1 | 0.32 | Upregulated: FASN | 0.33 |
| Downregulated: PPT2, MCAT, FASN, ELOVL6 | −0.44 | |||
| Fatty Acid Oxidation | Upregulated: HSD17B10, HADH, ECI1, ECHS1, ACAA2 | 0.27 | Upregulated: HADHB, ECHS1, CPT1C, SLC27A2, ECI1, HADHA | 0.28 |
| Ketogenesis | Upregulated: HMGCS2, BDH1, ACAT1, ACAA2 | 0.34 | ||
| Downregulated: HMGCS2, BDH2, HMGCS1 | −0.31 | |||
| Glycogenesis | Upregulated: HK2, GSK3A | 0.21 | Upregulated: PGM1, PGM2, GSK3B | 0.20 |
| Downregulated: PGM1, PGM2, GSK3B, GBE1, ADPGK | −0.39 | Downregulated: GCKR | −0.25 | |
| Glycogenolysis | Upregulated: G6PC1 | 0.20 | Upregulated: PGM1, PGM2 | 0.21 |
| Downregulated: PGM1, PGM2, AGL | −0.54 | |||
| Urea Cycle | Upregulated: ASS1, ASL, AGMAT | 0.44 | Upregulated: CAD | 0.24 |
| Downregulated: ARG2 | −0.18 | Downregulated: ASS1 | −0.65 | |
| Pentose Phosphate/Glutathione Pathways | Upregulated: TKT, TALDO1, SOD1, RPIA, HK2, GSS, GCLC | 0.26 | ||
| Downregulated: RPE, GPI | −0.29 | Downregulated: GSS, RPIA | −0.37 |
3. Discussion
4. Materials and Methods
4.1. Filtering Metabolic Pathways and Genes of Interest
4.2. Building the “Ketosis” Module within the Kaleidoscope Lookup Tool
4.3. Querying Gene Lists and Generating Data Tables from Kaleidoscope Lookup

4.4. Transcriptomic Analysis among Neuropsychiatric Illness and Ketosis Datasets
4.5. Analysis of the Effect of Antipsychotics and Mood Stabilizers on Metabolic Gene Expression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Whiteford, H.A.; Ferrari, A.J.; Degenhardt, L.; Feigin, V.; Vos, T. The global burden of mental, neurological and substance use disorders: An analysis from the Global Burden of Disease Study 2010. PLoS ONE 2015, 10, e0116820. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, Y.; Wang, L.; Tao, M.; Cao, H.; Yuan, H.; Ye, M.; Chen, X.; Wang, K.; Zhu, C. Changing trends in the global burden of mental disorders from 1990 to 2019 and predicted levels in 25 years. Epidemiol. Psychiatry Sci. 2023, 32, e63. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Alshaya, D.S. Genetic and epigenetic factors associated with depression: An updated overview. Saudi J. Biol. Sci. 2022, 29, 103311. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Andlauer, T.F.M.; Mühleisen, T.W.; Hoffstaedter, F.; Teumer, A.; Wittfeld, K.; Teuber, A.; Reinbold, C.S.; Grotegerd, D.; Bülow, R.; Caspers, S.; et al. Genetic factors influencing a neurobiological substrate for psychiatric disorders. Transl. Psychiatry 2021, 11, 192. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kolar, D.; Kleteckova, L.; Brozka, H.; Vales, K. Mini-review: Brain energy metabolism and its role in animal models of depression, bipolar disorder, schizophrenia and autism. Neurosci. Lett. 2021, 760, 136003. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.R.; O’Donovan, S.M.; McCullumsmith, R.E.; Ramsey, A. Defects in Bioenergetic Coupling in Schizophrenia. Biol. Psychiatry 2018, 83, 739–750. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zuccoli, G.S.; Saia-Cereda, V.M.; Nascimento, J.M.; Martins-de-Souza, D. The Energy Metabolism Dysfunction in Psychiatric Disorders Postmortem Brains: Focus on Proteomic Evidence. Front. Neurosci. 2017, 11, 493. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Danan, A.; Westman, E.C.; Saslow, L.R.; Ede, G. The Ketogenic Diet for Refractory Mental Illness: A Retrospective Analysis of 31 Inpatients. Front. Psychiatry 2022, 13, 951376. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dietch, D.M.; Kerr-Gaffney, J.; Hockey, M.; Marx, W.; Ruusunen, A.; Young, A.H.; Berk, M.; Mondelli, V. Efficacy of low carbohydrate and ketogenic diets in treating mood and anxiety disorders: Systematic review and implications for clinical practice. BJPsych Open 2023, 9, e70. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tillery, E.E.; Ellis, K.D.; Threatt, T.B.; Reyes, H.A.; Plummer, C.S.; Barney, L.R. The use of the ketogenic diet in the treatment of psychiatric disorders. Ment. Health Clin. 2021, 11, 211–219. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Swerdlow, R.H. Bioenergetic medicine. Br. J. Pharmacol. 2014, 171, 1854–1869. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bipolar Disorder and Schizophrenia Working Group of the Psychiatric Genomics Consortium. Genomic Dissection of Bipolar Disorder and Schizophrenia, Including 28 Subphenotypes. Cell 2018, 173, 1705–1715.e16. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gao, X.; Qin, Y.; Jiao, S.; Hao, J.; Zhao, J.; Wang, J.; Wen, Y.; Wang, T. Genetic evidence for the causal relations between metabolic syndrome and psychiatric disorders: A Mendelian randomization study. Transl. Psychiatry 2024, 14, 46. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ben-Shachar, D.; Karry, R. Neuroanatomical pattern of mitochondrial complex I pathology varies between schizophrenia, bipolar disorder and major depression. PLoS ONE 2008, 3, e3676. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Halim, N.D.; Lipska, B.K.; Hyde, T.M.; Deep-Soboslay, A.; Saylor, E.M.; Herman, M.M.; Thakar, J.; Verma, A.; Kleinman, J.E. Increased lactate levels and reduced pH in postmortem brains of schizophrenics: Medication confounds. J. Neurosci. Methods 2008, 169, 208–213. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Prabakaran, S.; Swatton, J.E.; Ryan, M.M.; Huffaker, S.J.; Huang, J.T.; Griffin, J.L.; Wayland, M.; Freeman, T.; Dudbridge, F.; Lilley, K.S.; et al. Mitochondrial dysfunction in schizophrenia: Evidence for compromised brain metabolism and oxidative stress. Mol. Psychiatry 2004, 9, 684–697. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.C.; Barksdale, K.A.; Roche, J.K.; Lahti, A.C. Decreased synaptic and mitochondrial density in the postmortem anterior cingulate cortex in schizophrenia. Schizophr. Res. 2015, 168, 543–553. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, X.; Wang, J.F.; Tseng, M.; Young, L.T. Downregulation in components of the mitochondrial electron transport chain in the postmortem frontal cortex of subjects with bipolar disorder. J. Psychiatry Neurosci. 2006, 31, 189–196. [Google Scholar] [PubMed] [PubMed Central]
- das Neves Duarte, J.M.; Kulak, A.; Gholam-Razaee, M.M.; Cuenod, M.; Gruetter, R.; Do, K.Q. N-acetylcysteine normalizes neurochemical changes in the glutathione-deficient schizophrenia mouse model during development. Biol. Psychiatry 2012, 71, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Smolensky, I.V.; Zajac-Bakri, K.; Gass, P.; Inta, D. Ketogenic diet for mood disorders from animal models to clinical application. J. Neural Transm. 2023, 130, 1195–1205. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Le Foll, C.; Levin, B.E. Fatty acid-induced astrocyte ketone production and the control of food intake. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R1186–R1192. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Longo, R.; Peri, C.; Cricrì, D.; Coppi, L.; Caruso, D.; Mitro, N.; De Fabiani, E.; Crestani, M. Ketogenic Diet: A New Light Shining on Old but Gold Biochemistry. Nutrients 2019, 11, 2497. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- LaManna, J.C.; Salem, N.; Puchowicz, M.; Erokwu, B.; Koppaka, S.; Flask, C.; Lee, Z. Ketones suppress brain glucose consumption. Adv. Exp. Med. Biol. 2009, 645, 301–306. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mattson, M.P.; Moehl, K.; Ghena, N.; Schmaedick, M.; Cheng, A. Intermittent metabolic switching, neuroplasticity and brain health. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Roberts, M.N.; Wallace, M.A.; Tomilov, A.A.; Zhou, Z.; Marcotte, G.R.; Tran, D.; Perez, G.; Gutierrez-Casado, E.; Koike, S.; Knotts, T.A.; et al. A Ketogenic Diet Extends Longevity and Healthspan in Adult Mice. Cell Metab. 2017, 26, 539–546.e5. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Newman, J.C.; Verdin, E. β-Hydroxybutyrate: A Signaling Metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kraeuter, A.K.; Loxton, H.; Lima, B.C.; Rudd, D.; Sarnyai, Z. Ketogenic diet reverses behavioral abnormalities in an acute NMDA receptor hypofunction model of schizophrenia. Schizophr. Res. 2015, 169, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Kraeuter, A.K.; Archambault, N.; van den Buuse, M.; Sarnyai, Z. Ketogenic diet and olanzapine treatment alone and in combination reduce a pharmacologically-induced prepulse inhibition deficit in female mice. Schizophr. Res. 2019, 212, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Kraeuter, A.K.; Mashavave, T.; Suvarna, A.; van den Buuse, M.; Sarnyai, Z. Effects of beta-hydroxybutyrate administration on MK-801-induced schizophrenia-like behaviour in mice. Psychopharmacology 2020, 237, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, A.J. NR1 knockdown mice as a representative model of the glutamate hypothesis of schizophrenia. Prog. Brain Res. 2009, 179, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Paula Farias Waltrick, A.; Henrique Bernardo de Lima Silva, A.; Cristina de Carvalho, M.; Aparecida Comotti de Oliveira, B.; Naliwaiko, K.; Maria da Cunha, J.; Menezes Zanoveli, J. Preventive treatment with fish oil facilitates the antidepressant-like effect of antidepressant drugs in type-1 diabetes mellitus rats: Implication of serotonergic system. Neurosci. Lett. 2022, 772, 136477. [Google Scholar] [CrossRef] [PubMed]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 2006, 60, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Phelps, J.R.; Siemers, S.V.; El-Mallakh, R.S. The ketogenic diet for type II bipolar disorder. Neurocase 2013, 19, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Kraft, B.D.; Westman, E.C. Schizophrenia, gluten, and low-carbohydrate, ketogenic diets: A case report and review of the literature. Nutr. Metab. 2009, 6, 10. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Palmer, C.M. Ketogenic diet in the treatment of schizoaffective disorder: Two case studies. Schizophr. Res. 2017, 189, 208–209. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.M.; Gilbert-Jaramillo, J.; Westman, E.C. The ketogenic diet and remission of psychotic symptoms in schizophrenia: Two case studies. Schizophr. Res. 2019, 208, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Kovács, Z.; D’Agostino, D.P.; Diamond, D.; Kindy, M.S.; Rogers, C.; Ari, C. Therapeutic Potential of Exogenous Ketone Supplement Induced Ketosis in the Treatment of Psychiatric Disorders: Review of Current Literature. Front. Psychiatry 2019, 10, 363. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kaidanovich-Beilin, O.; Cha, D.S.; McIntyre, R.S. Crosstalk between metabolic and neuropsychiatric disorders. F1000 Biol. Rep. 2012, 4, 14. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Campbell, I.H.; Campbell, H. The metabolic overdrive hypothesis: Hyperglycolysis and glutaminolysis in bipolar mania. Mol. Psychiatry 2024, 29, 1521–1527. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Ke, S.; Wang, Q.; Zhuang, T.; Xia, C.; Xu, Y.; Yang, L.; Zhou, M. Energy metabolism in major depressive disorder: Recent advances from omics technologies and imaging. Biomed. Pharmacother. 2021, 141, 111869. [Google Scholar] [CrossRef] [PubMed]
- Regenold, W.T.; Pratt, M.; Nekkalapu, S.; Shapiro, P.S.; Kristian, T.; Fiskum, G. Mitochondrial detachment of hexokinase 1 in mood and psychotic disorders: Implications for brain energy metabolism and neurotrophic signaling. J. Psychiatry Res. 2012, 46, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Shan, D.; Mount, D.; Moore, S.; Haroutunian, V.; Meador-Woodruff, J.H.; McCullumsmith, R.E. Abnormal partitioning of hexokinase 1 suggests disruption of a glutamate transport protein complex in schizophrenia. Schizophr. Res. 2014, 154, 1–13. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stork, C.; Renshaw, P.F. Mitochondrial dysfunction in bipolar disorder: Evidence from magnetic resonance spectroscopy research. Mol. Psychiatry 2005, 10, 900–919. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.R.; Mielnik, C.A.; O’Donovan, S.M.; Funk, A.J.; Bentea, E.; DePasquale, E.A.; Alganem, K.; Wen, Z.; Haroutunian, V.; Katsel, P.; et al. Connectivity Analyses of Bioenergetic Changes in Schizophrenia: Identification of Novel Treatments. Mol. Neurobiol. 2019, 56, 4492–4517. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sullivan, C.R.; Koene, R.H.; Hasselfeld, K.; O’Donovan, S.M.; Ramsey, A.; McCullumsmith, R.E. Neuron-specific deficits of bioenergetic processes in the dorsolateral prefrontal cortex in schizophrenia. Mol. Psychiatry 2019, 24, 1319–1328. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bubber, P.; Hartounian, V.; Gibson, G.E.; Blass, J.P. Abnormalities in the tricarboxylic acid (TCA) cycle in the brains of schizophrenia patients. Eur. Neuropsychopharmacol. 2011, 21, 254–260. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Büttiker, P.; Weissenberger, S.; Esch, T.; Anders, M.; Raboch, J.; Ptacek, R.; Kream, R.M.; Stefano, G.B. Dysfunctional mitochondrial processes contribute to energy perturbations in the brain and neuropsychiatric symptoms. Front. Pharmacol. 2022, 13, 1095923. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Middleton, F.A.; Mirnics, K.; Pierri, J.N.; Lewis, D.A.; Levitt, P. Gene expression profiling reveals alterations of specific metabolic pathways in schizophrenia. J. Neurosci. 2002, 22, 2718–2729. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Henkel, N.D.; Wu, X.; O’Donovan, S.M.; Devine, E.A.; Jiron, J.M.; Rowland, L.M.; Sarnyai, Z.; Ramsey, A.J.; Wen, Z.; Hahn, M.K.; et al. Schizophrenia: A disorder of broken brain bioenergetics. Mol. Psychiatry 2022, 27, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Kuang, H.; Duong, A.; Jeong, H.; Zachos, K.; Andreazza, A.C. Lactate in bipolar disorder: A systematic review and meta-analysis. Psychiatry Clin. Neurosci. 2018, 72, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Roman, C.; Egert, L.; Di Benedetto, B. Astrocytic-neuronal crosstalk gets jammed: Alternative perspectives on the onset of neuropsychiatric disorders. Eur. J. Neurosci. 2021, 54, 5717–5729. [Google Scholar] [CrossRef] [PubMed]
- Özaslan, M.S.; Balcı, N.; Demir, Y.; Gürbüz, M.; Küfrevioğlu, Ö. Inhibition effects of some antidepressant drugs on pentose phosphate pathway enzymes. Environ. Toxicol. Pharmacol. 2019, 72, 103244. [Google Scholar] [CrossRef] [PubMed]
- Sarandol, A.; Sarandol, E.; Eker, S.S.; Erdinc, S.; Vatansever, E.; Kirli, S. Major depressive disorder is accompanied with oxidative stress: Short-term antidepressant treatment does not alter oxidative-antioxidative systems. Hum. Psychopharmacol. 2007, 22, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Parnell, L.D.; McCaffrey, K.S.; Brooks, A.W.; Smith, C.E.; Lai, C.Q.; Christensen, J.J.; Wiley, C.D.; Ordovas, J.M. Rate-Limiting Enzymes in Cardiometabolic Health and Aging in Humans. Lifestyle Genom. 2023, 16, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Chasiotis, D.; Sahlin, K.; Hultman, E. Regulation of glycogenolysis in human muscle at rest and during exercise. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1982, 53, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Roach, P.J.; Depaoli-Roach, A.A.; Hurley, T.D.; Tagliabracci, V.S. Glycogen and its metabolism: Some new developments and old themes. Biochem. J. 2012, 441, 763–787. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Miller, T.B.; Larner, J. Mechanism of control of hepatic glycogenesis by insulin. J. Biol. Chem. 1973, 248, 3483–3488. [Google Scholar] [CrossRef] [PubMed]
- Eyre, J.A.; Stuart, A.G.; Forsyth, R.J.; Heaviside, D.; Bartlett, K. Glucose export from the brain in man: Evidence for a role for astrocytic glycogen as a reservoir of glucose for neural metabolism. Brain Res. 1994, 635, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, R.; Fray, A.; Boutelle, M.; Fillenz, M.; Middleditch, C.; Burchell, A. A role for astrocytes in glucose delivery to neurons? Dev. Neurosci. 1996, 18, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, R.; Varacallo, M. Biochemistry, Glycolysis; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar] [PubMed]
- Churchward, M.A.; Tchir, D.R.; Todd, K.G. Microglial Function during Glucose Deprivation: Inflammatory and Neuropsychiatric Implications. Mol. Neurobiol. 2018, 55, 1477–1487. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Morris, G.; Berk, M. The many roads to mitochondrial dysfunction in neuroimmune and neuropsychiatric disorders. BMC Med. 2015, 13, 68. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pei, L.; Wallace, D.C. Mitochondrial Etiology of Neuropsychiatric Disorders. Biol. Psychiatry 2018, 83, 722–730. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Eggleston, L.V.; Krebs, H.A. Regulation of the pentose phosphate cycle. Biochem. J. 1974, 138, 425–435. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ermakov, E.A.; Dmitrieva, E.M.; Parshukova, D.A.; Kazantseva, D.V.; Vasilieva, A.R.; Smirnova, L.P. Oxidative Stress-Related Mechanisms in Schizophrenia Pathogenesis and New Treatment Perspectives. Oxid. Med. Cell. Longev. 2021, 2021, 8881770. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free Radic. Biol. Med. 2012, 53, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. Brain lactate metabolism: The discoveries and the controversies. J. Cereb. Blood Flow Metab. 2012, 32, 1107–1138. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hagihara, H.; Shoji, H.; Hattori, S.; Sala, G.; Takamiya, Y.; Tanaka, M.; Ihara, M.; Shibutani, M.; Hatada, I.; Hori, K.; et al. Large-scale animal model study uncovers altered brain pH and lactate levels as a transdiagnostic endophenotype of neuropsychiatric disorders involving cognitive impairment. Elife 2024, 12, 89376. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jensen-Urstad, A.P.; Semenkovich, C.F. Fatty acid synthase and liver triglyceride metabolism: Housekeeper or messenger? Biochim. Biophys. Acta 2012, 1821, 747–753. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bartlett, K.; Eaton, S. Mitochondrial beta-oxidation. Eur. J. Biochem. 2004, 271, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Bhathena, S.J. Relationship between fatty acids and the endocrine and neuroendocrine system. Nutr. Neurosci. 2006, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Perica, M.M.; Delas, I. Essential fatty acids and psychiatric disorders. Nutr. Clin. Pract. 2011, 26, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Ross, B.M.; Seguin, J.; Sieswerda, L.E. Omega-3 fatty acids as treatments for mental illness: Which disorder and which fatty acid? Lipids Health Dis. 2007, 6, 21. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell. Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Krebs, H.A. Rate control of the tricarboxylic acid cycle. Adv. Enzym. Regul. 1970, 8, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Morella, I.M.; Brambilla, R.; Morè, L. Emerging roles of brain metabolism in cognitive impairment and neuropsychiatric disorders. Neurosci. Biobehav. Rev. 2022, 142, 104892. [Google Scholar] [CrossRef] [PubMed]
- Clay, H.B.; Sillivan, S.; Konradi, C. Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int. J. Dev. Neurosci. 2011, 29, 311–324. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- White, H.M. The Role of TCA Cycle Anaplerosis in Ketosis and Fatty Liver in Periparturient Dairy Cows. Animals 2015, 5, 793–802. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bostock, E.C.; Kirkby, K.C.; Taylor, B.V. The Current Status of the Ketogenic Diet in Psychiatry. Front. Psychiatry 2017, 8, 43. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brietzke, E.; Mansur, R.B.; Subramaniapillai, M.; Balanzá-Martínez, V.; Vinberg, M.; González-Pinto, A.; Rosenblat, J.D.; Ho, R.; McIntyre, R.S. Ketogenic diet as a metabolic therapy for mood disorders: Evidence and developments. Neurosci. Biobehav. Rev. 2018, 94, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Ford, J.M. The Role of Ketogenic Metabolic Therapy on the Brain in Serious Mental Illness: A Review. J. Psychiatry Brain Sci. 2022, 7, e220009. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002, 80, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Ramzan, R.; Kadenbach, B.; Vogt, S. Multiple Mechanisms Regulate Eukaryotic Cytochrome C Oxidase. Cells 2021, 10, 514. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Giménez-Palomo, A.; Dodd, S.; Anmella, G.; Carvalho, A.F.; Scaini, G.; Quevedo, J.; Pacchiarotti, I.; Vieta, E.; Berk, M. The Role of Mitochondria in Mood Disorders: From Physiology to Pathophysiology and to Treatment. Front. Psychiatry 2021, 12, 546801. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kam, P.C.; See, A.U. Cyclo-oxygenase isoenzymes: Physiological and pharmacological role. Anaesthesia 2000, 55, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol. 2015, 4, 381–398. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pinto, A.; Bonucci, A.; Maggi, E.; Corsi, M.; Businaro, R. Anti-Oxidant and Anti-Inflammatory Activity of Ketogenic Diet: New Perspectives for Neuroprotection in Alzheimer’s Disease. Antioxidants 2018, 7, 63. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sadasivudu, B.; Rao, T.I. Studies on functional and metabolic role of urea cycle intermediates in brain. J. Neurochem. 1976, 27, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, N.; Futamura, T.; Kakumoto, K.; Salehi, A.M.; Sellgren, C.M.; Holmén-Larsson, J.; Jakobsson, J.; Pålsson, E.; Landén, M.; Hashimoto, K. Blood metabolomics analysis identifies abnormalities in the citric acid cycle, urea cycle, and amino acid metabolism in bipolar disorder. BBA Clin. 2016, 5, 151–158. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- American Psychiatric Association. Practice guideline for the treatment of patients with schizophrenia. Am. J. Psychiatry 1997, 154 (Suppl. S4), 1–63. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Practice guideline for the treatment of patients with bipolar disorder (revision). Am. J. Psychiatry 2002, 159 (Suppl. S4), 1–50. [Google Scholar] [PubMed]
- Ghasemi, R.; Dargahi, L.; Haeri, A.; Moosavi, M.; Mohamed, Z.; Ahmadiani, A. Brain insulin dysregulation: Implication for neurological and neuropsychiatric disorders. Mol. Neurobiol. 2013, 47, 1045–1065. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.W.; Chen, L.C.; Huang, K.L.; Bai, Y.M.; Tsai, S.J.; Su, T.P.; Chen, M.H. Appetite hormone dysregulation and executive dysfunction among adolescents with bipolar disorder and disruptive mood dysregulation disorder. Eur. Child Adolesc. Psychiatry 2024, 33, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Kong, A.P.; Chan, N.N.; Chan, J.C. The role of adipocytokines and neurohormonal dysregulation in metabolic syndrome. Curr. Diabetes Rev. 2006, 2, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Sonsalla, P.K.; Pedata, F.; Melani, A.; Domenici, M.R.; Popoli, P.; Geiger, J.; Lopes, L.V.; de Mendonça, A. Adenosine A2A receptors and brain injury: Broad spectrum of neuroprotection, multifaceted actions and “fine tuning” modulation. Prog. Neurobiol. 2007, 83, 310–331. [Google Scholar] [CrossRef] [PubMed]
- Sahay, S.; Hamoud, A.-R.; Osman, M.; Pulvender, P.; McCullumsmith, R.E. Expression of WNT Signaling Genes in the Dorsolateral Prefrontal Cortex in Schizophrenia. Brain Sci. 2024, 14, 649. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, J.R.; Szeto, R.A.; Carvalho, V.M.A.; Muotri, A.R.; Papes, F. Transcription factor 4 and its association with psychiatric disorders. Transl. Psychiatry 2021, 11, 19. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kolar, D.; Krajcovic, B.; Kleteckova, L.; Kuncicka, D.; Vales, K.; Brozka, H. Review: Genes Involved in Mitochondrial Physiology within 22q11.2 Deleted Region and Their Relevance to Schizophrenia. Schizophr. Bull. 2023, 49, 1637–1653. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Alnafisah, R.; Lundh, A.; Asah, S.M.; Hoeflinger, J.; Wolfinger, A.; Hamoud, A.R.; McCullumsmith, R.E.; O’Donovan, S.M. Altered purinergic receptor expression in the frontal cortex in schizophrenia. Schizophrenia 2022, 8, 96. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- O’Donovan, S.M.; Sullivan, C.; Koene, R.; Devine, E.; Hasselfeld, K.; Moody, C.L.; McCullumsmith, R.E. Cell-subtype-specific changes in adenosine pathways in schizophrenia. Neuropsychopharmacology 2018, 43, 1667–1674. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sahay, S.; Devine, E.A.; McCullumsmith, R.E.; O’Donovan, S.M. Adenosine Receptor mRNA Expression in Frontal Cortical Neurons in Schizophrenia. Cells 2023, 13, 32. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Devine, E.A.; Imami, A.S.; Eby, H.; Hamoud, A.R.; Golchin, H.; Ryan, W.; Sahay, S.; Shedroff, E.A.; Arvay, T.; Joyce, A.W.; et al. Neuronal alterations in AKT isotype expression in schizophrenia. Res. Sq. 2024. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Funk, A.J.; McCullumsmith, R.E.; Haroutunian, V.; Meador-Woodruff, J.H. Abnormal activity of the MAPK- and cAMP-associated signaling pathways in frontal cortical areas in postmortem brain in schizophrenia. Neuropsychopharmacology 2012, 37, 896–905. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- McGuire, J.L.; Hammond, J.H.; Yates, S.D.; Chen, D.; Haroutunian, V.; Meador-Woodruff, J.H.; McCullumsmith, R.E. Altered serine/threonine kinase activity in schizophrenia. Brain Res. 2014, 1568, 42–54. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- McGuire, J.L.; Depasquale, E.A.; Funk, A.J.; O’Donnovan, S.M.; Hasselfeld, K.; Marwaha, S.; Hammond, J.H.; Hartounian, V.; Meador-Woodruff, J.H.; Meller, J.; et al. Abnormalities of signal transduction networks in chronic schizophrenia. NPJ Schizophr. 2017, 3, 30. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Moody, C.L.; Funk, A.J.; Devine, E.; Devore Homan, R.C.; Boison, D.; McCullumsmith, R.E.; O’Donovan, S.M. Adenosine Kinase Expression in the Frontal Cortex in Schizophrenia. Schizophr. Bull. 2020, 46, 690–698. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sahay, S.; Henkel, N.D.; Vargas, C.F.; McCullumsmith, R.E.; O’Donovan, S.M. Activity of Protein Kinase A in the Frontal Cortex in Schizophrenia. Brain Sci. 2023, 14, 13. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kokras, N.; Dalla, C. Sex differences in animal models of psychiatric disorders. Br. J. Pharmacol. 2014, 171, 4595–4619. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Labonté, B.; Engmann, O.; Purushothaman, I.; Menard, C.; Wang, J.; Tan, C.; Scarpa, J.R.; Moy, G.; Loh, Y.E.; Cahill, M.; et al. Sex-specific transcriptional signatures in human depression. Nat. Med. 2017, 23, 1102–1111. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Adeva-Andany, M.M.; Pérez-Felpete, N.; Fernández-Fernández, C.; Donapetry-García, C.; Pazos-García, C. Liver glucose metabolism in humans. Biosci. Rep. 2016, 36, e00416. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Judge, A.; Dodd, M.S. Metabolism. Essays Biochem. 2020, 64, 607–647. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nuyen, P.; Leray, V.; Diez, M.; Serisier, S.; Le Bloc’h, J.; Siliart, B.; Dumon, H. Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. 2008, 92, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Alganem, K.; Imami, A.S.; Sahay, S.; Eby, H.; Arvay, T.O.; Abel, M.; Zhang, X.; McIntyre, W.B.; Lee, J.; Au-Yeung, C.; et al. Kaleidoscope: A Bioinformatics Web Application for In Silico Exploration of Omics Data. J. Bioinform. Syst. Biol. 2023, 6, 327–338. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- The Galaxy Community. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 2022, 50, W345–W351. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: A genome-wide analysis. Lancet 2013, 381, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
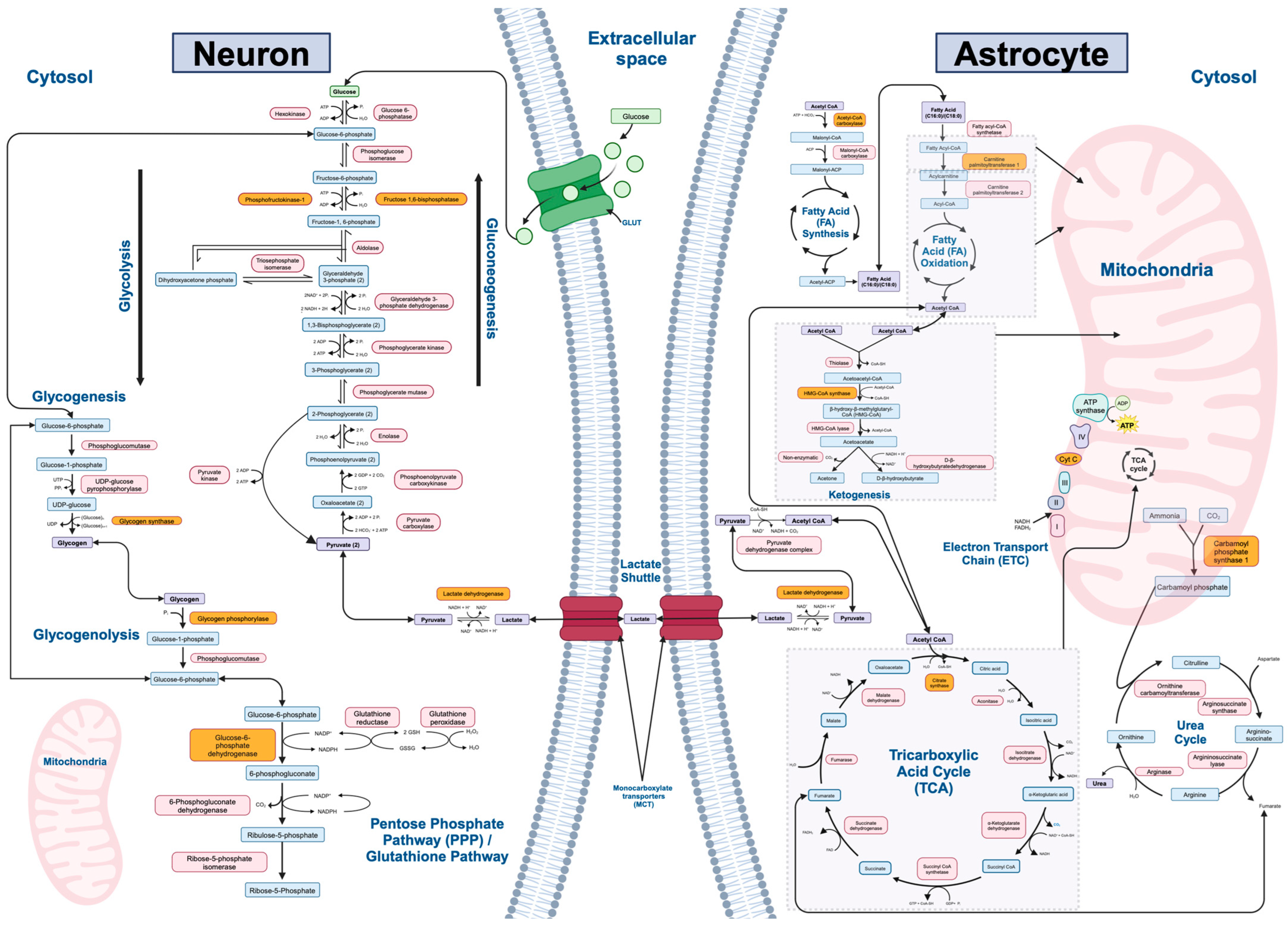
| Metabolic Pathway (n = 12) | Rate-Limiting Enzyme | Final Gene List (Output from Kaleidoscope STRING) | Number of Genes (n = 242) |
|---|---|---|---|
| Gluconeogenesis | Fructose-1,6- bisphosphate | PC, PCK1, PCK2, FBP1, FBP2, G6PC1, G6PC2, G6PC3, PFKFB2 | 9 |
| Glycolysis | Phosphofructokinase | HK1, HK2, HK3, GCK, GPI, PFKL, PFKM, PFKP, ALDOA, ALDOB, ALDOC, TPI1, GAPDH, GAPDHS, PGK1, PGK2, BPGM, PGAM1, PGAM2, PGAM4, ENO1, ENO2, ENO3, ENO4, PKLR, PKM | 27 |
| Lactate Shuttle (Neuron–Astrocyte) | Lactate dehydrogenase | SLC2A3, LDHA, LDHC, LDHD, SLC16A1, SLC16A3, GLS, GLS2, GLUL, SLC2A1, SLC2A10, SLC2A11, SLC2A12, SLC2A14, SLC1A1, SLC1A2, SLC1A3, SLC1A6 | 18 |
| Tricarboxylic Acid (TCA) Cycle | Citrate synthase | CS, ACLY, ACO1, ACO2, IREB2, IDH1, IDH2, IDH3A, IDH3B, IDH3G, OGDH, OGDHL, SUCLA2, SUCLG2, SUCLG1, SDHA, SDHB, SDHC, SDHD, SDHAF1, SDHAF4, ALDH5A1, FH, MDH1B, MDH2, PDHA1, PDHA2, DBT, DLAT, DLD | 30 |
| Electron Transport Chain (ETC) | Cytochrome C oxidase | NDUFA8, NDUFS4, NDUFV3, NDUFA11, NDUFS5, NDUFC1, NDUFC2, NDUFS1, NDUFV2, NDUFV1, NDUFA12, NDUFB5, MT-ND1, MT-ND2, MT-ND3, MT-ND4, MT-ND5, MT-ND6, MT-ND4L, SDHA, SDHB, SDHC, SDHD, SDHAF1, SDHAF2, SDHAF3, SDHAF4, UQCRB, UQCRQ, UQCRC1, UQCRC2, UQCR10, MT-CYB, CYC1, UQCRFS1, UQCRH, UQCR10, UQCR11, COX4I1, COX4I2, COX5A, COX5B, COX6A1, COX6A2, COX6B1, COX6B2, COX6C, COX7A1, COX7A2, COX7B, COX7B2, COX7C, COX8A, COX8C, MT-CO1, MT-CO2, MT-CO3, COQ4, COQ7, COQ10A, COQ10B, CYCS, ATP5F1A, ATP5F1B, ATP5F1C, ATP5F1D, ATP5F1E, MT-ATP6, MT-ATP8 | 69 |
| Fatty Acid Synthesis | Acetyl-CoA carboxylase | ACACA, ACACB, MCAT, FASN, OXSM, DECR1, HADH, ELOVL1, ELOVL3, ELOVL6, OLAH, PPT1, PPT2 | 13 |
| Fatty Acid Oxidation | Carnitine palmitoyltransferase I | SLC27A2, ACSBG2, ACSBG1, ECI1, ECHS1, HADHA, HSD17B10, HADH, HADHB, ACAA2, SCP2, VLCAD, SCAD, MCAD, LCAD, CPT1C, CPT2 | 17 |
| Ketogenesis | HMG-CoA synthase | ACAT1, ACAA1, ACAA2, HMGCS1, HMGCS2, HMGCL, HMGCLL1, BDH2, BDH1 | 9 |
| Glycogenesis | Glycogen synthase | ADPGK, GCKR, HK1, HK2, HK3, PGM1, PGM2, UGP2, GYS1, GYS2, GSK3A, GSK3B, GBE1 | 13 |
| Glycogenolysis | Glycogen phosphorylase | PYGL, PYGM, PYGB, AGL, PGM1, PGM2, G6PC1, G6PC2, G6PC3 | 9 |
| Urea Cycle | Carbamoyl phosphate synthetase I | OTC, OAT, CPS1, CAD, ASS1, ASL, ARG1, ARG2, AGMAT, NAGS, ACY1 | 11 |
| Pentose Phosphate/ Glutathione Pathways | Glucose-6-phosphate dehydrogenase | HK1, HK2, HK3, G6PD, PGD, TKT, TKTL1, TKTL2, TALDO1, GPI, GCLC, GSS, GSR, SOD1, RPE, RPIA, RGN | 17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahay, S.; Pulvender, P.; Rami Reddy, M.V.S.R.; McCullumsmith, R.E.; O’Donovan, S.M. Metabolic Insights into Neuropsychiatric Illnesses and Ketogenic Therapies: A Transcriptomic View. Int. J. Mol. Sci. 2024, 25, 8266. https://doi.org/10.3390/ijms25158266
Sahay S, Pulvender P, Rami Reddy MVSR, McCullumsmith RE, O’Donovan SM. Metabolic Insights into Neuropsychiatric Illnesses and Ketogenic Therapies: A Transcriptomic View. International Journal of Molecular Sciences. 2024; 25(15):8266. https://doi.org/10.3390/ijms25158266
Chicago/Turabian StyleSahay, Smita, Priyanka Pulvender, Madhu Vishnu Sankar Reddy Rami Reddy, Robert E. McCullumsmith, and Sinead M. O’Donovan. 2024. "Metabolic Insights into Neuropsychiatric Illnesses and Ketogenic Therapies: A Transcriptomic View" International Journal of Molecular Sciences 25, no. 15: 8266. https://doi.org/10.3390/ijms25158266
APA StyleSahay, S., Pulvender, P., Rami Reddy, M. V. S. R., McCullumsmith, R. E., & O’Donovan, S. M. (2024). Metabolic Insights into Neuropsychiatric Illnesses and Ketogenic Therapies: A Transcriptomic View. International Journal of Molecular Sciences, 25(15), 8266. https://doi.org/10.3390/ijms25158266