
Sphingosine 1-Phosphate Stimulates ER to Golgi Ceramide Traffic to Promote Survival in T98G Glioma Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
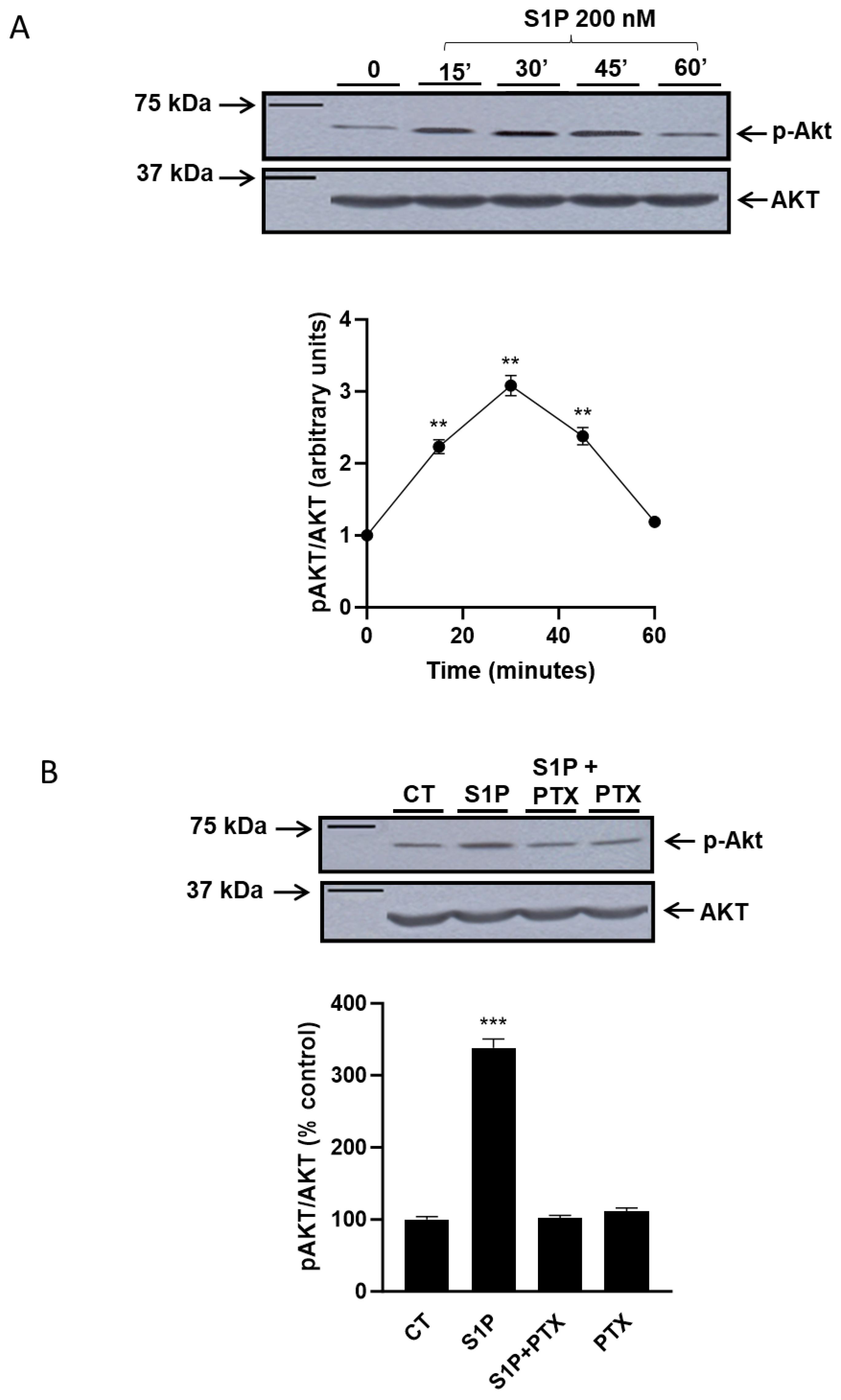
2.1. Role of Sphingosine-1-Phosphate in the Regulation of the PI3K/Akt Pathway
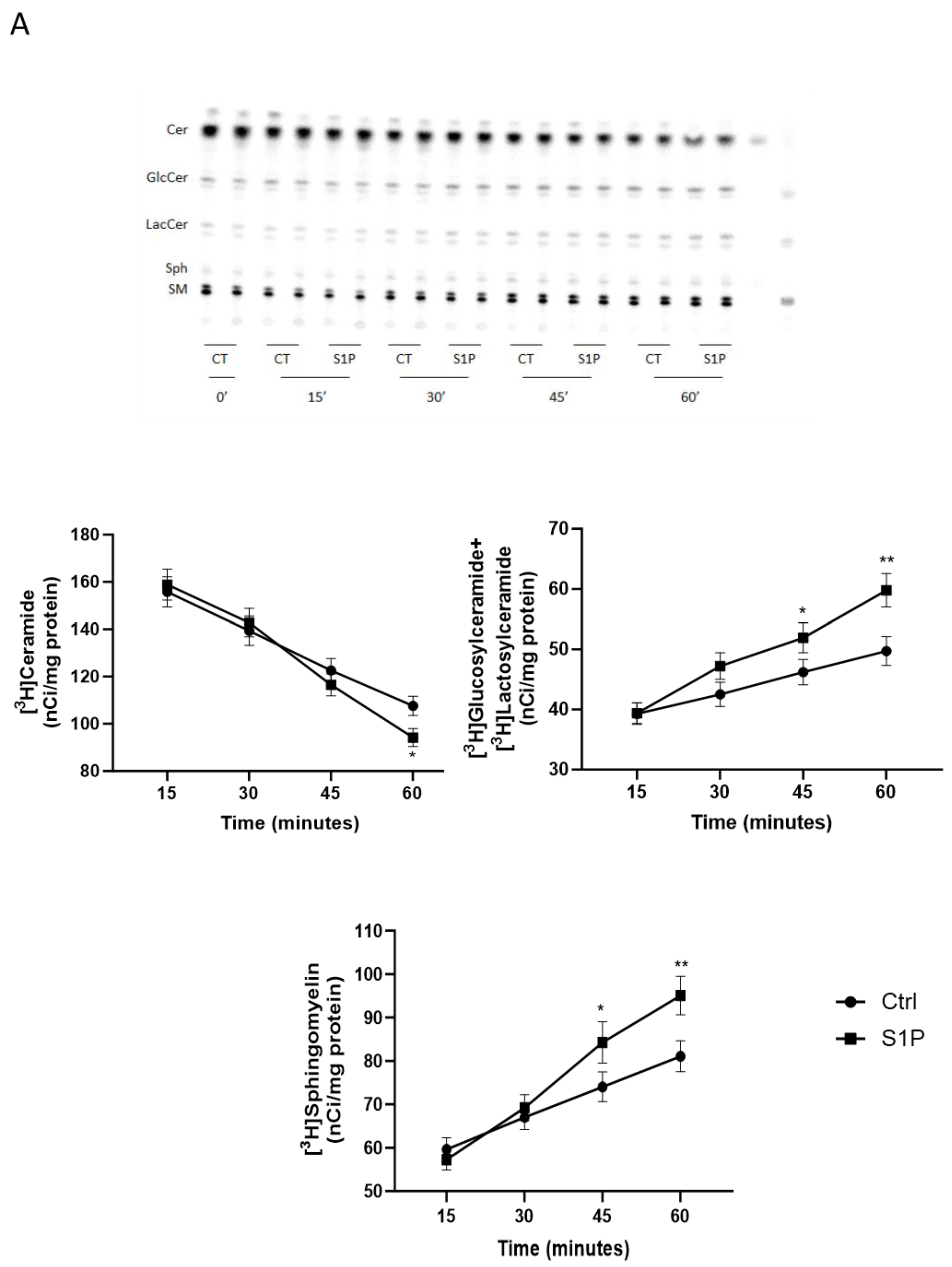
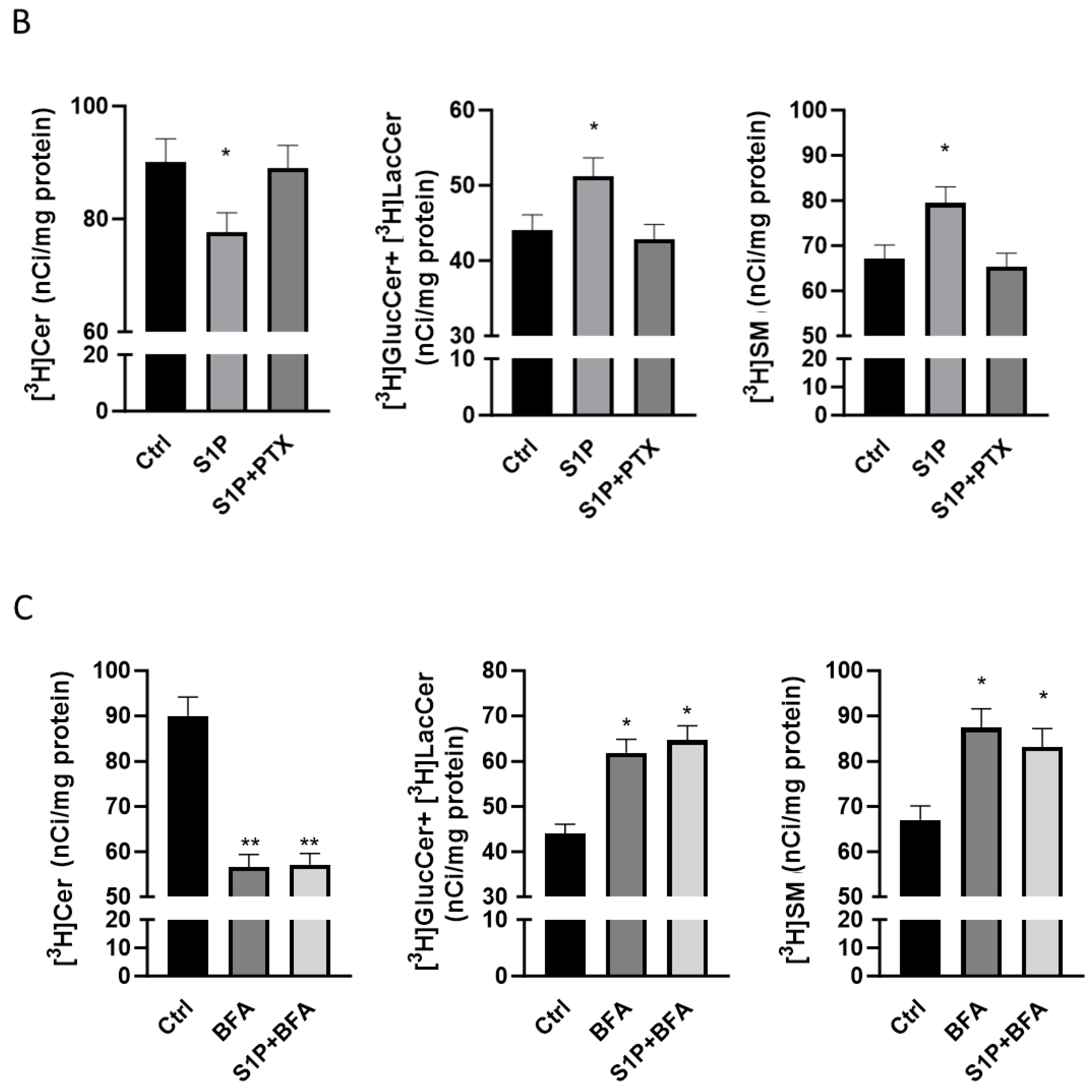
2.2. Role of S1P on Cer Utilization for Complex Sphingolipids Synthesis
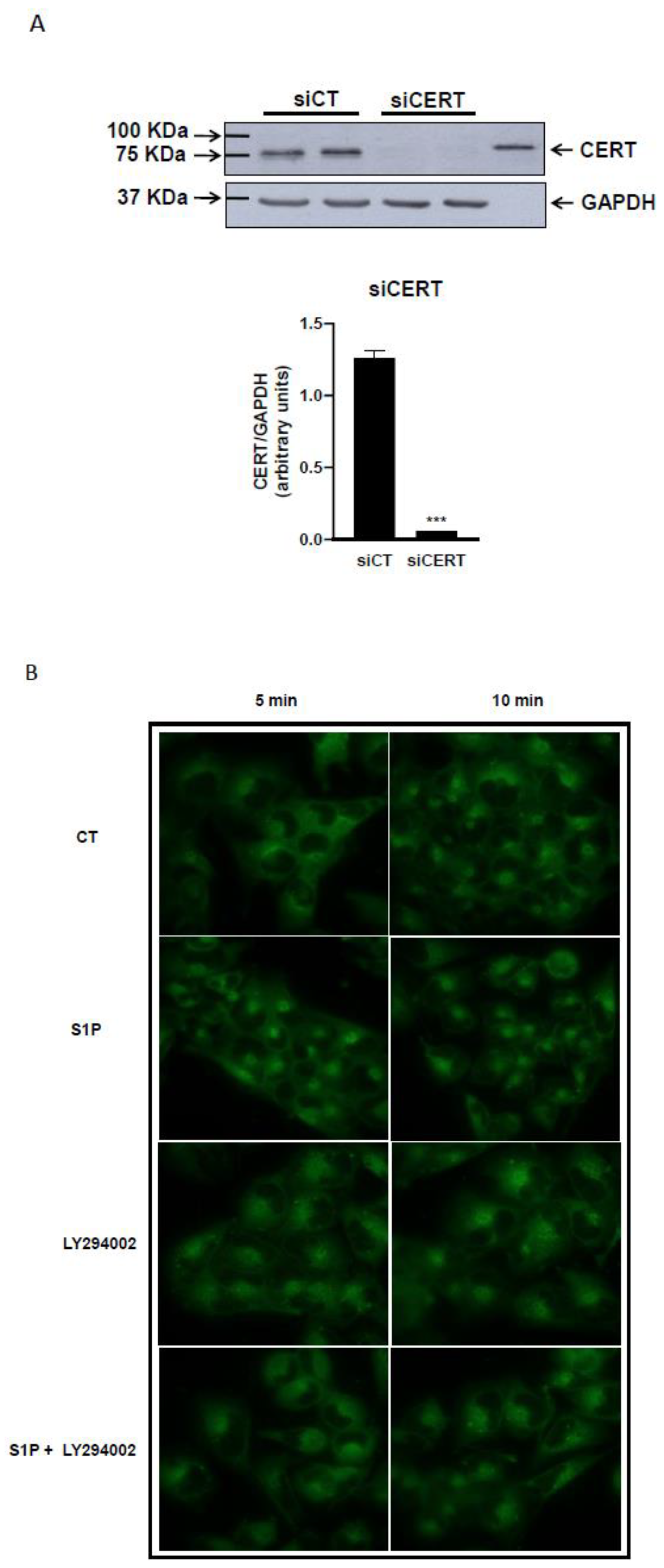
2.3. Role of S1P on Cer Transport in the ER-Golgi District
2.4. Role of Cer, Sphingosine-1-Phosphate and PI3K/Akt Pathway in the Etoposide-Induced Toxicity in T98G Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Treatment of Cultured T98G Cells
4.4. Immunoblotting
4.5. RNA Interference
4.6. [3H]Sphingosine Metabolism
4.7. [14C]Palmitate Metabolism
4.8. Analysis of the Intracellular Distribution of Fluorescent Ceramides
4.9. MTT Assays
4.10. Other Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Cer | Ceramide |
| ER | Endoplasmic reticulum |
| ETO | Etoposide |
| GBM | Glioblastoma multiforme |
| GlcCer | Glucosylceramide |
| GSL | Glycosphingolipids |
| PTX | Pertuxin Toxin |
| SM | Sphingomyelin |
| Sph | Sphingosine |
| S1P | Sphingosine-1-phosphate |
| PI3K | phosphatidylinositol 3-phosphate kinase |
| CERT | ceramide transfer protein |
| BFA | Brefeldin A |
| Fb1 | Fumonisin B1 |
| MYR | Myriocin |
References
- Davis, F.G.; McCarthy, B.J. Current Epidemiological Trends and Surveillance Issues in Brain Tumors. Expert Rev. Anticancer Ther. 2001, 1, 395–401. [Google Scholar] [CrossRef]
- Yalamarty, S.S.K.; Filipczak, N.; Li, X.; Subhan, M.A.; Parveen, F.; Ataide, J.A.; Rajmalani, B.A.; Torchilin, V.P. Mechanisms of Resistance and Current Treatment Options for Glioblastoma Multiforme (GBM). Cancers 2023, 15, 2116. [Google Scholar] [CrossRef]
- Rich, J.N.; Bigner, D.D. Development of novel targeted therapies in the treatment of malignant glioma. Nat. Rev. Drug Discov. 2004, 3, 430–446. [Google Scholar] [CrossRef]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Sh, U.S. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar]
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. [Google Scholar] [CrossRef]
- Truman, J.P.; Garcia-Barros, M.; Obeid, L.M.; Hannun, Y.A. Evolving concepts in cancer therapy through targeting sphingolipid metabolism. Biochim. Biophys. Acta 2014, 1841, 1174–1188. [Google Scholar] [CrossRef]
- Li, R.Z.; Wang, X.R.; Wang, J.; Xie, C.; Wang, X.X.; Pan, H.D.; Meng, W.Y.; Liang, T.L.; Li, J.X.; Yan, P.Y.; et al. The key role of sphingolipid metabolism in cancer: New therapeutic targets, diagnostic and prognostic values, and anti-tumor immunotherapy resistance. Front. Oncol. 2022, 12, 941643. [Google Scholar] [CrossRef]
- Kumagai, K.; Yasuda, S.; Okemoto, K.; Nishijima, M.; Kobayashi, S.; Hanada, K. CERT mediates intermembrane transfer of various molecular species of ceramides. J. Biol. Chem. 2005, 280, 6488–6495. [Google Scholar] [CrossRef]
- Turpin-Nolan, S.M.; Brüning, J.C. The role of ceramides in metabolic disorders: When size and localization matters. Nat. Rev. Endocrinol. 2020, 16, 224–233. [Google Scholar] [CrossRef]
- Hanada, K.; Kumagai, K.; Tomishige, N.; Yamaji, T. CERT-mediated trafficking of ceramide. Biochim. Biophys. Acta 2009, 1791, 684–691. [Google Scholar] [CrossRef]
- Giussani, P.; Colleoni, T.; Brioschi, L.; Bassi, R.; Hanada, K.; Tettamanti, G.; Riboni, L.; Viani, P. Ceramide traffic in C6 glioma cells: Evidence for CERT-dependent and independent transport from ER to the Golgi apparatus. Biochim. Biophys. Acta 2008, 1781, 40–51. [Google Scholar] [CrossRef]
- Hanada, K.; Kumagai, K.; Yasuda, S.; Miura, Y.; Kawano, M.; Fukasawa, M.; Nishijima, M. Molecular machinery for non-vesicular trafficking of ceramide. Nature 2003, 426, 803–809. [Google Scholar] [CrossRef]
- Riboni, L.; Giussani, P.; Viani, P. Sphingolipid transport. Adv. Exp. Med. Biol. 2010, 688, 24–45. [Google Scholar] [CrossRef]
- Kumagai, K.; Hanada, K. Structure, functions and regulation of CERT, a lipid-transfer protein for the delivery of ceramide at the ER-Golgi membrane contact sites. FEBS Lett. 2019, 593, 2366–2377. [Google Scholar] [CrossRef]
- Chung, L.H.; Liu, D.; Liu, X.T.; Qi, Y. Ceramide Transfer Protein (CERT): An Overlooked Molecular Player in Cancer. Int. J. Mol. Sci. 2021, 22, 13184. [Google Scholar] [CrossRef]
- Viani, P.; Giussani, P.; Brioschi, L.; Bassi, R.; Anelli, V.; Tettamanti, G.; Riboni, L. Ceramide in nitric oxide inhibition of glioma cell growth—Evidence for the involvement of ceramide traffic. J. Biol. Chem. 2003, 278, 9592–9601. [Google Scholar] [CrossRef]
- Gouaze-Andersson, V.; Cabot, M.C. Glycosphingolipids and drug resistance. Biochim. Biophys. Acta 2006, 1758, 2096–2103. [Google Scholar] [CrossRef]
- Maurer, B.J.; Metelitsa, L.S.; Seeger, R.C.; Cabot, M.C.; Reynolds, C.P. Increase of ceramide and induction of mixed apoptosis/necrosis by N-(4-hydroxyphenyl)- retinamide in neuroblastoma cell lines. J. Natl. Cancer Inst. 1999, 91, 1138–1146. [Google Scholar] [CrossRef]
- Moro, K.; Nagahashi, M.; Gabriel, E.; Takabe, K.; Wakai, T. Clinical application of ceramide in cancer treatment. Breast Cancer 2019, 26, 407–415. [Google Scholar] [CrossRef]
- Dimanche-Boitrel, M.T.; Rebillard, A.; Gulbins, E. Ceramide in chemotherapy of tumors. Recent Pat. Anti-Cancer Drug Discov. 2011, 6, 284–293. [Google Scholar] [CrossRef]
- Wajapeyee, N.; Beamon, T.C.; Gupta, R. Roles and therapeutic targeting of ceramide metabolism in cancer. Mol. Metab. 2024, 83, 101936. [Google Scholar] [CrossRef]
- Riboni, L.; Campanella, R.; Bassi, R.; Villani, R.; Gaini, S.M.; Martinelli-Boneschi, F.; Viani, P.; Tettamanti, G. Ceramide levels are inversely associated with malignant progression of human glial tumors. Glia 2002, 39, 105–113. [Google Scholar] [CrossRef]
- Yu, L.; He, L.; Gan, B.; Ti, R.; Xiao, Q.; Hu, H.; Zhu, L.; Wang, S.; Ren, R. Structural insights into sphingosine-1-phosphate receptor activation. Proc. Natl. Acad. Sci. USA 2022, 119, e2117716119. [Google Scholar] [CrossRef]
- Blaho, V.A.; Hla, T. An update on the biology of sphingosine 1-phosphate receptors. J. Lipid Res. 2014, 55, 1596–1608. [Google Scholar] [CrossRef]
- Wang, Z.; Wen, L.; Zhu, F.; Wang, Y.; Xie, Q.; Chen, Z.; Li, Y. Overexpression of ceramide synthase 1 increases C18-ceramide and leads to lethal autophagy in human glioma. Oncotarget 2017, 8, 104022–104036. [Google Scholar] [CrossRef]
- Le Stunff, H.; Milstien, S.; Spiegel, S. Generation and metabolism of bioactive sphingosine-1-phosphate. J. Cell. Biochem. 2004, 92, 882–899. [Google Scholar] [CrossRef]
- Strub, G.M.; Maceyka, M.; Hait, N.C.; Milstien, S.; Spiegel, S. Extracellular and intracellular actions of sphingosine-1-phosphate. Adv. Exp. Med. Biol. 2010, 688, 141–155. [Google Scholar]
- Van Brocklyn, J.R.; Young, N.; Roof, R. Sphingosine-1-phosphate stimulates motility and invasiveness of human glioblastoma multiforme cells. Cancer Lett. 2003, 199, 53–60. [Google Scholar] [CrossRef]
- Van Brocklyn, J.; Letterle, C.; Snyder, P.; Prior, T. Sphingosine-1-phosphate stimulates human glioma cell proliferation through Gi-coupled receptors: Role of ERK MAP kinase and phosphatidylinositol 3-kinase beta. Cancer Lett. 2002, 181, 195–204. [Google Scholar] [CrossRef]
- Mahajan-Thakur, S.; Bien-Moller, S.; Marx, S.; Schroeder, H.; Rauch, B.H. Sphingosine 1-phosphate (S1P) signaling in glioblastoma multiforme-A systematic review. Int. J. Mol. Sci. 2017, 18, 2448. [Google Scholar] [CrossRef]
- Wang, P.; Yuan, Y.; Lin, W.; Zhong, H.; Xu, K.; Qi, X. Roles of sphingosine-1-phosphate signaling in cancer. Cancer Cell Int. 2019, 19, 295. [Google Scholar] [CrossRef]
- Bassi, R.; Brambilla, S.; Tringali, C.; Giussani, P. Extracellular Sphingosine-1-Phosphate Downstream of EGFR Increases Human Glioblastoma Cell Survival. Int. J. Mol. Sci. 2021, 22, 6824. [Google Scholar] [CrossRef]
- Alkafaas, S.S.; Elsalahaty, M.I.; Ismail, D.F.; Radwan, M.A.; Elkafas, S.S.; Loutfy, S.A.; Elshazli, R.M.; Baazaoui, N.; Ahmed, A.E.; Hafez, W.; et al. The emerging roles of sphingosine 1-phosphate and SphK1 in cancer resistance: A promising therapeutic target. Cancer Cell Int. 2024, 24, 89. [Google Scholar] [CrossRef]
- Johnson, K.R.; Johnson, K.Y.; Becker, K.P.; Bielawski, J.; Mao, C.; Obeid, L.M. Role of human sphingosine-1-phosphate phosphatase 1 in the regulation of intra- and extracellular sphingosine-1-phosphate levels and cell viability. J. Biol. Chem. 2003, 278, 34541–34547. [Google Scholar] [CrossRef]
- Senchenkov, A.; Litvak, D.A.; Cabot, M.C. Targeting ceramide metabolism—A strategy for overcoming drug resistance. J. Natl. Cancer Inst. 2001, 93, 347–357. [Google Scholar] [CrossRef]
- Brachtendorf, S.; El-Hindi, K.; Grösch, S. WITHDRAWN: Ceramide synthases in cancer therapy and chemoresistance. Prog. Lipid Res. 2019, 74, 100992. [Google Scholar] [CrossRef]
- Bassi, R.; Cas, M.D.; Tringali, C.; Compostella, F.; Paroni, R.; Giussani, P. Ceramide Is Involved in Temozolomide Resistance in Human Glioblastoma U87MG Overexpressing EGFR. Int. J. Mol. Sci. 2023, 24, 15394. [Google Scholar] [CrossRef]
- Salazar, M.; Carracedo, A.; Salanueva, I.J.; Hernandez-Tiedra, S.; Lorente, M.; Egia, A.; Vazquez, P.; Blazquez, C.; Torres, S.; Garcia, S.; et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Investig. 2009, 119, 1359–1372. [Google Scholar] [CrossRef]
- Giussani, P.; Brioschi, L.; Bassi, R.; Riboni, L.; Viani, P. Phosphatidylinositol 3-Kinase/AKT Pathway Regulates the Endoplasmic Reticulum to Golgi Traffic of Ceramide in Glioma Cells A link between lipid signaling pathways involved in the control of cell survival. J. Biol. Chem. 2009, 284, 5088–5096. [Google Scholar] [CrossRef]
- Riboni, L.; Viani, P.; Bassi, R.; Giussani, P.; Tettamanti, G. Cultured granule cells and astrocytes from cerebellum differ in metabolizing sphingosine. J. Neurochem. 2000, 75, 503–510. [Google Scholar] [CrossRef]
- Pagano, R.E.; Martin, O.C.; Kang, H.C.; Haugland, R.P. A novel fluorescent ceramide analogue for studying membrane traffic in animal cells: Accumulation at the Golgi apparatus results in altered spectral properties of the sphingolipid precursor. J. Cell Biol. 1991, 113, 1267–1279. [Google Scholar] [CrossRef]
- Perry, D.K.; Carton, J.; Shah, A.K.; Meredith, F.; Uhlinger, D.J.; Hannun, Y.A. Serine palmitoyltransferase regulates de novo ceramide generation during etoposide-induced apoptosis. J. Biol. Chem. 2000, 275, 9078–9084. [Google Scholar] [CrossRef]
- Van Brocklyn, J.R. Sphingolipid signaling pathways as potential therapeutic targets in gliomas. Mini Rev. Med. Chem. 2007, 7, 984–990. [Google Scholar] [CrossRef]
- Gillard, B.K.; Harrell, R.G.; Marcus, D.M. Pathways of glycosphingolipid biosynthesis in SW13 cells in the presence and absence of vimentin intermediate filaments. Glycobiology 1996, 6, 33–42. [Google Scholar] [CrossRef]
- Tettamanti, G.; Bassi, R.; Viani, P.; Riboni, L. Salvage pathways in glycosphingolipid metabolism. Biochimie 2003, 85, 423–437. [Google Scholar] [CrossRef]
- Young, N.; Van Brocklyn, J.R. Roles of sphingosine-1-phosphate (S1P) receptors in malignant behavior of glioma cells. Differential effects of S1P2 on cell migration and invasiveness. Exp. Cell Res. 2007, 313, 1615–1627. [Google Scholar] [CrossRef]
- Van Brocklyn, J.R.; Jackson, C.A.; Pearl, D.K.; Kotur, M.S.; Snyder, P.J.; Prior, T.W. Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: Roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J. Neuropathol. Exp. Neurol. 2005, 64, 695–705. [Google Scholar] [CrossRef]
- Guan, H.; Song, L.; Cai, J.; Huang, Y.; Wu, J.; Yuan, J.; Li, J.; Li, M. Sphingosine kinase 1 regulates the Akt/FOXO3a/Bim pathway and contributes to apoptosis resistance in glioma cells. PLoS ONE 2011, 6, e19946. [Google Scholar] [CrossRef]
- Radeff-Huang, J.; Seasholtz, T.M.; Chang, J.W.; Smith, J.M.; Walsh, C.T.; Brown, J.H. Tumor necrosis factor-alpha-stimulated cell proliferation is mediated through sphingosine kinase-dependent Akt activation and cyclin D expression. J. Biol. Chem. 2007, 282, 863–870. [Google Scholar] [CrossRef]
- Osawa, Y.; Banno, Y.; Nagaki, M.; Brenner, D.A.; Naiki, T.; Nozawa, Y.; Nakashima, S.; Moriwaki, H. TNF-alpha-induced sphingosine 1-phosphate inhibits apoptosis through a phosphatidylinositol 3-kinase/Akt pathway in human hepatocytes. J. Immunol. 2001, 167, 173–180. [Google Scholar] [CrossRef]
- Gjoni, E.; Brioschi, L.; Cinque, A.; Coant, N.; Islam, M.N.; Ng, C.K.; Verderio, C.; Magnan, C.; Riboni, L.; Viani, P.; et al. Glucolipotoxicity Impairs Ceramide Flow from the Endoplasmic Reticulum to the Golgi Apparatus in INS-1 beta-Cells. PLoS ONE 2014, 9, e110875. [Google Scholar] [CrossRef]
- Giussani, P.; Maceyka, M.; Le Stunff, H.; Mikami, A.; Lepine, S.; Wang, E.; Kelly, S.; Merrill, A.H., Jr.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate phosphohydrolase regulates endoplasmic reticulum-to-golgi trafficking of ceramide. Mol. Cell. Biol. 2006, 26, 5055–5069. [Google Scholar] [CrossRef]
- Ermoian, R.P.; Furniss, C.S.; Lamborn, K.R.; Basila, D.; Berger, M.S.; Gottschalk, A.R.; Nicholas, M.K.; Stokoe, D.; Haas-Kogan, D.A. Dysregulation of PTEN and protein kinase B is associated with glioma histology and patient survival. Clin. Cancer Res. 2002, 8, 1100–1106. [Google Scholar]
- Chakravarti, A.; Zhai, G.; Suzuki, Y.; Sarkesh, S.; Black, P.M.; Muzikansky, A.; Loeffler, J.S. The prognostic significance of phosphatidylinositol 3-kinase pathway activation in human gliomas. J. Clin. Oncol. 2004, 22, 1926–1933. [Google Scholar] [CrossRef]
- Swanton, C.; Marani, M.; Pardo, O.; Warne, P.H.; Kelly, G.; Sahai, E.; Elustondo, F.; Chang, J.; Temple, J.; Ahmed, A.A.; et al. Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer Cell 2007, 11, 498–512. [Google Scholar] [CrossRef]
- Kolesnick, R.; Altieri, D.; Fuks, Z. A CERTain role for ceramide in taxane-induced cell death. Cancer Cell 2007, 11, 473–475. [Google Scholar] [CrossRef]
- Oskouian, B.; Saba, J.D. Cancer treatment strategies targeting sphingolipid metabolism. Adv. Exp. Med. Biol. 2010, 688, 185–205. [Google Scholar]
- Ponnusamy, S.; Meyers-Needham, M.; Senkal, C.E.; Saddoughi, S.A.; Sentelle, D.; Selvam, S.P.; Salas, A.; Ogretmen, B. Sphingolipids and cancer: Ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance. Future Oncol. 2010, 6, 1603–1624. [Google Scholar] [CrossRef]
- Lou, H.O.; Clausen, J.; Bierring, F. Phospholipids and glycolipids of tumours in the central nervous system. J. Neurochem. 1965, 12, 619–627. [Google Scholar]
- Giussani, P.; Bassi, R.; Anelli, V.; Brioschi, L.; De Zen, F.; Riccitelli, E.; Caroli, M.; Campanella, R.; Gaini, S.M.; Viani, P.; et al. Glucosylceramide synthase protects glioblastoma cells against autophagic and apoptotic death induced by temozolomide and Paclitaxel. Cancer Investig. 2012, 30, 27–37. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Exogenous and intracellularly generated sphingosine 1-phosphate can regulate cellular processes by divergent pathways. Biochem. Soc. Trans. 2003, 31, 1216–1219. [Google Scholar] [CrossRef]
- Morales, A.; Lee, H.; Goni, F.M.; Kolesnick, R.; Fernandez-Checa, J.C. Sphingolipids and cell death. Apoptosis 2007, 12, 923–939. [Google Scholar] [CrossRef]
- Riboni, L.; Viani, P.; Tettamanti, G. Estimating sphingolipid metabolism and trafficking in cultured cells using radiolabeled compounds. Methods Enzymol. 2000, 311, 656–682. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giussani, P.; Brioschi, L.; Gjoni, E.; Riccitelli, E.; Viani, P. Sphingosine 1-Phosphate Stimulates ER to Golgi Ceramide Traffic to Promote Survival in T98G Glioma Cells. Int. J. Mol. Sci. 2024, 25, 8270. https://doi.org/10.3390/ijms25158270
Giussani P, Brioschi L, Gjoni E, Riccitelli E, Viani P. Sphingosine 1-Phosphate Stimulates ER to Golgi Ceramide Traffic to Promote Survival in T98G Glioma Cells. International Journal of Molecular Sciences. 2024; 25(15):8270. https://doi.org/10.3390/ijms25158270
Chicago/Turabian StyleGiussani, Paola, Loredana Brioschi, Enida Gjoni, Elena Riccitelli, and Paola Viani. 2024. "Sphingosine 1-Phosphate Stimulates ER to Golgi Ceramide Traffic to Promote Survival in T98G Glioma Cells" International Journal of Molecular Sciences 25, no. 15: 8270. https://doi.org/10.3390/ijms25158270