
Identification of Variants of Uncertain Significance in the Genes Associated with Thoracic Aortic Disease in Russian Patients with Nonsyndromic Sporadic Subtypes of the Disorder
 , , , , , ,
, , , , , ,
Abstract
:
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Patient Characteristics
4.2. Virtual Gene Panel Analysis
4.3. Clinical Exome Sequencing
4.4. Sanger Sequencing
4.5. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
- GWAS catalog:
- https://www.ebi.ac.uk/gwas/search?query=thoracic%20aortic%20aneurysm, accessed on 15 January 2023
- https://www.ebi.ac.uk/gwas/genes/MYH11, accessed on 20 June 2024
- https://www.ebi.ac.uk/gwas/genes/FBN1, accessed on 20 June 2024
- ClinGen: https://clinicalgenome.org/, accessed on 15 January 2023
- VarSome The Human Genomics Community: https://varsome.com/, accessed on 20 June 2024
- PrimerQuest Tool: https://eu.idtdna.com/pages, accessed on 8 April 2023
- Primer Design and Search Tool: http://bisearch.enzim.hu/, accessed on 8 April 2023
- NCBI:
- https://www.ncbi.nlm.nih.gov/snp/rs1317299348, accessed on 20 June 2024
- https://www.ncbi.nlm.nih.gov/snp/rs371344739, accessed on 20 June 2024
- CADD: Combined Annotation Dependent Depletion: https://cadd.gs.washington.edu/snv, accessed on 20 June 2024
- gnomAD browser, v. 4.1.0: https://gnomad.broadinstitute.org, accessed on 20 June 2024
- RUSeq Browser: http://ruseq.ru/, accessed on 20 June 2024
- PVR package in R: https://cran.r-project.org/web/packages/pwr/, accessed on 1 February 2023
References
- Cecchi, A.C.; Drake, M.; Campos, C.; Howitt, J.; Medina, J.; Damrauer, S.M.; Shalhub, S.; Milewicz, D.M. Aortic Dissection Collaborative. Current state and future directions of genomic medicine in aortic dissection: A path to prevention and personalized care. Semin. Vasc. Surg. 2022, 35, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Milewicz, D.M.; Guo, D.; Hostetler, E.; Marin, I.; Pinard, A.C.; Cecchi, A.C. Update on the genetic risk for thoracic aortic aneurysms and acute aortic dissections: Implications for clinical care. J. Cardiovasc. Surg. 2021, 62, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, P.; Hanna, N.; Benarroch, L.; Aubart, M.; Bal, L.; Bouvagnet, P.; Busa, T.; Dulac, Y.; Dupuis-Girod, S.; Edouard, T.; et al. Genetic diversity and pathogenic variants as possible predictors of severity in a French sample of nonsyndromic heritable thoracic aortic aneurysms and dissections (nshTAAD). Genet. Med. 2019, 21, 2015–2024. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Cecchi, A.C.; Prakash, S.K.; Milewicz, D.M. Risk factors for thoracic aortic dissection. Genes 2022, 13, 1814. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.K.; LeMaire, S.A.; Guo, D.C.; Russell, L.; Regalado, E.S.; Golabbakhsh, H.; Johnson, R.J.; Safi, H.J.; Estrera, A.L.; Coselli, J.S.; et al. Rare copy number variants disrupt genes regulating vascular smooth muscle cell adhesion and contractility in sporadic thoracic aortic aneurysms and dissections. Am. J. Hum. Genet. 2010, 87, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.; Kuang, S.Q.; GenTAC Registry Investigators; Regalado, E.; Guo, D.; Milewicz, D. Recurrent Rare Genomic Copy Number Variants and Bicuspid Aortic Valve Are Enriched in Early Onset Thoracic Aortic Aneurysms and Dissections. PLoS ONE 2016, 11, e0153543. [Google Scholar] [CrossRef] [PubMed]
- LeMaire, S.A.; McDonald, M.L.; Guo, D.C.; Russell, L.; Miller, C.C.; Johnson, R.J.; Bekheirnia, M.R.; Franco, L.M.; Nguyen, M.; Pyeritz, R.E.; et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat. Genet. 2011, 43, 996–1000. [Google Scholar] [CrossRef]
- Meester, J.A.N.; Hebert, A.; Loeys, B.L. Structural genomic variants in thoracic aortic disease. Curr. Opin. Cardiol. 2023, 38, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.C.; Hostetler, E.M.; Fan, Y.; Kulmacz, R.J.; Zhang, D.; GenTAC Investigators; Nickerson, D.A.; Leal, S.M.; LeMaire, S.A.; Regalado, E.S.; et al. Heritable Thoracic Aortic Disease Genes in Sporadic Aortic Dissection. J. Am. Coll. Cardiol. 2017, 70, 2728–2730. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhuang, X.; Chen, B.; Wen, J.; Peng, F.; Liu, X.; Wei, M. 99-Case Study of Sporadic Aortic Dissection by Whole Exome Sequencing Indicated Novel Disease-Associated Genes and Variants in Chinese Population. Biomed. Res. Int. 2020, 2020, 7857043. [Google Scholar] [CrossRef]
- Chen, Z.R.; Bao, M.H.; Wang, X.Y.; Yang, Y.M.; Huang, B.; Han, Z.L.; Cai, J.; Fan, X.H. Genetic variants in Chinese patients with sporadic Stanford type A aortic dissection. J. Thorac. Dis. 2021, 13, 4008–4022. [Google Scholar] [CrossRef] [PubMed]
- Brownstein, A.J.; Kostiuk, V.; Ziganshin, B.A.; Zafar, M.A.; Kuivaniemi, H.; Body, S.C.; Bale, A.E.; Elefteriades, J.A. Genes Associated with Thoracic Aortic Aneurysm and Dissection: 2018 Update and Clinical Implications. Aorta 2018, 6, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Renard, M.; Francis, C.; Ghosh, R.; Scott, A.F.; Witmer, P.D.; Adès, L.C.; Andelfinger, G.U.; Arnaud, P.; Boileau, C.; Callewaert, B.L.; et al. Clinical Validity of Genes for Heritable Thoracic Aortic Aneurysm and Dissection. J. Am. Coll. Cardiol. 2018, 72, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Ostberg, N.P.; Zafar, M.A.; Ziganshin, B.A.; Elefteriades, J.A. The Genetics of Thoracic Aortic Aneurysms and Dissection: A Clinical Perspective. Biomolecules. 2020, 10, 182. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Du, P.; Pei, Y.; Yang, J.; Li, S.; Chang, S.; Sun, H.; He, X.; Dong, J.; Zhou, J.; et al. Whole-Exome Sequencing Identified Genes Responsible for Thoracic Aortic Aneurysms and Dissections in three Chinese Families. Front. Genet. 2022, 13, 910932. [Google Scholar] [CrossRef]
- Krywanczyk, A.; Rodriguez, E.R.; Tan, C.D.; Gilson, T. Thoracic aortic aneurysm and dissection: Review and recommendations for evaluation. Am. J. Forensic Med. Pathol. 2023, 44, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gao, S.; Han, Y.; Song, L.; Kong, Y.; Jiao, Y.; Huang, S.; Du, J.; Li, Y. Variants of Focal Adhesion Scaffold Genes Cause Thoracic Aortic Aneurysm. Circ. Res. 2021, 128, 8–23. [Google Scholar] [CrossRef] [PubMed]
- De Backer, J.; Bondue, A.; Budts, W.; Evangelista, A.; Gallego, P.; Jondeau, G.; Loeys, B.; Peña, M.L.; Teixido-Tura, G.; van de Laar, I.; et al. Genetic counselling and testing in adults with congenital heart disease: A consensus document of the ESC Working Group of Grown-Up Congenital Heart Disease, the ESC Working Group on Aorta and Peripheral Vascular Disease and the European Society of Human Genetics. Eur. J. Prev. Cardiol. 2020, 27, 1423–1435. [Google Scholar] [PubMed]
- Isselbacher, E.M.; Preventza, O.; Hamilton Black, J.; Augoustides, J.G.; Beck, A.W.; Bolen, M.A.; Braverman, A.C.; Bray, B.E.; Brown-Zimmerman, M.M.; Chen, E.P.; et al. 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation 2022, 146, e334–e482. [Google Scholar] [CrossRef] [PubMed]
- Mariscalco, G.; Debiec, R.; Elefteriades, J.A.; Samani, N.J.; Murphy, G.J. Systematic review of studies that have evaluated screening tests in relatives of patients affected by nonsyndromic thoracic aortic disease. J. Am. Heart Assoc. 2018, 7, e009302. [Google Scholar] [CrossRef] [PubMed]
- Weerakkody, R.; Ross, D.; Parry, D.A.; Ziganshin, B.; Vandrovcova, J.; Gampawar, P.; Abdullah, A.; Biggs, J.; Dumfarth, J.; Ibrahim, Y.; et al. Targeted genetic analysis in a large cohort of familial and sporadic cases of aneurysm or dissection of the thoracic aorta. Genet. Med. 2018, 20, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Guo, J.; Huang, L.; Wu, Q.; Yin, K.; Wang, L.; Zhang, T.; Quan, L.; Zhao, Q.; Cheng, J. Genetic diagnosis of acute aortic dissection in South China Han population using next-generation sequencing. Int. J. Legal Med. 2018, 132, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Xiong, J.; Lai, Z.; Zhong, Y.; Tian, C.; Du, Z.; Luo, Z.; Yu, J.; Li, W.; Xu, W.; et al. Analysis of the genetic contribution to thoracic aortic aneurysm or dissection in a prospective cohort of patients with familial and sporadic cases in East China. Orphanet J. Rare Dis. 2023, 18, 251. [Google Scholar] [CrossRef] [PubMed]
- Semyachkina, A.N.; Adyan, T.A.; Kharabadze, M.N.; Novikov, P.V.; Polyakov, A.V. Clinical and genetic characteristics of Russian Marfan patients. Russ. J. Genet. 2015, 51, 695–701. [Google Scholar] [CrossRef]
- Zhukov, V.A.; Zhernakov, A.I.; Gavrilyuk, N.D.; Pinaev, A.G.; Andronov, E.E.; Uspensky, V.E.; Irtyuga, O.B.; Moiseeva, O.M. Polymorphism analysis of the ACTA2 gene by pyrosequencing in patients with non-familial thoracic aortic aneurysms. Mol. Med. 2016, 14, 45–49. [Google Scholar]
- Irtyuga, O.B.; Freilikhman, O.A.; Krivonosov, D.S.; Malashicheva, A.B.; Tarnovskaya, S.I.; Uspensky, V.E.; Gordeev, M.L.; Rotar, O.P.; Kostareva, A.A.; Moiseeva, O.M. Role of the NOTCH1 gene in formation of aortic aneurysm. Russ. J. Cardiol. 2018, 23, 53–59. [Google Scholar] [CrossRef]
- Schubert, J.A.; Landis, B.J.; Shikany, A.R.; Hinton, R.B.; Ware, S.M. Clinically relevant variants identified in thoracic aortic aneurysm patients by research exome sequencing. Am. J. Med. Genet. A 2016, 170, 1288–1294. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-Y.; Song, J.; Liu, Y.; Song, C.-X.; Yi, C. Mapping the epigenetic modifications of DNA and RNA. Protein Cell 2020, 11, 792–808. [Google Scholar] [CrossRef] [PubMed]
- Elbitar, S.; Renard, M.; Arnaud, P.; Hanna, N.; Jacob, M.P.; Guo, D.C.; Tsutsui, K.; Gross, M.S.; Kessler, K.; Tosolini, L.; et al. Pathogenic variants in THSD4, encoding the ADAMTS-like 6 protein, predispose to inherited thoracic aortic aneurysm. Genet. Med. 2021, 23, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Ke, T.; Han, M.; Zhao, M.; Wang, Q.K.; Zhang, H.; Zhao, Y.; Ruan, X.; Li, H.; Xu, C.; Sun, T. Alpha-actin-2 mutations in Chinese patients with a non-syndromatic thoracic aortic aneurysm. BMC Med. Genet. 2016, 17, 45. [Google Scholar] [CrossRef]
- Tashima, Y.; Iwakoshi, S.; Inoue, T.; Nakamura, N.; Sano, T.; Kimura, N.; Inoue, T.; Adachi, K.; Yamaguchi, A. Aortic Agatston score correlates with the progression of acute type A aortic dissection. PLoS ONE 2022, 17, e0263881. [Google Scholar] [CrossRef] [PubMed]
- Hirono, K.; Hata, Y.; Miyao, N.; Okabe, M.; Takarada, S.; Nakaoka, H.; Ibuki, K.; Ozawa, S.; Origasa, H.; Nishida, N.; et al. LVNC study collaborates. Increased burden of ion channel gene variants is related to distinct phenotypes in pediatric patients with left ventricular noncompaction. Circ. Genom. Precis. Med. 2020, 13, e002940. [Google Scholar] [CrossRef] [PubMed]
- Lui, M.M.; Shadrina, M.; Gelb, B.D.; Kontorovich, A.R. Features of vascular Ehlers-Danlos syndrome among biobank participants harboring predicted high-risk COL3A1 genotypes. Circ. Genom. Precis. Med. 2023, 16, e003864. [Google Scholar] [CrossRef] [PubMed]
- Harakalova, M.; van der Smagt, J.; de Kovel, C.G.; Van’t Slot, R.; Poot, M.; Nijman, I.J.; Medic, J.; Joziasse, I.; Deckers, J.; Roos-Hesselink, J.W.; et al. Incomplete segregation of MYH11 variants with thoracic aortic aneurysms and dissections and patent ductus arteriosus. Eur. J. Hum. Genet. 2013, 21, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Kashtan, C.E.; Segal, Y.; Flinter, F.; Makanjuola, D.; Gan, J.S.; Watnick, T. Aortic abnormalities in males with Alport syndrome. Nephrol. Dial. Transplant. 2010, 25, 3554–3560. [Google Scholar] [CrossRef] [PubMed]
- Ewans, L.J.; Colley, A.; Gaston-Massuet, C.; Gualtieri, A.; Cowley, M.J.; McCabe, M.J.; Anand, D.; Lachke, S.A.; Scietti, L.; Forneris, F.; et al. Pathogenic variants in PLOD3 result in a Stickler syndrome-like connective tissue disorder with vascular complications. J. Med. Genet. 2019, 56, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Lee, J.K.; Lee, J.O.; Kwon, B.; Seo, E.J.; Suh, D.C. Whole exome sequencing in patients with phenotypically associated familial intracranial aneurysm. Korean J. Radiol. 2022, 23, 101–111. [Google Scholar] [CrossRef]
- Torres-Juan, L.; Rico, Y.; Fortuny, E.; Pons, J.; Ramos, R.; Santos-Simarro, F.; Asensio, V.; Martinez, I.; Heine-Suñer, D. NOTCH1 gene as a novel cause of thoracic aortic aneurysm in patients with tricuspid aortic valve: Two cases reported. Int. J. Mol. Sci. 2023, 24, 8644. [Google Scholar] [CrossRef] [PubMed]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG-AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Kwartler, C.S.; Gong, L.; Chen, J.; Wang, S.; Kulmacz, R.; Duan, X.Y.; Janda, A.; Huang, J.; Kamm, K.E.; Stull, J.T.; et al. Variants of unknown significance in genes associated with heritable thoracic aortic disease can be low penetrant ‘risk variants’. Am. J. Hum. Genet. 2018, 103, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Irtyuga, O.; Malashicheva, A.; Zhiduleva, E.; Freylikhman, O.; Rotar, O.; Bäck, M.; Tarnovskaya, S.; Kostareva, A.; Moiseeva, O. NOTCH1 mutations in aortic stenosis: Association with osteoprotegerin/RANK/RANKL. Biomed. Res. Int. 2017, 6917907. [Google Scholar] [CrossRef]
- Meloni, I.; Rubegni, P.; De Aloe, G.; Bruttini, M.; Pianigiani, E.; Cusano, R.; Seri, M.; Mondillo, S.; Federico, A.; Bardelli, A.M.; et al. Pseudoxanthoma elasticum: Point mutations in the ABCC6 gene and a large deletion including also ABCC1 and MYH11. Hum. Mutat. 2001, 18, 85. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Ito, K.; Terao, C.; Akiyama, M.; Horikoshi, M.; Momozawa, Y.; Matsunaga, H.; Ieki, H.; Ozaki, K.; Onouchi, Y.; et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat. Genet. 2020, 52, 1169–1177. [Google Scholar] [CrossRef]
- Klarin, D.; Devineni, P.; Sendamarai, A.K.; Angueira, A.R.; Graham, S.E.; Shen, Y.H.; Levin, M.G.; Pirruccello, J.P.; Surakka, I.; Karnam, P.R.; et al. Genome-wide association study of thoracic aortic aneurysm and dissection in the Million Veteran Program. Nat. Genet. 2023, 55, 1106–1115. [Google Scholar] [CrossRef]
- Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F.; Casey, D.E.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; Kazerooni, E.A.; et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 2010, 121, e266–e369. [Google Scholar] [PubMed]
- Grahame, R.; Bird, H.A.; Child, A. The revised (Brighton 1998) criteria for the diagnosis of benign joint hypermobility syndrome (BJHS). J. Rheumatol. 2000, 27, 1777–1779. [Google Scholar]
- Loeys, B.L.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.L.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010, 47, 476–485. [Google Scholar] [CrossRef]
- Rodrigues Bento, J.; Meester, J.; Luyckx, I.; Peeters, S.; Verstraeten, A.; Loeys, B. The genetics and typical traits of thoracic aortic aneurysm and dissection. Annu. Rev. Genom. Hum. Genet. 2022, 23, 223–253. [Google Scholar] [CrossRef]
- Hamandi, M.; Bolin, M.L.; Fan, J.; Lanfear, A.T.; Woolbert, S.K.; Baxter, R.D.; DiMaio, J.M.; Brinkman, W.T. A newly discovered genetic disorder associated with life-threatening aortic disease in a 6-year-old boy. J. Investig. Med. High. Impact Case Rep. 2020, 8, 2324709620909234. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Purification of nucleic acids by extraction with phenol: Chloroform. CSH Protoc. 2006, 2006, pdb.prot4455. [Google Scholar]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; UGENE team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
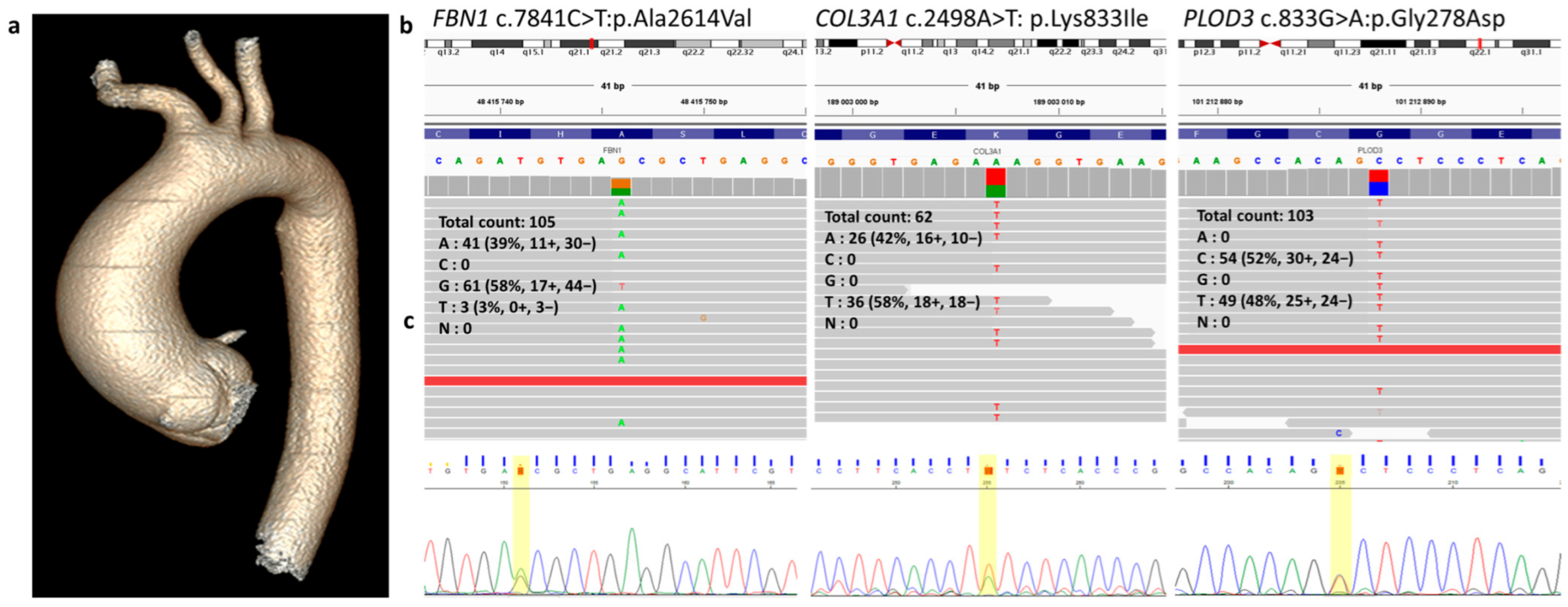


{kind=link}
{kind=link}
{kind=link}
| Parameters | All Patients (n = 41) | Males (n = 27) | Females (n = 14) | p-Value |
|---|---|---|---|---|
| Age, years (Median (Q1; Q3)) | 54(48; 64) | 58.0 (48.0; 62.5) | 53.0 (48.8; 68.5) | 0.563 |
| Mid-ascending aorta diameter, mm (Median (Q1; Q3)) | 52 (49; 54) | 51.9 (46.5; 54.0) | 52.0 (50.3; 52.9) | 0.535 |
| Sinotubular junction diameter, mm (Median (Q1; Q3)) | 42 (38.2; 46) | 44.0 (39.9; 52.3) | 39.0 (37.0; 40.9) | 0.012 |
| Proximal aortic arch diameter, mm (Median (Q1; Q3)) | 39.5 (36; 42.5) | 39.5 (36.0; 41.8) | 39.5 (36.0; 43.0) | 0.976 |
| Bicuspid aortic valve, n (%) | 25 (61.0) | 14 (51.8) | 11 (78.6) | 0.105 |
| BMI, kg/m2 (Median (Q1; Q3)) | 29 (26.4; 31.6) | 29 (27.3; 31.4) | 29.1 (23.6; 32.3) | 0.530 |
| Arterial hypertension, n (%) | 27 (64.3) | 18 (64.3) | 9 (64.3) | 0.879 |
| Hyperlipidemia, n (%) | 10 (23.8) | 7 (25.0) | 3 (21.4) | 0.751 |
| Obesity (BMI (kg/m2) > 30), n (%) | 18 (42.9) | 12 (42.9) | 6 (42.9) | 1 |
| T2DM or glucose intolerance, n (%) | 3 (7.1) | 2 (7.1) | 1 (7.1) | 1 |
| Angina pectoris, CAD or MI, n (%) | 13 (30.9) | 12 (42.8) | 1 (7.1) | 0.039 |
| Atherosclerosis, coronary, carotid, renal or femoral, n (%) | 21 (50) | 16 (59.2) | 4 (28.6) | 0.055 |
| Calcification measurements (number of patients) | 26 | 18 | 8 | - |
| Ca-score in coronary arteries (Agatston index) | 71.2 | 94.9 | 17.9 | 0.414 |
| Ca-score in aorta (Agatston index) | 107 | 121 | 75.5 | 0.404 |
| Characteristics ID | Patient 13 | Patient 36 | Patient 43 | Patient 54 | |||
|---|---|---|---|---|---|---|---|
| Age (years) | 58 | 67 | 72 | 48 | |||
| Sex | m | f | m | m | |||
| Gene (category, (13)) | COL4A5 (C) | NOTCH1 (B) | MYH11 (A) | FBN1 (A) | COL3A1 (A) | PLOD3 (D) | |
| Genome coordinates (hg38/exon) | chrX: 108582900/ exon 17 | chr9: 136518726/ exon 6 | chr16: 15719674/ exon 35 | chr15: 48415746/ exon 64 | chr2: 189003007/ exon 36 | chr7: 101212888/ exon 8 | |
| Nucleotide:aminoacid change | c.953C>G: p.Pro318Arg | c.964G>A: p.Val322Met | c.4993C>T: p.Arg1665Cys | 7841C>T: p.Ala2614Val | c.2498A>T: p.Lys833Ile | c.833G>A: p.Gly278Asp | |
| SNP ID MAF * | rs1449979085 8.3 × 10−6/- | - 2.1 × 10−6/- | rs768569707 4.4 × 10−5/- | - 6.8 × 10−7/- | - - | rs1041461490 6.8 × 10−6/5.9 × 10−4 | |
| In silico predictors ** | 3/4/16 16.9 | 8/10/1 25.0 | 11/9/1 32.0 | 1/7/15 23.6 | 8/12/3 28.5 | 0/1/0 25.6 | |
| Aortic dimensions *** | 70/45/45/34 | 36/62/45/35 | 41/69/48/36 | 45/50/35/30 | |||
| Ca-score (Agatston index) | Coronary arteries | no data | 49 | 423 | 0 | ||
| Aorta | no data | 237 | 937 | 0 | |||
| Aortic valve | BAV | TAV | TAV | TAV | |||
| CAD | no | yes | yes | no | |||
| CVD risk factors (HTN/T2DM/HLD/O) | yes/no/yes/yes | yes/no/-/no | yes/no/yes/no | yes/no/no/no | |||
| Atherosclerosis | no | CarA:up to 20% | CorA: OMA 60%; LAD 40%; DA 40%; CarA: up to 20% | no | |||
| Gene | Exon | Sequence |
|---|---|---|
| COL3A1 NC_000002.12 | 36 | F: 5′-GCTGAGAGATTGCTGTTG-3′ R: 5′-GGTGCTGAGATTCATACTTG-3′ |
| FBN1 NC_000015.10 | 64 | F: 5′-GACAGCCACACAGGTAA-3′ R: 5′-CATAGCAAGAAGCCACATC-3′ |
| MYH11 NC_000016.10 | 35 | F: 5′-CAGAGGAGGACGAAATGA-3′ R: 5′-TGTGCAAAGCTGAACTG-3′ |
| COL4A5 NC_000023.11 | 17 | F: 5′-CCAGTATTCTCATTGCTTCTAT-3′ R: 5′-TATTTCTGCAACATGGACTG-3′ |
| NOTCH1 NC_000009.12 | 6 | F: 5′-GGACACTCGCAGTAGAA-3′ R: 5′-TCCACAGAGCACAAAGA-3′ |
| PLOD3 NC_000007.14 | 8 | F: 5′-GCTGGAAGATGCAACAC-3′ R: 5′-GGAAACGGTCCCACTAA-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goncharova, I.A.; Shipulina, S.A.; Sleptcov, A.A.; Zarubin, A.A.; Valiakhmetov, N.R.; Panfilov, D.S.; Lelik, E.V.; Saushkin, V.V.; Kozlov, B.N.; Nazarenko, L.P.; et al. Identification of Variants of Uncertain Significance in the Genes Associated with Thoracic Aortic Disease in Russian Patients with Nonsyndromic Sporadic Subtypes of the Disorder. Int. J. Mol. Sci. 2024, 25, 8315. https://doi.org/10.3390/ijms25158315
Goncharova IA, Shipulina SA, Sleptcov AA, Zarubin AA, Valiakhmetov NR, Panfilov DS, Lelik EV, Saushkin VV, Kozlov BN, Nazarenko LP, et al. Identification of Variants of Uncertain Significance in the Genes Associated with Thoracic Aortic Disease in Russian Patients with Nonsyndromic Sporadic Subtypes of the Disorder. International Journal of Molecular Sciences. 2024; 25(15):8315. https://doi.org/10.3390/ijms25158315
Chicago/Turabian StyleGoncharova, Irina A., Sofia A. Shipulina, Aleksei A. Sleptcov, Aleksei A. Zarubin, Nail R. Valiakhmetov, Dmitry S. Panfilov, Evgeniya V. Lelik, Viktor V. Saushkin, Boris N. Kozlov, Ludmila P. Nazarenko, and et al. 2024. "Identification of Variants of Uncertain Significance in the Genes Associated with Thoracic Aortic Disease in Russian Patients with Nonsyndromic Sporadic Subtypes of the Disorder" International Journal of Molecular Sciences 25, no. 15: 8315. https://doi.org/10.3390/ijms25158315