
Sequence-Based Protein Design: A Review of Using Statistical Models to Characterize Coevolutionary Traits for Developing Hybrid Proteins as Genetic Sensors

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Biological and Structural Properties of Allosterically Regulated Transcriptional Regulators
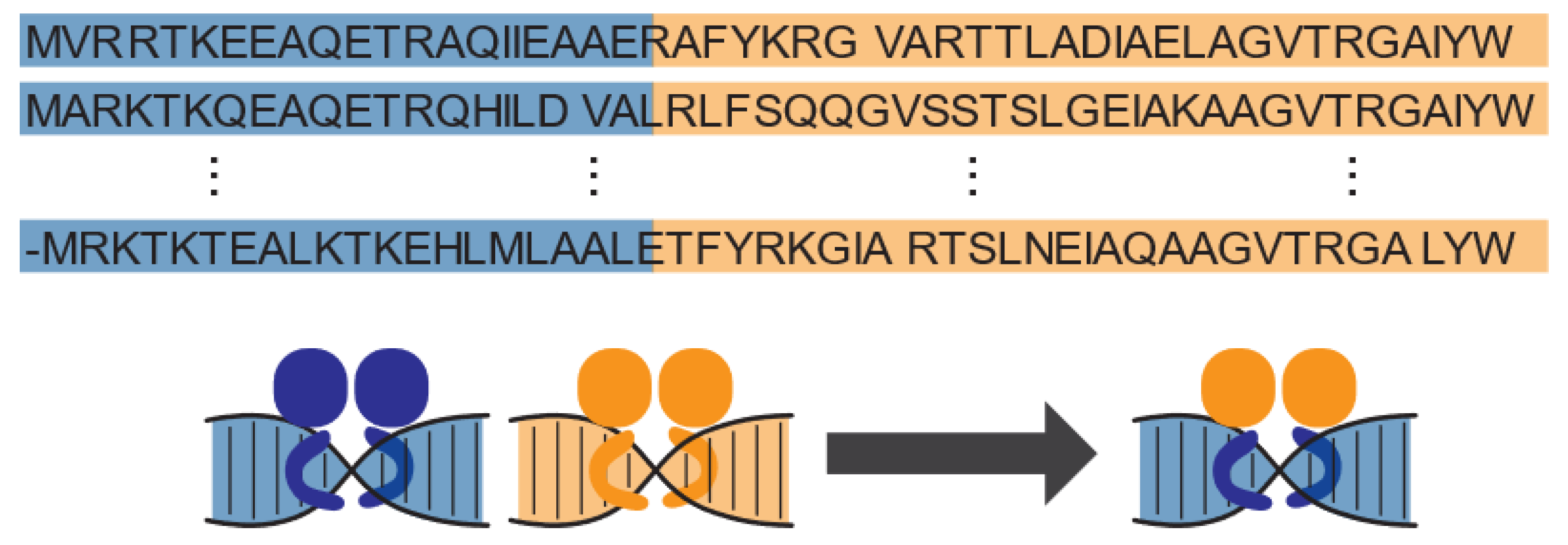
2. Previous Development of Hybrid Regulators
3. Genetic Circuit Development with Hybrid Regulators
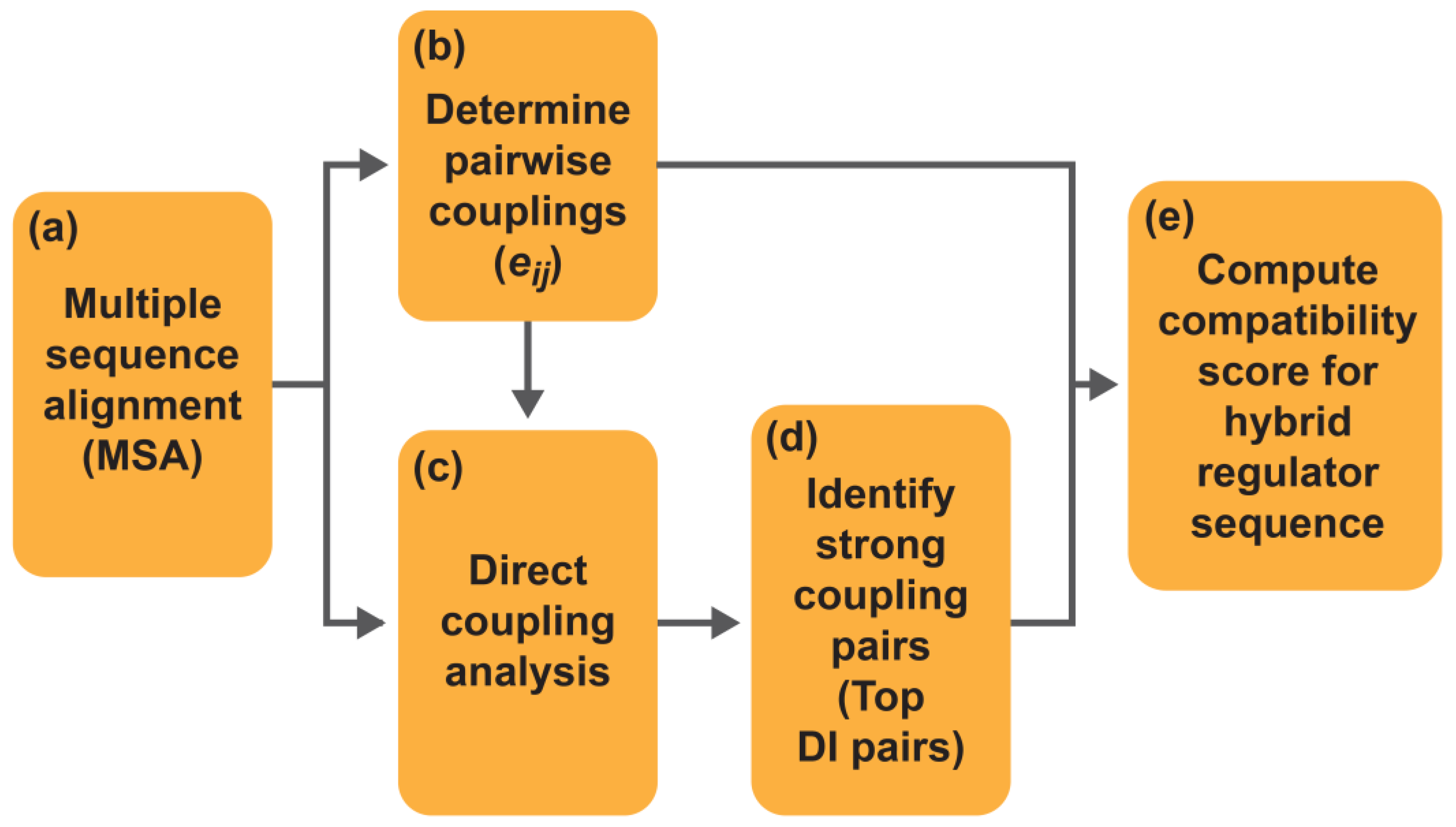
4. Using Direct Coupling Analysis and Direct Information Methods to Understand Coevolution among Proteins
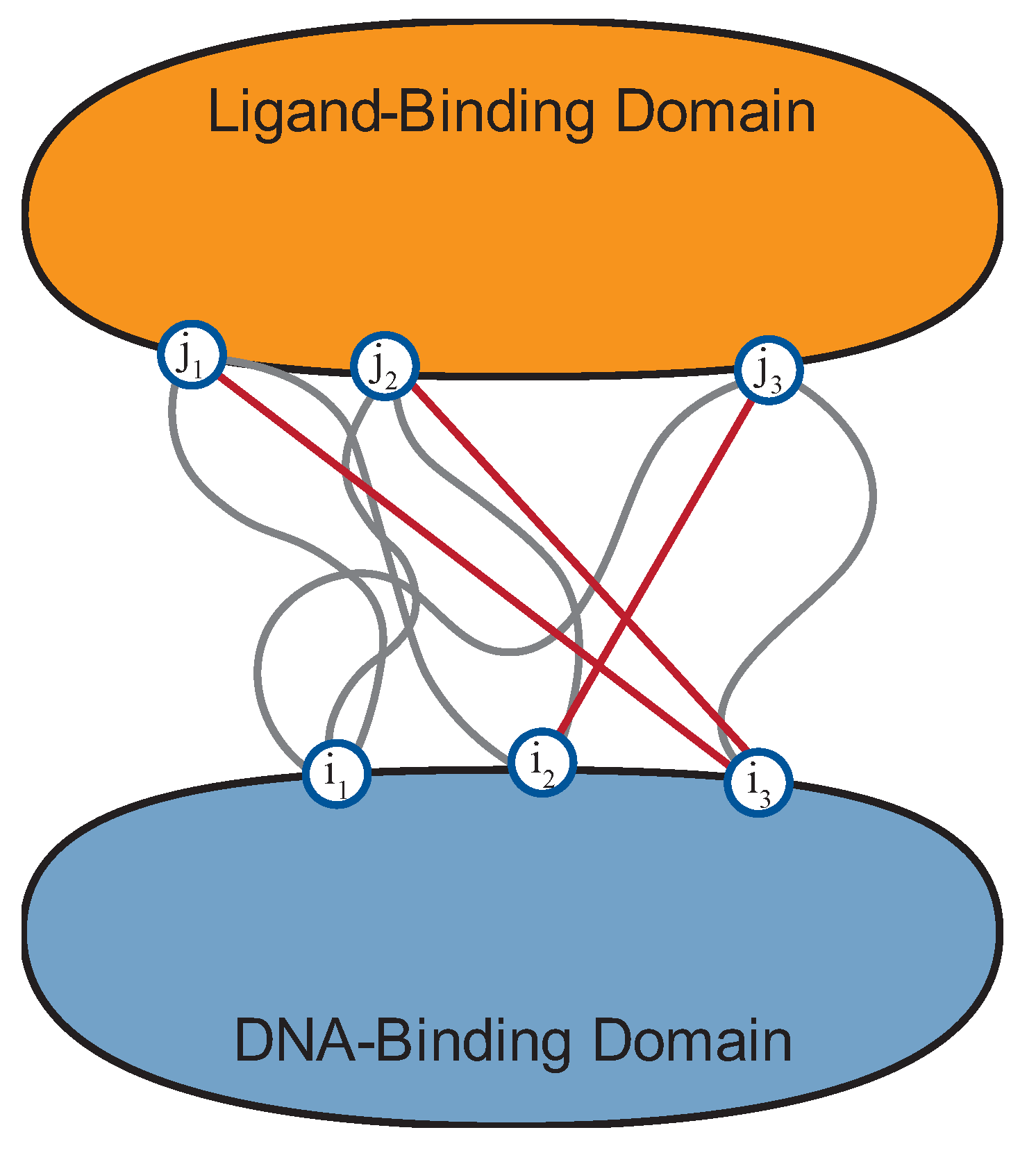
5. Using Statistical Modelling of Coevolution to Reveal Key DBM–LBM Interactions
6. Predictive Model Development for Evaluating DBM–LBM Compatibility
7. Hybrid Regulator Rescue Using the Coevolutionary Predictive Model Approach
8. Limitations of the Statistical Model Approach and Its Future Development
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ramos, J.L.; Martinez-Bueno, M.; Molina-Henares, A.J.; Teran, W.; Watanabe, K.; Zhang, X.; Gallegos, M.T.; Brennan, R.; Tobes, R. The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 2005, 69, 326–356. [Google Scholar] [CrossRef] [PubMed]
- Romero-Rodriguez, A.; Robledo-Casados, I.; Sanchez, S. An overview on transcriptional regulators in Streptomyces. Biochim. Biophys. Acta 2015, 1849, 1017–1039. [Google Scholar] [CrossRef] [PubMed]
- Baugh, A.C.; Momany, C.; Neidle, E.L. Versatility and Complexity: Common and Uncommon Facets of LysR-Type Transcriptional Regulators. Annu. Rev. Microbiol. 2023, 77, 317–339. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, L.; Nodwell, J.R. The TetR family of regulators. Microbiol. Mol. Biol. Rev. 2013, 77, 440–475. [Google Scholar] [CrossRef] [PubMed]
- Novichkov, P.S.; Kazakov, A.E.; Ravcheev, D.A.; Leyn, S.A.; Kovaleva, G.Y.; Sutormin, R.A.; Kazanov, M.D.; Riehl, W.; Arkin, A.P.; Dubchak, I.; et al. RegPrecise 3.0—A resource for genome-scale exploration of transcriptional regulation in bacteria. BMC Genom. 2013, 14, 745. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Huffman, J.L.; Brennan, R.G. Prokaryotic transcription regulators: More than just the helix-turn-helix motif. Curr. Opin. Struct. Biol. 2002, 12, 98–106. [Google Scholar] [CrossRef]
- Yu, Z.; Reichheld, S.E.; Savchenko, A.; Parkinson, J.; Davidson, A.R. A comprehensive analysis of structural and sequence conservation in the TetR family transcriptional regulators. J. Mol. Biol. 2010, 400, 847–864. [Google Scholar] [CrossRef]
- Chan, C.T.Y.; Kennedy, V.; Kinshuk, S. A domain swapping strategy to create modular transcriptional regulators for novel topology in genetic network. Biotechnol. Adv. 2024, 72, 108345. [Google Scholar] [CrossRef]
- Deng, W.; Li, C.; Xie, J. The underling mechanism of bacterial TetR/AcrR family transcriptional repressors. Cell Signal. 2013, 25, 1608–1613. [Google Scholar] [CrossRef]
- Herde, Z.D.; Short, A.E.; Kay, V.E.; Huang, B.D.; Realff, M.J.; Wilson, C.J. Engineering allosteric communication. Curr. Opin. Struct. Biol. 2020, 63, 115–122. [Google Scholar] [CrossRef]
- Swint-Kruse, L.; Matthews, K.S. Allostery in the LacI/GalR family: Variations on a theme. Curr. Opin. Microbiol. 2009, 12, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Sauer, R.T. Lac repressor at last. Structure 1996, 4, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.E.; Lewis, M. A closer view of the conformation of the Lac repressor bound to operator. Nat. Struct. Biol. 2000, 7, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Hersey, A.N.; Kay, V.E.; Lee, S.; Realff, M.J.; Wilson, C.J. Engineering allosteric transcription factors guided by the LacI topology. Cell Syst. 2023, 14, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M. The lac repressor. Comptes Rendus Biol. 2005, 328, 521–548. [Google Scholar] [CrossRef] [PubMed]
- Saenger, W.; Orth, P.; Kisker, C.; Hillen, W.; Hinrichs, W. The Tetracycline Repressor—A Paradigm for a Biological Switch. Angew. Chem. Int. Ed. Engl. 2000, 39, 2042–2052. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Arkin, A.P. Sequestration-based bistability enables tuning of the switching boundaries and design of a latch. Mol. Syst. Biol. 2012, 8, 620. [Google Scholar] [CrossRef]
- Tigges, M.; Denervaud, N.; Greber, D.; Stelling, J.; Fussenegger, M. A synthetic low-frequency mammalian oscillator. Nucleic Acids Res. 2010, 38, 2702–2711. [Google Scholar] [CrossRef]
- Elowitz, M.B.; Leibler, S. A synthetic oscillatory network of transcriptional regulators. Nature 2000, 403, 335–338. [Google Scholar] [CrossRef]
- Friedland, A.E.; Lu, T.K.; Wang, X.; Shi, D.; Church, G.; Collins, J.J. Synthetic gene networks that count. Science 2009, 324, 1199–1202. [Google Scholar] [CrossRef] [PubMed]
- Kiel, C.; Yus, E.; Serrano, L. Engineering signal transduction pathways. Cell 2010, 140, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Rowland, M.A.; Fontana, W.; Deeds, E.J. Crosstalk and competition in signaling networks. Biophys. J. 2012, 103, 2389–2398. [Google Scholar] [CrossRef] [PubMed]
- Vert, G.; Chory, J. Crosstalk in cellular signaling: Background noise or the real thing? Dev. Cell. 2011, 21, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Meinhardt, S.; Manley, M.W., Jr.; Becker, N.A.; Hessman, J.A.; Maher, L.J., 3rd; Swint-Kruse, L. Novel insights from hybrid LacI/GalR proteins: Family-wide functional attributes and biologically significant variation in transcription repression. Nucleic Acids Res. 2012, 40, 11139–11154. [Google Scholar] [CrossRef] [PubMed]
- Tungtur, S.; Egan, S.M.; Swint-Kruse, L. Functional consequences of exchanging domains between LacI and PurR are mediated by the intervening linker sequence. Proteins 2007, 68, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Groseclose, T.M.; Hersey, A.N.; Huang, B.D.; Realff, M.J.; Wilson, C.J. Biological signal processing filters via engineering allosteric transcription factors. Proc. Natl. Acad. Sci. USA 2021, 118, e2111450118. [Google Scholar] [CrossRef]
- Dimas, R.P.; Jiang, X.L.; Alberto de la Paz, J.; Morcos, F.; Chan, C.T.Y. Engineering repressors with coevolutionary cues facilitates toggle switches with a master reset. Nucleic Acids Res. 2019, 47, 5449–5463. [Google Scholar] [CrossRef] [PubMed]
- Rondon, R.E.; Wilson, C.J. Engineering a New Class of Anti-LacI Transcription Factors with Alternate DNA Recognition. ACS Synth. Biol. 2019, 8, 307–317. [Google Scholar] [CrossRef]
- Dimas, R.P.; Jordan, B.R.; Jiang, X.L.; Martini, C.; Glavy, J.S.; Patterson, D.P.; Morcos, F.; Chan, C.T.Y. Engineering DNA recognition and allosteric response properties of TetR family proteins by using a module-swapping strategy. Nucleic Acids Res. 2019, 47, 8913–8925. [Google Scholar] [CrossRef]
- Mukherji, R.; Zhang, S.; Chowdhury, S.; Stallforth, P. Chimeric LuxR Transcription Factors Rewire Natural Product Regulation. Angew. Chem. Int. Ed. Engl. 2020, 59, 6192–6195. [Google Scholar] [CrossRef]
- Schmidl, S.R.; Ekness, F.; Sofjan, K.; Daeffler, K.N.; Brink, K.R.; Landry, B.P.; Gerhardt, K.P.; Dyulgyarov, N.; Sheth, R.U.; Tabor, J.J. Rewiring bacterial two-component systems by modular DNA-binding domain swapping. Nat. Chem. Biol. 2019, 15, 690–698. [Google Scholar] [CrossRef]
- Ghataora, J.S.; Gebhard, S.; Reeksting, B.J. Chimeric MerR-Family Regulators and Logic Elements for the Design of Metal Sensitive Genetic Circuits in Bacillus subtilis. ACS Synth. Biol. 2023, 12, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Shis, D.L.; Hussain, F.; Meinhardt, S.; Swint-Kruse, L.; Bennett, M.R. Modular, multi-input transcriptional logic gating with orthogonal LacI/GalR family chimeras. ACS Synth. Biol. 2014, 3, 645–651. [Google Scholar] [CrossRef]
- Milner, P.T.; Zhang, Z.; Herde, Z.D.; Vedire, N.R.; Zhang, F.; Realff, M.J.; Wilson, C.J. Performance Prediction of Fundamental Transcriptional Programs. ACS Synth. Biol. 2023, 12, 1094–1108. [Google Scholar] [CrossRef]
- Chan, C.T.; Lee, J.W.; Cameron, D.E.; Bashor, C.J.; Collins, J.J. ‘Deadman’ and ‘Passcode’ microbial kill switches for bacterial containment. Nat. Chem. Biol. 2016, 12, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.L.; Dimas, R.P.; Chan, C.T.Y.; Morcos, F. Coevolutionary methods enable robust design of modular repressors by reestablishing intra-protein interactions. Nat. Commun. 2021, 12, 5592. [Google Scholar] [CrossRef] [PubMed]
- Morcos, F.; Pagnani, A.; Lunt, B.; Bertolino, A.; Marks, D.S.; Sander, C.; Zecchina, R.; Onuchic, J.N.; Hwa, T.; Weigt, M. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. USA 2011, 108, E1293–E1301. [Google Scholar] [CrossRef] [PubMed]
- Dinan, J.C.; McCormick, J.W.; Reynolds, K.A. Engineering Proteins Using Statistical Models of Coevolutionary Sequence Information. Cold Spring Harb. Perspect. Biol. 2024, 16, a041463. [Google Scholar] [CrossRef]
- Kamisetty, H.; Ovchinnikov, S.; Baker, D. Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era. Proc. Natl. Acad. Sci. USA 2013, 110, 15674–15679. [Google Scholar] [CrossRef]
- Marks, D.S.; Colwell, L.J.; Sheridan, R.; Hopf, T.A.; Pagnani, A.; Zecchina, R.; Sander, C. Protein 3D structure computed from evolutionary sequence variation. PLoS ONE 2011, 6, e28766. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, R.N.; Ferrari, A.J.R.; de Jesus, H.C.R.; Gozzo, F.C.; Morcos, F.; Martinez, L. Enhancing protein fold determination by exploring the complementary information of chemical cross-linking and coevolutionary signals. Bioinformatics 2018, 34, 2201–2208. [Google Scholar] [CrossRef] [PubMed]
- Morcos, F.; Schafer, N.P.; Cheng, R.R.; Onuchic, J.N.; Wolynes, P.G. Coevolutionary information, protein folding landscapes, and the thermodynamics of natural selection. Proc. Natl. Acad. Sci. USA 2014, 111, 12408–12413. [Google Scholar] [CrossRef] [PubMed]
- Sulkowska, J.I.; Morcos, F.; Weigt, M.; Hwa, T.; Onuchic, J.N. Genomics-aided structure prediction. Proc. Natl. Acad. Sci. USA 2012, 109, 10340–10345. [Google Scholar] [CrossRef]
- Dos Santos, R.N.; Khan, S.; Morcos, F. Characterization of C-ring component assembly in flagellar motors from amino acid coevolution. R. Soc. Open Sci. 2018, 5, 171854. [Google Scholar] [CrossRef]
- Weinreb, C.; Riesselman, A.J.; Ingraham, J.B.; Gross, T.; Sander, C.; Marks, D.S. 3D RNA and Functional Interactions from Evolutionary Couplings. Cell 2016, 165, 963–975. [Google Scholar] [CrossRef]
- Feinauer, C.; Szurmant, H.; Weigt, M.; Pagnani, A. Inter-Protein Sequence Co-Evolution Predicts Known Physical Interactions in Bacterial Ribosomes and the Trp Operon. PLoS ONE 2016, 11, e0149166. [Google Scholar] [CrossRef]
- Uguzzoni, G.; John Lovis, S.; Oteri, F.; Schug, A.; Szurmant, H.; Weigt, M. Large-scale identification of coevolution signals across homo-oligomeric protein interfaces by direct coupling analysis. Proc. Natl. Acad. Sci. USA 2017, 114, E2662–E2671. [Google Scholar] [CrossRef]
- Croce, G.; Gueudre, T.; Ruiz Cuevas, M.V.; Keidel, V.; Figliuzzi, M.; Szurmant, H.; Weigt, M. A multi-scale coevolutionary approach to predict interactions between protein domains. PLoS Comput. Biol. 2019, 15, e1006891. [Google Scholar] [CrossRef]
- Cheng, R.R.; Morcos, F.; Levine, H.; Onuchic, J.N. Toward rationally redesigning bacterial two-component signaling systems using coevolutionary information. Proc. Natl. Acad. Sci. USA 2014, 111, E563–E571. [Google Scholar] [CrossRef]
- Figliuzzi, M.; Barrat-Charlaix, P.; Weigt, M. How Pairwise Coevolutionary Models Capture the Collective Residue Variability in Proteins? Mol. Biol. Evol. 2018, 35, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Russ, W.P.; Figliuzzi, M.; Stocker, C.; Barrat-Charlaix, P.; Socolich, M.; Kast, P.; Hilvert, D.; Monasson, R.; Cocco, S.; Weigt, M.; et al. An evolution-based model for designing chorismate mutase enzymes. Science 2020, 369, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Leman, J.K.; Weitzner, B.D.; Lewis, S.M.; Adolf-Bryfogle, J.; Alam, N.; Alford, R.F.; Aprahamian, M.; Baker, D.; Barlow, K.A.; Barth, P.; et al. Macromolecular modeling and design in Rosetta: Recent methods and frameworks. Nat. Methods 2020, 17, 665–680. [Google Scholar] [CrossRef]
- Simonson, T.; Archontis, G.; Karplus, M. Free energy simulations come of age: Protein-ligand recognition. Acc. Chem. Res. 2002, 35, 430–437. [Google Scholar] [CrossRef]
- Childers, M.C.; Daggett, V. Insights from molecular dynamics simulations for computational protein design. Mol. Syst. Des. Eng. 2017, 2, 9–33. [Google Scholar] [CrossRef] [PubMed]
- Rouhani, M.; Khodabakhsh, F.; Norouzian, D.; Cohan, R.A.; Valizadeh, V. Molecular dynamics simulation for rational protein engineering: Present and future prospectus. J. Mol. Graph. Model. 2018, 84, 43–53. [Google Scholar] [CrossRef]
- Giuliani, A.; Benigni, R.; Colafranceschi, M.; Chandrashekar, I.; Cowsik, S.M. Large contact surface interactions between proteins detected by time series analysis methods: Case study on C-phycocyanins. Proteins 2003, 51, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Bruni, R.; Costantino, A.; Tritarelli, E.; Marcantonio, C.; Ciccozzi, M.; Rapicetta, M.; El Sawaf, G.; Giuliani, A.; Ciccaglione, A.R. A computational approach identifies two regions of Hepatitis C Virus E1 protein as interacting domains involved in viral fusion process. BMC Struct. Biol. 2009, 9, 48. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Do, C.B.; Mahabhashyam, M.S.; Brudno, M.; Batzoglou, S. ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome Res. 2005, 15, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Gromiha, M.M.; Selvaraj, S. Comparison between long-range interactions and contact order in determining the folding rate of two-state proteins: Application of long-range order to folding rate prediction. J. Mol. Biol. 2001, 310, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Li, Z. Inter-residue interactions in protein structures exhibit power-law behavior. Biopolymers 2008, 89, 1174–1178. [Google Scholar] [CrossRef] [PubMed]
- Vymetal, J.; Jakubec, D.; Galgonek, J.; Vondrasek, J. Amino Acid Interactions (INTAA) web server v2.0: A single service for computation of energetics and conservation in biomolecular 3D structures. Nucleic Acids Res. 2021, 49, W15–W20. [Google Scholar] [CrossRef] [PubMed]
- Nakata, S.; Mori, Y.; Tanaka, S. End-to-end protein-ligand complex structure generation with diffusion-based generative models. BMC Bioinform. 2023, 24, 233. [Google Scholar] [CrossRef]
- Townshend, R.J.L.; Eismann, S.; Watkins, A.M.; Rangan, R.; Karelina, M.; Das, R.; Dror, R.O. Geometric deep learning of RNA structure. Science 2021, 373, 1047–1051. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinshuk, S.; Li, L.; Meckes, B.; Chan, C.T.Y. Sequence-Based Protein Design: A Review of Using Statistical Models to Characterize Coevolutionary Traits for Developing Hybrid Proteins as Genetic Sensors. Int. J. Mol. Sci. 2024, 25, 8320. https://doi.org/10.3390/ijms25158320
Kinshuk S, Li L, Meckes B, Chan CTY. Sequence-Based Protein Design: A Review of Using Statistical Models to Characterize Coevolutionary Traits for Developing Hybrid Proteins as Genetic Sensors. International Journal of Molecular Sciences. 2024; 25(15):8320. https://doi.org/10.3390/ijms25158320
Chicago/Turabian StyleKinshuk, Sahaj, Lin Li, Brian Meckes, and Clement T. Y. Chan. 2024. "Sequence-Based Protein Design: A Review of Using Statistical Models to Characterize Coevolutionary Traits for Developing Hybrid Proteins as Genetic Sensors" International Journal of Molecular Sciences 25, no. 15: 8320. https://doi.org/10.3390/ijms25158320