Abstract
Chronic pain is a prevalent condition with a multifaceted pathogenesis, where epigenetic modifications, particularly DNA methylation, might play an important role. This review delves into the intricate mechanisms by which DNA methylation and demethylation regulate genes associated with nociception and pain perception in nociceptive pathways. We explore the dynamic nature of these epigenetic processes, mediated by DNA methyltransferases (DNMTs) and ten-eleven translocation (TET) enzymes, which modulate the expression of pro- and anti-nociceptive genes. Aberrant DNA methylation profiles have been observed in patients with various chronic pain syndromes, correlating with hypersensitivity to painful stimuli, neuronal hyperexcitability, and inflammatory responses. Genome-wide analyses shed light on differentially methylated regions and genes that could serve as potential biomarkers for chronic pain in the epigenetic landscape. The transition from acute to chronic pain is marked by rapid DNA methylation reprogramming, suggesting its potential role in pain chronicity. This review highlights the importance of understanding the temporal dynamics of DNA methylation during this transition to develop targeted therapeutic interventions. Reversing pathological DNA methylation patterns through epigenetic therapies emerges as a promising strategy for pain management.
1. Introduction
Chronic pain is a primary reason individuals seek medical care, accounting for severe disability and socio-economic burdens worldwide [1]. Among the leading causes of years lost to disability, three (back pain, musculoskeletal disorders, and neck pain) are chronic pain conditions [2]. Affecting an estimated 11% to 40% of the global population [3,4], the costs for chronic pain are much larger than the annual costs of heart disease, cancer, and diabetes combined [5]. The lack of precise diagnostic tools and effective treatment options often leads to frustration for both patients and clinicians [6], with some patients developing resistance to conventional opioid analgesics [7]. Furthermore, existing strategies for pain prevention and management are largely inadequate and unsatisfactory, leading to a reduced quality of life.
Ground-breaking research has led the World Health Organization to recognize chronic pain as a disease characterized by intricate functional and structural alterations in the brain, neuroinflammation, and increased sensitivity of the central nervous system (CNS) to nociceptive stimuli [8,9,10,11,12]. This condition is further complicated by aberrant gene expression within neural cells responsible for nociceptive signal processing [12,13]. While genetic mutations provide some insight, the emerging field of epigenetics offers a more dynamic understanding by clarifying gene–environment interactions and gene expression patterns associated with chronic pain [14].
Epigenetic mechanisms refer to a series of physiological processes for stable control over gene expression to establish tissue- and cell-specific phenotypes [15]. DNA methylation, a key epigenetic mechanism, allows for dynamic and reversible gene regulation without altering the DNA sequence. Pathological DNA methylation patterns, observed in conditions such as cancer, neurological disorders, and CNS dysregulation, can lead to aberrant gene expression, thereby affecting pathogenesis [16,17,18]. Recent findings indicate that DNA methylation modulates the expression of pro-nociceptive and anti-nociceptive genes in the nociceptive pathways [19,20,21]. Patients with chronic pain syndromes, such as fibromyalgia [22,23], chronic widespread pain [24,25], chronic low back pain (CLBP) [26,27], migraine [28], and chronic postoperative pain [29,30], display altered global DNA methylation profiles compared to healthy individuals. Moreover, specific gene/locus DNA methylation alterations have been correlated with pain hypersensitivity, neuronal hyperexcitability, central sensitization, and immune/inflammatory responses [20,31].
Epigenetics has become one of the most promising fields for unravelling the gene expression patterns of many diseases, leading to ground-breaking discoveries and innovative therapies, including novel cancer treatments [32,33]. Chronic pain is also considered a reversible process substantially shaped by the interplay between noxious stimuli and the neuropsychological environment [34]. Therefore, investigating the role of DNA methylation in pain progression and identifying biomarker signatures closely related to chronic pain onset and evolution are imperative [35].
Here, this narrative review offers an up-to-date exploration of the potential involvement of DNA methylation in the pathogenesis and symptomatology of chronic pain. We highlight how DNA (de)methylation modifications regulate genes associated with nociception and pain perception in pain circuitry. By evaluating DNA methylation patterns as potential diagnostic markers for chronic pain, we underscore the need for comprehensive strategies to unravel the complex transition from acute to chronic pain. In contrast to previous reviews, this review addresses the scarcity of clinical studies involving human subjects with chronic pain, providing a comprehensive overview of the current landscape in chronic pain research and potential therapeutic avenues. Finally, we summarize unresolved questions in pain epigenetics, emphasizing areas that require further investigation.
2. Search Strategy and Selection Criteria
A comprehensive search of peer-reviewed articles was completed to outline the dynamics of DNA methylation under chronic pain conditions. We searched the PubMed, MEDLINE, Embase, and Web of Science databases, considering publications up to June 2024. The aim was to identify randomized clinical trials, observational studies, preclinical studies, and narrative reviews in English for inclusion in the text and tables. The literature search used the following terms: DNA methylation, demethylation, DNA methyltransferase, ten-eleven translocation, Methyl-CpG-binding domain, pain, and pain model. The search was limited to English-language publications. A preliminary screening of articles was conducted based on their title and abstract to ensure relevance. This was followed by a thorough screening based on inclusion criteria and quality assessment. Additionally, reference lists of retrieved articles were examined to identify potentially relevant studies not found in the initial search, further supplementing the discussion. The included studies focused on primary clinical research on DNA methylation in human subjects with various chronic pain conditions and preclinical studies on DNA methylation in animal pain models with specific investigation time points.
3. DNA Methylation Dynamics in Chronic Pain
DNA methylation involves the transfer of a methyl group (-CH₃) from S-adenosyl-methionine (SAM) to the fifth carbon of the cytosine ring, resulting in the formation of 5-methylcytosine (5-mC) [36]. This modification primarily occurs at cytosine-phosphate-guanosine (CpG) sites of double-stranded DNA, which tend to cluster into ‘CpG islands’—regions often encompassing gene promoters [37]. Around 75% of protein-encoding genes in humans present this enrichment at their promoters [38]. The influence of DNA methylation on transcriptional activity is profound and location-dependent. In promoter regions, DNA methylation generally suppresses gene expression by hindering transcription factor binding, recruiting transcription repressors, and prompting chromatin remodelling. Consequently, this modification serves as a transcriptional barrier, preventing RNA polymerase from initiating gene transcription. Indeed, DNA methylation fulfils two essential roles in maintaining genomic integrity: (i) silencing potentially harmful elements such as transposons, viral DNA, and genes that should not be expressed; and (ii) preventing transcription factor binding to specific sites in promoter regions or recruiting transcription repressors.
The intricate relationship between DNA methylation and gene expression is a cornerstone of genomic regulation. Notably, the orchestration of DNA methylation patterns is managed by a series of specialized enzymes, each with a distinct function: (i) writers: enzymes that add the modifications to the DNA, establishing the methylation landscape; (ii) erasers: enzymes that remove the methyl marks, allowing for the dynamic regulation of gene expression; and (iii) readers: enzymes that identify and interpret the methylation patterns, translating methylation marks into biological outcomes.
3.1. DNMTs: Writers of DNA Methylation in Pain Chronicity
DNA methyltransferases (DNMTs) are responsible for adding methyl groups to cytosine nucleotides in DNA, resulting in 5-mC [39,40]. The DNMT family comprises two main classes: the de novo DNMTs (DNMT3a and DNMT3b), which introduce new methylation marks to previously unmethylated DNA, and the maintenance DNMT (DNMT1), which perpetuates already established methylation patterns and facilitates their repair [41,42] (Figure 1). During DNA replication, DNMT1 recognizes hemi-methylated DNA and ensures the new strand is appropriately methylated, thus preserving the epigenetic information through cell division and in response to DNA damage [43].
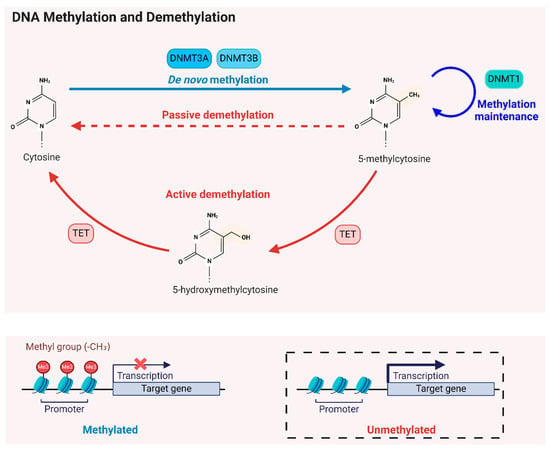
Figure 1.
DNA methylation and demethylation modifications. DNA methylation and demethylation are regulated by several key enzymes. de novo DNMTs (DNMT3a and DNMT3b) introduce new methylation marks to previously unmethylated DNA, while maintenance DNMT (DNMT1) perpetuates existing methylation patterns and facilitates their repair. TET enzymes convert 5-mC to 5-hydroxymethylcytosine (5-hmC) within the CpG dinucleotide, initiating active DNA demethylation. Passive DNA demethylation occurs during DNA replication, resulting in the gradual removal of methylation marks if maintenance of methylation on hemi-methylated DNA is hindered, such as by reduced DNMT activity or a deficiency of SAM (methyl donor). When methylation occurs at CpG islands in gene promoters, it generally represses gene expression. The removal of methyl groups from CpG sites can lead to the activation of gene expression.
Recent studies have implied a pro-nociceptive role of DNMTs in almost all major stations in the nociceptive pathway, including the dorsal root ganglion (DRG) [44,45], spinal cord [46,47], and brain regions such as the amygdala [48] and prefrontal cortex (PFC) [49], thereby contributing to chronic pain. Alterations in DNA methylation patterns at these stations can significantly influence gene expression, thereby affecting an individual’s normal physiological functions and processing of pain.
In neuropathic pain models, significant upregulations of DNMT1 [50] and DNMT3a [51] in DRG neurons were correlated with nociceptive hypersensitivity. These upregulations are associated with DNA methylation alteration at the Kcna2 promoter, leading to the epigenetic silencing of the Kcna2 gene, which encodes the Kv1.2 potassium channel. Reduced Kv1.2 levels diminish voltage-dependent potassium currents, depolarize the resting membrane potential, and promote neuronal hyperexcitability in DRG neurons, thereby amplifying spinal cord sensitization and neuropathic pain manifestations [52,53]. Moreover, DNMT3a-mediated epigenetic silencing of Kv1.2 in the spinal dorsal horn is also implicated in bone cancer pain [54].
In addition, a study demonstrated that DNMT3a-mediated downregulation of the K2p1.1 gene might contribute to paclitaxel-induced neuropathic pain [55]. Paclitaxel, a chemotherapy drug often used in ovarian cancer, induces substantial transcriptional changes in DRG neurons, leading to chemotherapy-induced neuropathic pain [56,57]. Following paclitaxel treatment, DNMT3a levels rise while K2p1.1 mRNA and protein levels reduce, resulting in a decrease in outward potassium currents and an increase in DRG neuron excitability [58], which ultimately lead to mechanical allodynia and thermal hyperalgesia. Conversely, inhibition of DNMT3a via RG108, a specific non-nucleoside DNMT inhibitor, significantly reduces paclitaxel-induced nociceptive hypersensitivity, suggesting the pro-nociceptive role of DNMT3a in neuropathic pain development [55].
Furthermore, DNMT3a upregulation in the spinal cord [47] and DRG [59] following peripheral nerve injury also orchestrate the epigenetic silencing of the the mu-1 opioid receptor gene (Oprm1) gene, which encodes the mu-opioid receptor (MOR). MOR’s presence on presynaptic axon terminals in the spinal cord reduces neurotransmitter release by inhibiting voltage-gated calcium channels, thereby attenuating nociceptive input to the CNS [60]. MOR is also located post-synaptically, where it activates G protein-coupled inwardly, rectifying potassium channels and reducing neuronal excitability [60]. Moreover, it is well established that decreased MOR protein levels reduce the efficacy of opioid analgesics [61,62]. Consistent with this, inhibition of DNMT3a in vivo can prevent the spinal nerve ligation (SNL)-induced DNA methylation of Oprm1 in the DRG, restore the analgesic effects of morphine or loperamide, and reduce the development of analgesic tolerance [59].
Notably, Jiang et al. [63,64] reported that DNMT3b downregulation following SNL prevents DNA methylation maintenance, leading to decreased DNA methylation levels of the G-protein-coupled receptor 151 (GPR151) and chemokine receptor CXCR3 genes, significantly increasing their expression in spinal neurons. The upregulation of these genes may contribute to neuropathic pain through activation of the mitogen-activated protein kinase (MAPK) signalling pathway [65]. In addition, in inflammatory pain models, significant hyperalgesia is observed, which correlates with increased CXCR4 mRNA/protein expression in the DRG [66]. This upregulation pattern is paralleled by significant demethylation at the CXCR4 gene promoter and a decrease in DNMT3b levels in the DRG.
3.2. MBD: Readers of DNA Methylation in Pain Modulation
Methyl-CpG-binding domain (MBD) proteins are key interpreters of DNA methylation, selectively recognizing and binding to methylated CpG sites within the genome. This binding is a critical step in transducing the methylation signal, which in turn regulates gene expression. MBD proteins achieve this by recruiting proteins with chromatin remodelling capacities, transcription factors, or repressors to methylated DNA regions [67,68]. At promoter regions, these recruited factors often modify chromatin structure into a more condensed state, leading to transcriptional repression. Therefore, MBD proteins are essential for the regulation of gene activity that is initiated by DNA methylation, and can function as either a transcriptional activator or repressor depending on their protein partners and target genes [69].
In neuropathic pain models, MBD1-deficient mice display reduced responses to acute sensory stimuli after nerve injury [70]. Correspondingly, MBD1-deficient mice show increased levels of Oprm1 and Kcna2 in the DRG. In contrast, MBD1 overexpression in the DRG induces spontaneous pain and evoked nociceptive hypersensitivity in MBD1-deficient mice. This effect is likely due to MBD1’s suppression of Oprm1 and Kcna2 expression, potentially downregulating MOR and Kv1.2 proteins in nociceptive pathways [51,70]. Supporting this, acute knockout of MBD1 via siRNA-MBD1 boosts MOR and Kv1.2 protein abundance, reducing pain sensitivity [70].
Previous studies have also reported a potential pro-nociceptive role of Methyl-CpG-binding protein 2 (MeCP2) in nociceptive pathways [71]. For example, mutations in the MeCP2 gene cause Rett syndrome [72], where decreased pain perception is commonly reported [73,74]. Moreover, an increase in both global DNA methylation and MeCP2 expression in the spinal cord two weeks after chronic constriction injury (CCI) correlates with mechanical allodynia and thermal hyperalgesia [75]. Conversely, inhibition of DNA methylation through 5-azacytidine treatment can reverse these effects, reducing CCI-induced hyperalgesia [75,76]. In addition, MeCP2 mutant mice also show reduced nociceptive sensitivity [71]. These findings indicate that a reduction in functional MeCP2 could contribute either directly or indirectly to reduced nociceptive sensitivity.
Recent studies have however challenged this view, indicating that MeCP2 expression is reduced in a cell-type-specific manner after nerve injury [77,78,79], and MeCP2-knockout mice show hypersensitivity to noxious stimuli [80]. Increased MeCP2 expression attenuated spared nerve injury (SNI)-induced mechanical and thermal hypersensitivity, suggesting that it weakens the development of neuropathic pain [81]. Additionally, MeCP2 deletion in peripheral sensory neurons causes nociceptive hypersensitivity, likely due to the loss of gamma-aminobutyric acid (GABA) receptors on sensory neuron terminals and subsequent diminished presynaptic inhibition. This loss may directly increase neuronal excitability, potentially promoting central sensitization within nociceptive pathways [77].
3.3. TET: Erasers of DNA Methylation in Chronic Pain
DNA methylation typically suppresses gene transcription, while DNA demethylation can (re-)activate it through either active or passive processes [82,83]. The ten-eleven translocation (TET) enzymes—including TET1, TET2, and TET3—function as ‘erasers’ of DNA methylation by converting 5-mC to 5-hydroxymethylcytosine (5-hmC) within the CpG dinucleotide, thereby initiating active DNA demethylation [84,85]. Passive DNA demethylation, on the other hand, occurs during DNA replication and can lead to the gradual removal of the methylation marks over time if the maintenance of methylation on hemi-methylated DNA is hindered, such as by reduced DNMT activity or a deficiency of SAM (methyl donor) [86]. Unlike active DNA demethylation, which is an enzymatic process that can occur independently of DNA replication, passive demethylation does not require direct enzymatic action [87]. 5-hmC markers are widely distributed across mammalian DNA, and TET enzymes are expressed in diverse tissues, including the brain and blood [88].
Since their discovery, TET enzymes have become a central focus of pain epigenetics research. In models of osteoarthritis [89], visceral pain [90], inflammatory pain [91,92], and neuropathic pain [93,94,95], DNA demethylation patterns modulated by TET enzymes are associated with nociceptive hypersensitivity and allodynia. An increase in TET expression and 5-hmC enrichment in the spinal cord and DRG post-injury highlights the link between TET-mediated demethylation and the onset of chronic pain. Moreover, intrathecal administration of a TET1 vector induces long-lasting allodynia and hyperalgesia in naive rats, starting on day 7, peaking at day 14, and lasting for at least 4 weeks [96]. Conversely, TET1 knockdown mitigates established pain behaviours and downregulates TET1 in dorsal horn neurons.
Notably, several studies have identified alterations in specific gene expressions due to DNA demethylation regulation, suggesting a potential role in chronic pain. For example, in neuropathic pain models, SNL enhances TET1 expression in dorsal horn neurons, leading to 5-hmC enrichment at the brain derived neurotrophic factor (BDNF) promoter, which in turn increases spinal BDNF expression and neuronal excitability [97]. Moreover, one week after SNL, TET1 overexpression also hinders DNMTs (DNMT1, DNMT3A, and DNMT3B) from binding to the BDNF promoter, thereby preventing transcriptional silencing via DNA methylation. Conversely, knockdown of Tet1 not only decreased TET1 binding and 5-hmC enrichment at the BDNF promoter, but also reduced BDNF expression and alleviated allodynia and nociceptive hypersensitivity [97]. Hence, spinal TET1 enhances BDNF transcription by promoting TET1-mediated demethylation and inhibiting DNMT-dependent methylation. In addition, inhibiting TET enzymes, such as TET1 and TET2, significantly reduces nociceptive behaviours by blocking the upregulation of Stat3 [92] and NLRP3 [95] expression, respectively.
However, in another neuropathic pain model, TET1 overexpression via full length-TET1 mRNA microinjection into the DRG significantly alleviated SNL-induced nociceptive hypersensitivity during both development and maintenance phases without affecting acute pain [98]. This intervention restores morphine analgesia and reduced morphine tolerance following SNL. Moreover, Tet1 overexpression rescues the expression of MOR and Kv1.2 by decreasing 5-mC and enhancing 5-hmC levels at the Oprm1 (encoding MOR) and Kcna2 (encoding Kv1.2) promoters in the DRG. Peripheral nerve injury is known to suppress MOR and Kv1.2 expression by DNA methylation-mediated epigenetic silence at these sites, thereby contributing to neuropathic pain. TET1-mediated DNA demethylation may counteract this effect.
4. Altered DNA Methylation Patterns in Patients with Chronic Pain: DNA Hypermethylation and Hypomethylation
While the previous parts of this narrative review were based on preclinical studies, this third section focusses on epigenetic studies in patients with chronic pain conditions (Table 1). Advancements in genome-scale mapping have revolutionized our ability to evaluate DNA methylation levels and sites under various conditions. Unbiased genome-wide analyses through digital restriction enzyme analysis of methylation (DREAM) and reduced representation bisulfite sequencing (RRBS) have revealed dynamic DNA methylation changes in many chronic pain disorders. These techniques overcome potential biases and increase the accuracy of methylation quantitative measurements, facilitating the detection of differentially methylated regions (DMRs) and cytosines (DMCs) that could serve as biomarkers for chronic pain. DNA extracted from peripheral blood samples can be used to develop non-invasive molecular tests, as methylation analysis in peripheral blood cells often reflects methylation levels in the target tissue [99,100], as supported by evidence of blood–brain methylation correspondence [101,102]. For example, specific overlapping DNA methylation signatures of chronic pain have been found in both the PFC and peripheral T cells [103].
The interplay between genes and the environment is pivotal in chronic pain. Investigations into global DNA methylation differences between chronic pain patients and healthy individuals have scrutinized methylation patterns of pain-related genes to identify diagnostic and therapeutic markers. For instance, studies involving patients with fibromyalgia identified 1610 promoter regions with differentially methylated CpG sites, most (69%) of which were hypomethylated compared to healthy controls [22,23,104]. In a large cohort study of twin pairs with and without chronic widespread pain as well as non-related individuals, with repeated measurements over three years, significant DNA methylation differences were observed between patients with chronic widespread pain and healthy subjects, with epigenetic factors accounting for 6% of the variance in the pain phenotype of chronic widespread pain [24]. This study highlights methylation enrichment in neurological pathways and the potential involvement of genes such as RE1-Silencing Transcription Factor (REST), Monoamine Oxidase B (MAOB), and Collagen Type I Alpha 2 Chain (COL1A2) in immune function, inflammatory responses, and central sensitization [25,105].
In patients with non-specific CLBP, 159 differentially methylated positions were identified, mostly hypomethylated [27]. Individuals who developed CLBP showed lower global DNA methylation levels than those with resolved acute low back pain or healthy controls, suggesting that DNA hypomethylation might contribute to pain chronicity [106]. Additionally, significant differences were observed between individuals with early vs. advanced intervertebral disc degeneration [107], with around 98% of CpG sites being hypermethylated in advanced stages, although their direct association with pain was not evaluated.
In addition, a large cohort study of patients with a period of 3–18 months post-traumatic limb amputation, revealed that those who developed complex regional pain syndrome (CRPS) had 48 differentially methylated CpG sites compared to those with non-CRPS neuropathic pain, with 85% being hypomethylated [108]. Together with the above-described study regarding the transition from acute to chronic low back pain, this highlights the potential diagnostic and therapeutic importance of global DNA methylation in the progression from acute to chronic pain.
Global DNA methylation serves as an indicator of the overall state of the DNA methylation machinery, influencing genome function and organization on a large scale [109]. In other diseases, such as cancer, altered DNA methylation landscapes affect thousands of genes [110] and intergenic regions [111]. DNA methylation reprogramming involves both global changes in genome methylation and gene-specific alterations targeting discrete regulatory regions, thereby producing lasting effects on the chromatin structure, and transcriptome, ultimately influencing many aspects of cellular function.
In addition to these global changes, recent clinical studies have identified transcriptional changes in specific genes due to DNA methylation in chronic pain sufferers. Pro- and anti-nociceptive genes, which promote or inhibit pain sensation, respectively, are particularly prone to DNA methylation changes. For instance, a positive correlation has been established between a DNA hypermethylation pattern of the transient receptor potential ankyrin 1 (TRPA1) and pain symptoms in patients with CLBP, postherpetic neuralgia, and Crohn’s disease [26,112,113]. The TRPA1 channel, known for detecting various noxious stimuli [114], is increasingly recognized as a key player in nociceptive modulation [115]. DNA methylation alterations of the tumor necrosis factor (TNF) gene may influence the risk of developing chronic breast pain in patients with breast cancer undergoing surgery [29]. Additionally, increased DNA methylation of the secreted protein, acidic and rich in cysteine (SPARC) gene, and its subsequent downregulation, have been observed in animal models and patients with CLBP [116]. SPARC, a matricellular protein important for tissue remodelling and injury response [117], may have its activity and gene transcription repressed through DNA hypermethylation, potentially contributing to the development of chronic pain.

Table 1.
DNA methylation reprogramming in patients with chronic pain.
Table 1.
DNA methylation reprogramming in patients with chronic pain.
| Pain Conditions | Study Design | Tissue | Global DNA Methylation | Specific Genes | Functional Genomic Analyses Methods | Functional Enrichment Analysis | Reference |
|---|---|---|---|---|---|---|---|
| Acute low back pain (n = 14), Chronic low back pain (n = 15), Healthy controls (n = 16) | Cross-sectional | Blood tissue | DNA hypomethylation (genome-wide) | BDNF, CX3CR1, TNF, and many others | \ | \ | [106] |
| Non-specific chronic low back pain (n = 50), Healthy controls (n = 48) | Cross-sectional | Blood tissue | DNA hypomethylation (genome-wide) | CELSR1, KIF11, NAV1, and many others | Gene Ontology (GO) pathway enrichment analyses | Immune signalling, endochondral ossification, and G-protein-coupled transmissions | [27] |
| Early-stage disc degeneration (n = 8), Advanced-stage disc degeneration (n = 8) | Cohort study | Nucleus pulpous tissues from intervertebral disc | DNA hypermethylation (genome-wide) | CARD14, GNL3, MAPKAPK5, and many others | Gene Ontology (GO) pathway enrichment analyses | Hemophilic cell adhesion and cell–cell adhesion | [107] |
| Fibromyalgia (n = 42), Healthy controls (n = 42) | Cross-sectional | Blood tissue | DNA hypermethylation (genome-wide) | GCSAML, GRM2, TRPA1, and many others | \ | \ | [105] |
| Fibromyalgia (n = 24), Healthy controls (n = 24) | Cross-sectional | Blood tissue | DNA hypomethylation (genome-wide) | \ | MetaCore network analysis | MAPK signalling pathway and actin cytoskeleton regulation | [23] |
| Fibromyalgia (n = 10), Healthy controls (n = 42) | Cross-sectional | Blood tissue | DNA hypermethylation (genome-wide) | BDNF, NAT15, HDAC4, and many others | Gene Ontology (GO) pathway enrichment analyses | Neuron differentiation and nervous system development | [22] |
| Chronic widespread pain (n = 50), Twins without chronic widespread pain (n = 50), Healthy controls (n = 1608) | Cohort study | Blood tissue | \ | \ | Gene Ontology (GO) pathway enrichment analyses | Neurological pathways | [24] |
| Chronic fatigue syndrome and comorbid fibromyalgia (n = 28), Healthy controls (n = 26) | Cross-sectional | Blood tissue | DNA hypomethylation (gene-specific) | BDNF | \ | \ | [20] |
| Chronic fatigue syndrome and comorbid fibromyalgia (n = 28), Healthy controls (n = 26) | Cross-sectional | Blood tissue | DNA hypermethylation (gene-specific) | COMT | \ | \ | [31] |
| Chronic nociceptive pain (n = 18), Chronic neuropathic pain (n = 19), Healthy controls (n = 20) | Cohort study | Blood tissue | \ | RAB10, RBFOX-1/CGRP, MAGI2, and many others | STRING (https://string-db.org/) | Neuro-musculoskeletal system, immune response, and inflammation | [118] |
| Knee osteoarthritis pain (n = 182), Healthy controls (n = 31) | Cross-sectional | Blood tissue | DNA hypermethylation (genome-wide) | RNF39, KCNC1, ZFP57, and many others | Ingenuity Pathway Analysis | Immune response and inflammation | [119] |
| Musculoskeletal pain (n = 20), Healthy controls (n = 9) | Cross-sectional | Blood tissue | DNA hypomethylation (genome-wide) | PM20D1, LCLAT1, GNE, and many others | Ingenuity Pathway Analysis | Immune response and GABA receptor signalling | [120] |
| Complex regional pain syndrome (n = 8), Neuropathic pain (n = 38) | Cross-sectional | Blood tissue | DNA hypomethylation (genome-wide) | COL11A1, GPR75, GPR75, and many others | Gene Ontology (GO) pathway enrichment analyses | Immune function | [108] |
| Multisomatoform disorder (n = 136), Healthy controls (n = 145) | Cohort study | Blood tissue | DNA hypomethylation (gene-specific) | Leptin promoter | \ | \ | [121] |
| Chronic back pain or postherpetic neuralgia (n = 12) | Observational study | Blood tissue | DNA hypermethylation (gene-specific) | TRPA1 | \ | \ | [26] |
5. Long-Lasting Dynamic DNA Methylation Reprogramming: The Key to the Transition from Acute to Chronic Pain States?
Recent preclinical studies have revealed rapid DNA methylation reprogramming in response to peripheral nerve injury, suggesting a potential causal role in the transition from acute to chronic pain [44,49] (Table 2). These DNA methylation alterations persist as transition occurs, indicating the significance of DNA methylation in the development of chronic pain. For instance, in neuropathic pain models, SNL upregulates TET1 expression in dorsal horn neurons. This leads to 5-hmC enrichment at the BDNF promoter, which in turn increases spinal BDNF expression and subsequent neuronal excitability [97]. TET1 upregulation begins on day 3 post-SNL, peaks at day 7, and persists through days 14 and 21. Correspondingly, SNL induces nociceptive hypersensitivity at these time points, mirroring the pattern of TET1 expression enhancement. Similarly, intrathecal administration of a TET1 vector in naive rats triggers long-lasting allodynia and hyperalgesia, starting on day 7, peaking at day 14, and lasting for at least four weeks [96]. Notably, increased global DNA methylation and MeCP2 expression in the spinal cord two weeks following CCI also correlate with mechanical allodynia and thermal hyperalgesia [75]. However, critical unanswered questions concern the origin and long-term temporal progression of these alterations, as well as their external validity to humans suffering from (chronic) neuropathic pain.
To track epigenetic reprogramming in acute and chronic pain models, several studies have examined DNA methylation profiles in the brain over time, from one day to one year after SNI [49,122,123]. Initially, DNA methylation reprogramming in the PFC begins as early as day 1 post-injury, indicating that methylation alterations precede chronic pain manifestation [49]. The PFC, part of cortical and subcortical networks activated during pain experiences, undergoes structural and functional changes over time in chronic pain [124], including grey matter loss, reorganization of synaptic connections, and gene expression modulation [125,126,127,128,129,130]. Six months after SNI, hyperalgesia and increased anxiety were accompanied by a DNA hypomethylation pattern in the PFC and amygdala but not in the visual cortex and thalamus [122,123]. Furthermore, SNI triggers a series of dynamic DNA methylation changes that persist up to one-year post-injury in the PFC, suggesting that these alterations are concurrent with the onset and establishment of chronic pain phenotypes [49].
During this period, hundreds of differentially methylated genes and gene ontologies were identified, emphasizing the need to consider the time point-specific differential methylation of pain-related genes [49,122,123]. Most pain-related genes exhibit differential methylation at a single time point, indicating a time-dependent recruitment or dysregulation of normal functions. These genes influence pain modulation across several domains such as ion channel activity, inflammatory responses, and neurotransmitter regulation. While some genes may be relevant only at a specific time point, persistent differentially methylated genes could embed a genomic memory of the initial injury, necessary for the development of chronic pain, and can influence long-term gene regulation associated with chronic pain [131].
The temporal dynamics of differential gene methylation and ontology enrichment highlight the significance of understanding these processes during the transition to chronic pain. At one day post-injury, differential DNA methylation is likely driven by tonic sensory afferent signalling and the systemic impact of peripheral inflammation. As pain progresses to a sub-chronic state two weeks post-injury, central mechanisms such as central sensitization and neuroinflammation trigger changes in synaptic signalling and neuron–glia interactions within nociceptive pathways. By three months and one-year post-injury under a chronic pain state, genes associated with immune function become predominant. Chronic pain is characterized by the dysregulation of descending inhibitory and facilitatory nociceptive pathways, persistent low-grade neuroinflammation, and maladaptive neuroplasticity. Epigenetic processes are particularly capable of influencing chromatin structure and transcription, thereby supporting lasting change and acting as mediators of adaptive processes. Indeed, epigenetic mechanism has been implicated in learning, memory, and pain, with DNA methylation reprogramming fitting well into this picture [14,132].
There are few human studies on specific time points and longitudinal changes in response to acute or chronic pain. To date, only one human study, spanning six months, has reported that individuals who developed CLBP exhibited lower global DNA methylation levels compared to those with resolved acute low back pain and healthy controls [106]. This suggests that DNA hypomethylation and subsequent changes in candidate gene expression may contribute to pain chronicity. These patterns may reflect underlying differences between acute, sub-chronic, and chronic pain, implying that therapeutic interventions might also vary in efficacy based on the duration of pain. Thus, it is imperative for future research to conduct longitudinal studies that investigate the temporal dynamics of DNA methylation among individuals suffering from various chronic pain conditions.
Table 2.
DNA methylation modifications after injury in pain models.
Table 2.
DNA methylation modifications after injury in pain models.
| Pain Models | Methylation Marker/Enzyme | When and Time Course | Tissue | DNA Methylation Regulation | Gene-Specific Expression Regulation | Inhibition (Inhibitors) | Nociceptive Behaviour Response to Inhibitors | Reference |
|---|---|---|---|---|---|---|---|---|
| SNI-induced neuropathic pain model | \ | Starting on day 1, lasting for 1 year post-SNI | Prefrontal cortex | \ | MAPKBP1, Icos, Unc5cl, and many others | \ | \ | [49] |
| Partial SNL-induced neuropathic pain model | DNMT3a ↓ | 30 days post-SNI | Amygdala | \ | \ | \ | \ | [48] |
| SNI-induced neuropathic pain model | \ | 9 months post-SNI | Prefrontal cortex, T cells | \ | KCNAB3, KCNC4, DNMT1, and many others | \ | \ | [103] |
| SNI-induced neuropathic pain model | \ | 6 months post-SNI | Prefrontal cortex, amygdala | \ | \ | Environmental manipulation | Nociceptive hypersensitivity ↓ | [123] |
| SNL-induced neuropathic pain model | DNMT3b ↓ | Starting on day 1, lasting for 14 days post-SNL | Spinal dorsal horn | DNA hypomethylation (gene-specific) | GPR151 ↑ CXCR3 ↑ | SiRNA-DNMT3b | Nociceptive hypersensitivity ↓ | [63,64] |
| SNL-induced neuropathic pain model | DNMT3a ↑ | 7 days post-SNL | Spinal dorsal horn | DNA hypermethylation (gene-specific) | Kcna2 (Kv1.2) ↓ | AAV5-Dnmt3a shRNA | Nociceptive hypersensitivity ↓ | [51] |
| Bone cancer pain model | DNMT3a ↑ | Starting on day 3, lasting for 12 days post | Spinal dorsal horn | DNA hypermethylation (gene-specific) | Kcna2 (Kv1.2) ↓ | Decitabine | Nociceptive hypersensitivity ↓ | [54] |
| SNL-induced neuropathic pain model | TET1 ↑ 5-hmC ↑ | Starting on day 3, lasting for 21 days post-SNL | Spinal dorsal horn | DNA hypomethylation (gene-specific) | BDNF ↑ | SiRNA-TET1 | Nociceptive hypersensitivity ↓ | [97] |
| CFA-induced inflammatory pain model | TET1 ↑ TET3 ↑ 5-hmC ↑ | Starting on day 3, lasting for 14 days post-CFA | Spinal cord, Blood tissue | DNA hypomethylation (gene-specific) | STAT3 ↑ | Lenti-T1-siRNA, Lenti-T3-siRNA | Nociceptive hypersensitivity ↓ | [92] |
| Paclitaxel-induced neuropathic pain model | DNMT3a ↑ | Starting on day 7, lasting for 21 days post-paclitaxel | DRG | DNA hypermethylation (gene-specific) | K2P.1.1 ↓ | RG108 | Nociceptive hypersensitivity ↓ | [55] |
| SNL-induced neuropathic pain model | DNMT1 ↑ | Starting on day 3, lasting for 14 days post-SNL | DRG | DNA hypermethylation (gene-specific) | Kcna2 (Kv1.2) ↓ | RG108 | Nociceptive hypersensitivity ↓ | [50] |
| SNL-induced neuropathic pain model | DNMT3a ↑ MBD1 ↑ | 7 days post-SNL | DRG | DNA hypermethylation (gene-specific) | Oprm1 (MOR) ↓ | ShRNA-DNMT3a | Nociceptive hypersensitivity ↓, Analgesic effects of morphine ↑, Analgesic tolerance of morphine ↓ | [59] |
| CFA-induced inflammatory pain model | TET1 ↑ | Starting on day 3, lasting for 14 days post-CFA | DRG | DNA hypomethylation (gene-specific) | TRPV1 ↑ | Bobcat339 hydrochloride | Nociceptive hypersensitivity ↓ | [91] |
| Bone cancer pain model | TET1 ↑ | Starting on day 3, lasting for 21 days post-tumour cell inoculation | DRG | DNA hypomethylation (gene-specific) | TRPV4 ↑ | Bobcat339 hydrochloride | Nociceptive hypersensitivity ↓ | [133] |
| Diabetic neuropathic pain model | TET2 ↑ | 28 days post | DRG | DNA hypomethylation (gene-specific) | TXNIP ↑ NLRP3 ↑ | SiRNA-TET2 | Nociceptive hypersensitivity ↓ | [95] |
| SNL-induced neuropathic pain model | MBD1 ↑ | \ | DRG | DNA Hypermethylation (gene-specific) | MOR and KV.1.2 ↓ | SiRNA-MBD1 | Nociceptive hypersensitivity ↓ | [70] |
| SNI-induced neuropathic pain model | MeCP2 ↑ | 28 days post-SNI | DRG | DNA hypomethylation (gene-specific) | BDNF ↑ | MeCP2-null | Nociceptive hypersensitivity ↓ | [70] |
| Oral cancer pain model | \ | \ | DRG | DNA hypermethylation (gene-specific) | Oprm1 (MOR) ↓ | Decitabine | Nociceptive hypersensitivity ↓ | [134] |
SNL, spinal nerve ligation; SNI, spared nerve injury; CFA, complete Freund’s adjuvant; siRNA, small-interfering RNA; DRG, dorsal root ganglion; DNMT, DNA methyltransferases; MBD, methyl-CpG-binding domain; MeCP2, methyl-CpG-binding protein 2; TET, ten-eleven translocation.
6. DNA Methylation Modification in Patients with Chronic Pain: Links to Central Sensitization
Chronic pain is not merely an extended version of acute pain; it involves complex and long-lasting structural and functional plasticity within the CNS [135]. These changes go beyond a simple pain–damage correlation, involving intricate forms of maladaptive plasticity at molecular, cellular, and systemic levels. Central sensitization, characterized by increased neuronal excitability, maladaptive neuroplasticity, and amplified response to nociceptive and non-nociceptive stimulus, is crucial in the pathogenesis of chronic pain [8]. The induction and maintenance of central sensitization depend on maladaptive alterations in the expression, distribution, and activity of ion channels, receptors, and inflammatory mediators [135,136]. Many of these long-lasting adaptations are sustained via the modulation of transcriptional responses, with DNA methylation—a key modulator of transcription—potentially contributing to these maladaptive processes in chronic pain.
In patients with chronic musculoskeletal pain [24,120] and fibromyalgia [23,104], differentially methylated regions associated with pain were enriched across neurological pathways, such as GABA receptor signalling and MAPK signalling pathways [24,120]. Both pathways play critical roles in the development and maintenance of central sensitization [136]. Disruptions in GABAergic inhibition [137] and activation of MAPK pathways [65] both contribute to neuronal hyperexcitability and persistent pain. Consistently, in neuropathic pain models, increased DNA methylation following nerve injury may impair the descending inhibitory nociceptive modulation system by downregulating α5-GABA(A) receptor, which diminishes the inhibitory action of GABAergic neurons [138], thereby contributing to central sensitization and enhanced chronic pain states (Figure 2).
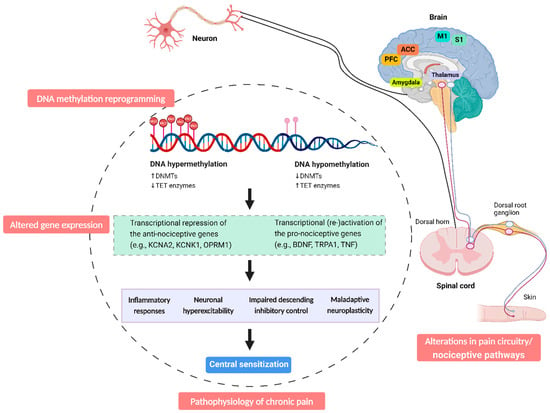
Figure 2.
DNA methylation reprogramming in chronic pain. DNA methylation and demethylation are controlled by multiple enzymes. The expression levels of these enzymes can alter the methylation state of specific gene promoters. An increase in DNMTs facilitates hypermethylation, while a decrease in DNMT expression is associated with hypomethylation. Conversely, a reduction in TET proteins promotes hypermethylation, while increased TET expression can lead to hypomethylation. DNA hypermethylation typically leads to gene silencing, while hypomethylation in promoter regions generally results in gene activation. These changes in DNA methylation levels affect the expression of various pro- and anti-nociceptive genes, facilitating the long-lasting plasticity of the pain circuitry both centrally and peripherally, ultimately contributing to the development and maintenance of chronic pain. In addition, the coexistence of both hypermethylated and hypomethylated genes in different chronic pain conditions suggests a focused regulatory mechanism rather than a uniform change in methylation levels. While DNA methylation reprogramming does not always correspond with gene expression changes, the methylation patterns of pain-related genes can significantly influence the pain phenotype. Abbreviation: ACC, anterior cingulate cortex; M1, primary motor cortex; PFC, prefrontal cortex; S1, primary somatosensory cortex.
We previously reported that patients with chronic fatigue syndrome and comorbid fibromyalgia exhibited lower DNA methylation levels in the BDNF gene compared to healthy individuals, correlating with increased serum BDNF levels and hyperalgesia [20]. Similarly, high levels of biopsychosocial complexity were also linked to lower DNA methylation levels of BDNF, leading to gene upregulation in patients with chronic musculoskeletal pain [21]. Preclinical studies have shown that TET-dependent DNA demethylation promotes 5-hmC enrichment at the BDNF promoter, inducing BDNF overexpression and neuronal hyperexcitability after SNL, while inhibition of TET expression reversed these effects and alleviated nociceptive hypersensitivity [97].
In any chronic disease, identifying perpetuating factors is crucial for identifying therapeutic targets to develop preventive strategies. BDNF, a key regulator of neuroplasticity, has been proposed to play a pro-nociceptive role in mediating central sensitization [139]. Human studies consistently report higher cerebrospinal fluid [140], plasma [141,142,143], and serum [144,145,146,147] levels of BDNF in patients with chronic pain compared to healthy individuals, with these levels positively correlating with more severe pain symptoms. DNA hypomethylation tends to enhance excitatory synaptic transmission and cortical excitability by upregulating BDNF expression, contributing to central sensitization. This suggests that persistent painful stimuli at an early stage may trigger DNA methylation reprogramming, thereby promoting maladaptive transcriptional processes and central sensitization which facilitate the transition from acute to chronic pain. Notably, if this indirect sensitization occurs, DNA methylation manipulation might also reverse pro-nociceptive epigenetic marks, thereby reducing pain.
7. DNA Methylation Patterns in Chronic Pain: Epigenetic Interventions in a Broader Picture
The exploration of DNA methylation’s role in chronic pain is shedding light on the mechanisms of pain progression and unveiling new therapeutic possibilities, especially given the generally poor efficacy of traditional analgesics such as opioids and NSAIDs. Many genes with differential DNA methylation have been identified, playing crucial roles in nociceptive pathways with either pro- or anti-nociceptive effects. These genes may play upstream roles in the hierarchy of epigenetic events involved in chronic pain, supporting a causal role. However, the specific genes or gene networks that could be targets to reverse the epigenetic landscape associated with chronic pain remain underexplored. DNA methylation modifications within gene networks can influence the activity of associated cellular pathways, contributing to the development of chronic pain. This complex landscape of changes poses challenges for establishing causality and designing interventions to reverse chronic pain. Although rather non-specific in nature, a potential strategy involves targeting entire pathways through therapeutic combinations that focus on “hub” proteins, such as DNMTs and TET enzymes, rather than individual candidate genes.
There is limited preclinical evidence to suggest that interventions targeting these enzymes might be effective for treating chronic pain in animal models (Table 2). For instance, the nucleoside analogues 5-azacytidine and decitabine (5-aza-deoxycytidine) are established inhibitors of DNMTs, used in clinical trials to reverse epigenetic mutations in cancer and myelodysplastic syndrome patients [148,149]. Despite initial challenges with response rates and high toxicity, these compounds have shown promise in pain management contexts, such as animal models for osteoarthritis [150] and cancer-related pain [151]. Both 5-azacytidine [75,76] and decitabine [138,152] have the potential to restore the expression of hypermethylated/silenced genes and reduce pain. However, we are unaware of studies exploring the potential of these compounds in patients with chronic pain.
Additionally, RG108 effectively blocks DNMTs’ active sites. In neuropathic pain models, RG108 administration has been shown to block the increase in DNA methylation of pain-related genes by downregulating DNMT1 and DNMT3a, thereby alleviating pain symptoms [47,50,153]. Moreover, chronic systemic administration of SAM has been observed to alleviate SNI-induced nociceptive hypersensitivity, pain avoidance behaviours, and cognitive deficits. SAM also partially reverses the reduction in global DNA methylation induced by SNI [122,154]. Marketed as a dietary supplement, SAM may offer pain relief by reducing inflammation and exerting direct analgesic effects [155,156,157]. Again, studies exploring the potential of these compounds in patients with chronic pain are needed.
Epigenetic modifications of gene expression in response to environmental changes or external stimuli are dynamic and reversible [123]. This aligns with findings in other fields where reversing disease-specific DNA methylation patterns follows successful conservative treatment [158,159,160]. However, while these interventions can modify pain-associated differential DNA methylation, they generally do not restore all alterations to a pre-chronic pain state.
Although several novel DNMT inhibitors have been identified, the number of such compounds remains limited. For many of these compounds, DNA methylation inhibition has been identified as a secondary characteristic, raising concerns about their specificity [161]. For instance, while pharmacological inhibition of DNMTS offers new therapeutic avenues for cancer treatment, the lack of specificity hampers the effectiveness of these compounds. Advances in understanding the functional properties of these enzymes will pave the way for more targeted drug design. The strategic development of DNMT inhibitors could be a feasible alternative, and the creation of molecular models for screening will aid in the identification of more specific molecules. Future research is expected to deepen our knowledge of the activity and structure of DNMTs and TET enzymes, facilitating the creation of targeted inhibitors.
8. Conclusions and Future Direction
Biologically, an organism’s reaction to external stimuli is reflected through epigenetic modifications. These alterations can affect brain activity, leading to behavioural changes [162]. Acute pain after injury may trigger rapid and persistent DNA methylation reprogramming that evolves as pain becomes chronic. DNA methylation reprogramming occurs early during the transition from acute to chronic pain, suggesting its potential causal role. Over the past decade, the concept of neuro-epigenetics [163] has highlighted the importance of epigenetic processes in neuro-adaptive phenomena, including neuronal differentiation, synaptic plasticity, memory consolidation, and cognitive adaptability [164,165,166,167,168]. Therefore, modifications in DNA methylation may represent one mechanism in the transition of pain from an acute sensation to the pathological states of neuroinflammation, central sensitization, and, eventually, chronic pain [14].
The intricate relationship between DNA methylation and gene expression is a cornerstone in the regulation of pain. DNMTs are responsible for adding methyl groups to DNA, which can interfere with gene transcription by blocking the binding of transcription factors to gene promoters. Conversely, TET enzymes facilitate gene expression through demethylation. In addition, transcriptional regulators such as MBD proteins and MeCP2 are indispensable for modulating gene transcription. However, interpretation of DNA methylation changes warrants caution, as they do not always mirror transcriptional activity. DNA methylation can occur across various genomic regions, and not all genes are equally regulated by CpG islands [169]. Moreover, DNA methylation does not have a linear relationship with gene expression; other epigenetic mechanisms such as histone acetylation and non-coding RNAs also play significant roles. Hence, future research should integrate gene expression data for a more comprehensive analysis.
In addition, current evidence highlights that broader epigenetic interventions, beyond single gene or protein targets, are required. The strategic development of DNMT inhibitors could be a feasible alternative, and the creation of molecular models for screening will aid in the identification of more specific molecules. Further research should delve into how chronic pain is embedded in the epigenome and identify epigenetic markers for therapeutic strategies.
Finally, while associations between DNA methylation changes and pain have been established, several critical questions remain unanswered:
- (i)
- Causal Relationship: Is there a causal relationship between DNA methylation and the occurrence of chronic pain in humans? Do DNA methylation changes result from chronic pain or precede its emergence? Are DNA methylation alterations merely stochastic footprints downstream of underlying pain neuropathology in patients with chronic pain conditions?
- (ii)
- Temporal Dynamics: How rapidly do DNA methylation changes occur, and are they persistent during chronic pain? How does epigenetic dysregulation at the onset of acute pain progress as pain transitions from acute to chronic in humans?
- (iii)
- Gene-Specific vs. Domain-Wide Changes: What is the extent of DNA methylation alterations in the brain after chronic pain induction, and are these changes specific to pain-related genes or broader domains and/or gene families?
- (iv)
- Translation to Clinical Practise: How can we translate epidemiological findings to the clinic? How can findings at the population level be applied to individual patients? What is the potential impact on pain perception, and to what extent can we influence this through therapy?
- (v)
- Therapeutic Potential: is DNA methylation reversible and targetable by existing or proposed treatments? Which of these processes should be targeted for therapeutic intervention?
Author Contributions
Conceptualization, H.-Y.X., A.P. and J.N.; methodology, A.P. and H.-Y.X.; validation, J.N., A.P., E.D.B. and S.S.; resources, H.-Y.X.; writing—original draft preparation, H.-Y.X.; writing—review and editing, J.N., S.S., A.P., E.D.B., L.G., J.H., A.W., J.V.C. and H.-Y.X.; visualization, H.-Y.X.; supervision, J.N., A.P., L.G. and S.S. All authors have read and agreed to the published version of the manuscript.
Funding
H.-Y.X. was funded by the China Scholarship Council. A.P. was funded by the Research Foundation—Flanders (FWO; FWOTM1051). J.H. was funded by the Research Foundation—Flanders (FWO; FWOTM1069). A.W. was funded by the Berekuyl Academy (The Netherlands) chair awarded to the Vrije Universiteit Brussel. J.V.C. was funded by the ME Research UK. This work was also supported by the Strategic Research Program SRP90 (‘Pain Never Sleeps: Unravelling the Sleep-Pain Interaction in Patients with Chronic Pain’) funded by the research council of the Vrije Universiteit Brussel, Brussels, Belgium.
Acknowledgments
The figures were created with BioRender.com, accessed on 30 June 2024.
Conflicts of Interest
J.N. and the Vrije Universiteit Brussel received lecturing/teaching fees from various professional associations and educational organizations.
References
- Cohen, S.P.; Vase, L.; Hooten, W.M. Chronic pain: An update on burden, best practices, and new advances. Lancet 2021, 397, 2082–2097. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.J.; Atkinson, C.; Bhalla, K.; Birbeck, G.; Burstein, R.; Chou, D.; Dellavalle, R.; Danaei, G.; Ezzati, M.; Fahimi, A.; et al. The state of US health, 1990-2010: Burden of diseases, injuries, and risk factors. JAMA 2013, 310, 591–608. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, Z.; Fraser, K.; Grol-Prokopczyk, H.; Zajacova, A. A global study of pain prevalence across 52 countries: Examining the role of country-level contextual factors. Pain 2022, 163, 1740–1750. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.; Thomas, S.; Stabile, V.; Han, X.; Shotwell, M.; McQueen, K. Prevalence of chronic pain in low-income and middle-income countries: A systematic review and meta-analysis. Lancet 2015, 385 (Suppl. S2), S10. [Google Scholar] [CrossRef] [PubMed]
- Gaskin, D.J.; Richard, P. The economic costs of pain in the United States. J. Pain. 2012, 13, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.H.; Fors, E.A.; Korwisi, B.; Barke, A.; Cameron, P.; Colvin, L.; Richardson, C.; Rief, W.; Treede, R.D.; The IASP Taskforce for the Classification of Chronic Pain. The IASP classification of chronic pain for ICD-11: Applicability in primary care. Pain 2019, 160, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Chou, R.; Deyo, R.; Friedly, J.; Skelly, A.; Weimer, M.; Fu, R.; Dana, T.; Kraegel, P.; Griffin, J.; Grusing, S. Systemic Pharmacologic Therapies for Low Back Pain: A Systematic Review for an American College of Physicians Clinical Practice Guideline. Ann. Intern. Med. 2017, 166, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Nijs, J.; George, S.Z.; Clauw, D.J.; Fernández-de-las-Peñas, C.; Kosek, E.; Ickmans, K.; Fernández-Carnero, J.; Polli, A.; Kapreli, E.; Huysmans, E.; et al. Central sensitisation in chronic pain conditions: Latest discoveries and their potential for precision medicine. Lancet Rheumatol. 2021, 3, e383–e392. [Google Scholar] [CrossRef] [PubMed]
- Kregel, J.; Meeus, M.; Malfliet, A.; Dolphens, M.; Danneels, L.; Nijs, J.; Cagnie, B. Structural and functional brain abnormalities in chronic low back pain: A systematic review. Semin. Arthritis Rheum. 2015, 45, 229–237. [Google Scholar] [CrossRef]
- Albrecht, D.S.; Forsberg, A.; Sandstrom, A.; Bergan, C.; Kadetoff, D.; Protsenko, E.; Lampa, J.; Lee, Y.C.; Hoglund, C.O.; Catana, C.; et al. Brain glial activation in fibromyalgia—A multi-site positron emission tomography investigation. Brain Behav. Immun. 2019, 75, 72–83. [Google Scholar] [CrossRef]
- Ji, R.R.; Xu, Z.Z.; Gao, Y.J. Emerging targets in neuroinflammation-driven chronic pain. Nat. Rev. Drug Discov. 2014, 13, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Knotkova, H.; Hamani, C.; Sivanesan, E.; Le Beuffe, M.F.E.; Moon, J.Y.; Cohen, S.P.; Huntoon, M.A. Neuromodulation for chronic pain. Lancet 2021, 397, 2111–2124. [Google Scholar] [CrossRef] [PubMed]
- Crunkhorn, S. Gene therapy for chronic pain. Nat. Rev. Drug Discov. 2021, 20, 344. [Google Scholar] [CrossRef] [PubMed]
- Descalzi, G.; Ikegami, D.; Ushijima, T.; Nestler, E.J.; Zachariou, V.; Narita, M. Epigenetic mechanisms of chronic pain. Trends Neurosci. 2015, 38, 237–246. [Google Scholar] [CrossRef]
- Gershman, A.; Sauria, M.E.G.; Guitart, X.; Vollger, M.R.; Hook, P.W.; Hoyt, S.J.; Jain, M.; Shumate, A.; Razaghi, R.; Koren, S.; et al. Epigenetic patterns in a complete human genome. Science 2022, 376, eabj5089. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Tamura, G.; Waki, T.; Kawata, S.; Terashima, M.; Nishizuka, S.; Motoyama, T. Demethylation of MAGE promoters during gastric cancer progression. Br. J. Cancer 2004, 90, 838–843. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M. DNA hypermethylation in disease: Mechanisms and clinical relevance. Epigenetics 2019, 14, 1141–1163. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Hajkova, P.; Ecker, J.R. Dynamic DNA methylation: In the right place at the right time. Science 2018, 361, 1336–1340. [Google Scholar] [CrossRef] [PubMed]
- Moller Johansen, L.; Gerra, M.C.; Arendt-Nielsen, L. Time course of DNA methylation in pain conditions: From experimental models to humans. Eur. J. Pain. 2021, 25, 296–312. [Google Scholar] [CrossRef]
- Polli, A.; Ghosh, M.; Bakusic, J.; Ickmans, K.; Monteyne, D.; Velkeniers, B.; Bekaert, B.; Godderis, L.; Nijs, J. DNA Methylation and Brain-Derived Neurotrophic Factor Expression Account for Symptoms and Widespread Hyperalgesia in Patients with Chronic Fatigue Syndrome and Comorbid Fibromyalgia. Arthritis Rheumatol. 2020, 72, 1936–1944. [Google Scholar] [CrossRef]
- Paoloni-Giacobino, A.; Luthi, F.; Stenz, L.; Le Carre, J.; Vuistiner, P.; Leger, B. Altered BDNF Methylation in Patients with Chronic Musculoskeletal Pain and High Biopsychosocial Complexity. J. Pain. Res. 2020, 13, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Menzies, V.; Lyon, D.E.; Archer, K.J.; Zhou, Q.; Brumelle, J.; Jones, K.H.; Gao, G.; York, T.P.; Jackson-Cook, C. Epigenetic alterations and an increased frequency of micronuclei in women with fibromyalgia. Nurs. Res. Pract. 2013, 2013, 795784. [Google Scholar] [CrossRef] [PubMed]
- Ciampi de Andrade, D.; Maschietto, M.; Galhardoni, R.; Gouveia, G.; Chile, T.; Victorino Krepischi, A.C.; Dale, C.S.; Brunoni, A.R.; Parravano, D.C.; Cueva Moscoso, A.S.; et al. Epigenetics insights into chronic pain: DNA hypomethylation in fibromyalgia-a controlled pilot-study. Pain 2017, 158, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Livshits, G.; Malkin, I.; Freidin, M.B.; Xia, Y.; Gao, F.; Wang, J.; Spector, T.D.; MacGregor, A.; Bell, J.T.; Williams, F.M.K. Genome-wide methylation analysis of a large population sample shows neurological pathways involvement in chronic widespread musculoskeletal pain. Pain 2017, 158, 1053–1062. [Google Scholar] [CrossRef]
- Burri, A.; Marinova, Z.; Robinson, M.D.; Kuhnel, B.; Waldenberger, M.; Wahl, S.; Kunze, S.; Gieger, C.; Livshits, G.; Williams, F. Are Epigenetic Factors Implicated in Chronic Widespread Pain? PLoS ONE 2016, 11, e0165548. [Google Scholar] [CrossRef] [PubMed]
- Sukenaga, N.; Ikeda-Miyagawa, Y.; Tanada, D.; Tunetoh, T.; Nakano, S.; Inui, T.; Satoh, K.; Okutani, H.; Noguchi, K.; Hirose, M. Correlation between DNA Methylation of TRPA1 and Chronic Pain States in Human Whole Blood Cells. Pain. Med. 2016, 17, 1906–1910. [Google Scholar] [CrossRef] [PubMed]
- Aroke, E.N.; Overstreet, D.S.; Penn, T.M.; Crossman, D.K.; Jackson, P.; Tollefsbol, T.O.; Quinn, T.L.; Yi, N.; Goodin, B.R. Identification of DNA methylation associated enrichment pathways in adults with non-specific chronic low back pain. Mol. Pain. 2020, 16. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; de Boer, I.; Sutherland, H.G.; Pijpers, J.A.; Bron, C.; Bainomugisa, C.; Haupt, L.M.; van den Maagdenberg, A.; Griffiths, L.R.; Nyholt, D.R.; et al. Alterations in DNA methylation associate with reduced migraine and headache days after medication withdrawal treatment in chronic migraine patients: A longitudinal study. Clin. Epigenetics 2023, 15, 190. [Google Scholar] [CrossRef] [PubMed]
- Stephens, K.E.; Levine, J.D.; Aouizerat, B.E.; Paul, S.M.; Abrams, G.; Conley, Y.P.; Miaskowski, C. Associations between genetic and epigenetic variations in cytokine genes and mild persistent breast pain in women following breast cancer surgery. Cytokine 2017, 99, 203–213. [Google Scholar] [CrossRef]
- Chidambaran, V.; Zhang, X.; Martin, L.J.; Ding, L.; Weirauch, M.T.; Geisler, K.; Stubbeman, B.L.; Sadhasivam, S.; Ji, H. DNA methylation at the mu-1 opioid receptor gene (OPRM1) promoter predicts preoperative, acute, and chronic postsurgical pain after spine fusion. Pharmgenomics Pers. Med. 2017, 10, 157–168. [Google Scholar] [CrossRef]
- Polli, A.; Hendrix, J.; Ickmans, K.; Bakusic, J.; Ghosh, M.; Monteyne, D.; Velkeniers, B.; Bekaert, B.; Nijs, J.; Godderis, L. Genetic and epigenetic regulation of Catechol-O-methyltransferase in relation to inflammation in chronic fatigue syndrome and Fibromyalgia. J. Transl. Med. 2022, 20, 487. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.E. Epigenetic Therapies for Cancer. N. Engl. J. Med. 2020, 383, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Topper, M.J.; Vaz, M.; Marrone, K.A.; Brahmer, J.R.; Baylin, S.B. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Fitzcharles, M.A.; Cohen, S.P.; Clauw, D.J.; Littlejohn, G.; Usui, C.; Hauser, W. Nociplastic pain: Towards an understanding of prevalent pain conditions. Lancet 2021, 397, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Denk, F.; McMahon, S.B. Chronic pain: Emerging evidence for the involvement of epigenetics. Neuron 2012, 73, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Spector, T.D.; Deloukas, P.; Bell, J.T.; Engelhardt, B.E. Predicting genome-wide DNA methylation using methylation marks, genomic position, and DNA regulatory elements. Genome Biol. 2015, 16, 14. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Fatemi, M.; Pao, M.M.; Jeong, S.; Gal-Yam, E.N.; Egger, G.; Weisenberger, D.J.; Jones, P.A. Footprinting of mammalian promoters: Use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level. Nucleic Acids Res. 2005, 33, e176. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Michalak, E.M.; Burr, M.L.; Bannister, A.J.; Dawson, M.A. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 573–589. [Google Scholar] [CrossRef]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [PubMed]
- Mortusewicz, O.; Schermelleh, L.; Walter, J.; Cardoso, M.C.; Leonhardt, H. Recruitment of DNA methyltransferase I to DNA repair sites. Proc. Natl. Acad. Sci. USA 2005, 102, 8905–8909. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Garriga, J.; Laumet, G.; Chen, S.R.; Zhang, Y.; Madzo, J.; Issa, J.J.; Pan, H.L.; Jelinek, J. Nerve Injury-Induced Chronic Pain Is Associated with Persistent DNA Methylation Reprogramming in Dorsal Root Ganglion. J. Neurosci. 2018, 38, 6090–6101. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xu, D.; Wang, T.; Zhang, Y.; Yang, X.; Wang, X.; Tang, Y. Epigenetic reduction of miR-214-3p upregulates astrocytic colony-stimulating factor-1 and contributes to neuropathic pain induced by nerve injury. Pain 2020, 161, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, Z.P.; Zheng, H.Z.; Zhang, S.; Zhang, Z.L.; Chen, Y.; You, Y.S.; Yang, M.H. Abnormal DNA methylation in the lumbar spinal cord following chronic constriction injury in rats. Neurosci. Lett. 2016, 610, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Gao, Y.; Jin, D.; Xu, X.; Tan, S.; Yu, H.; Zhao, Q.; Zhao, L.; Wang, W.; Wang, D. DNMT3a methylation in neuropathic pain. J. Pain. Res. 2017, 10, 2253–2262. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, C.; Cai, Y.; Pan, Z.Z. Pain vulnerability and DNA methyltransferase 3a involved in the affective dimension of chronic pain. Mol. Pain. 2017, 13. [Google Scholar] [CrossRef]
- Topham, L.; Gregoire, S.; Kang, H.; Salmon-Divon, M.; Lax, E.; Millecamps, M.; Szyf, M.; Stone, L.S. The transition from acute to chronic pain: Dynamic epigenetic reprogramming of the mouse prefrontal cortex up to 1 year after nerve injury. Pain 2020, 161, 2394–2409. [Google Scholar] [CrossRef]
- Sun, L.; Gu, X.; Pan, Z.; Guo, X.; Liu, J.; Atianjoh, F.E.; Wu, S.; Mo, K.; Xu, B.; Liang, L.; et al. Contribution of DNMT1 to Neuropathic Pain Genesis Partially through Epigenetically Repressing Kcna2 in Primary Afferent Neurons. J. Neurosci. 2019, 39, 6595–6607. [Google Scholar] [CrossRef]
- Zhao, J.Y.; Liang, L.; Gu, X.; Li, Z.; Wu, S.; Sun, L.; Atianjoh, F.E.; Feng, J.; Mo, K.; Jia, S.; et al. DNA methyltransferase DNMT3a contributes to neuropathic pain by repressing Kcna2 in primary afferent neurons. Nat. Commun. 2017, 8, 14712. [Google Scholar] [CrossRef]
- Fan, L.; Guan, X.; Wang, W.; Zhao, J.Y.; Zhang, H.; Tiwari, V.; Hoffman, P.N.; Li, M.; Tao, Y.X. Impaired neuropathic pain and preserved acute pain in rats overexpressing voltage-gated potassium channel subunit Kv1.2 in primary afferent neurons. Mol. Pain. 2014, 10, 8. [Google Scholar] [CrossRef]
- Zhao, X.; Tang, Z.; Zhang, H.; Atianjoh, F.E.; Zhao, J.Y.; Liang, L.; Wang, W.; Guan, X.; Kao, S.C.; Tiwari, V.; et al. A long noncoding RNA contributes to neuropathic pain by silencing Kcna2 in primary afferent neurons. Nat. Neurosci. 2013, 16, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.R.; Fan, L.C.; Wu, S.; Mao, Q.; Li, Z.; Lutz, B.; Xu, J.T.; Lu, Z.; Tao, Y.X. DNMT3a contributes to the development and maintenance of bone cancer pain by silencing Kv1.2 expression in spinal cord dorsal horn. Mol. Pain. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Wu, S.; Gu, X.; Du, S.; Mo, K.; Sun, L.; Cao, J.; Bekker, A.; Chen, L.; Tao, Y.X. DNMT3a-triggered downregulation of K(2p) 1.1 gene in primary sensory neurons contributes to paclitaxel-induced neuropathic pain. Int. J. Cancer 2019, 145, 2122–2134. [Google Scholar] [CrossRef]
- Li, Y.; Tatsui, C.E.; Rhines, L.D.; North, R.Y.; Harrison, D.S.; Cassidy, R.M.; Johansson, C.A.; Kosturakis, A.K.; Edwards, D.D.; Zhang, H.; et al. Dorsal root ganglion neurons become hyperexcitable and increase expression of voltage-gated T-type calcium channels (Cav3.2) in paclitaxel-induced peripheral neuropathy. Pain 2017, 158, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; North, R.Y.; Rhines, L.D.; Tatsui, C.E.; Rao, G.; Edwards, D.D.; Cassidy, R.M.; Harrison, D.S.; Johansson, C.A.; Zhang, H.; et al. DRG Voltage-Gated Sodium Channel 1.7 Is Upregulated in Paclitaxel-Induced Neuropathy in Rats and in Humans with Neuropathic Pain. J. Neurosci. 2018, 38, 1124–1136. [Google Scholar] [CrossRef]
- Kindler, C.H.; Yost, C.S. Two-pore domain potassium channels: New sites of local anesthetic action and toxicity. Reg. Anesth. Pain. Med. 2005, 30, 260–274. [Google Scholar] [CrossRef]
- Sun, L.; Zhao, J.Y.; Gu, X.; Liang, L.; Wu, S.; Mo, K.; Feng, J.; Guo, W.; Zhang, J.; Bekker, A.; et al. Nerve injury-induced epigenetic silencing of opioid receptors controlled by DNMT3a in primary afferent neurons. Pain 2017, 158, 1153–1165. [Google Scholar] [CrossRef]
- Woolf, C.J. Mu and delta opioid receptors diverge. Cell 2009, 137, 987–988. [Google Scholar] [CrossRef]
- Obara, I.; Parkitna, J.R.; Korostynski, M.; Makuch, W.; Kaminska, D.; Przewlocka, B.; Przewlocki, R. Local peripheral opioid effects and expression of opioid genes in the spinal cord and dorsal root ganglia in neuropathic and inflammatory pain. Pain 2009, 141, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Back, S.K.; Lee, J.; Hong, S.K.; Na, H.S. Loss of spinal mu-opioid receptor is associated with mechanical allodynia in a rat model of peripheral neuropathy. Pain 2006, 123, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.C.; Zhang, W.W.; Yang, T.; Guo, C.Y.; Cao, D.L.; Zhang, Z.J.; Gao, Y.J. Demethylation of G-Protein-Coupled Receptor 151 Promoter Facilitates the Binding of Kruppel-Like Factor 5 and Enhances Neuropathic Pain after Nerve Injury in Mice. J. Neurosci. 2018, 38, 10535–10551. [Google Scholar] [CrossRef]
- Jiang, B.C.; He, L.N.; Wu, X.B.; Shi, H.; Zhang, W.W.; Zhang, Z.J.; Cao, D.L.; Li, C.H.; Gu, J.; Gao, Y.J. Promoted Interaction of C/EBPalpha with Demethylated Cxcr3 Gene Promoter Contributes to Neuropathic Pain in Mice. J. Neurosci. 2017, 37, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Obata, K.; Noguchi, K. MAPK activation in nociceptive neurons and pain hypersensitivity. Life Sci. 2004, 74, 2643–2653. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Xue, Z.Y.; Yuan, Y.; Huang, S.S.; Fan, Y.H.; Zhu, X.; Wei, L. Upregulation of CXCR4 through promoter demethylation contributes to inflammatory hyperalgesia in rats. CNS Neurosci. Ther. 2018, 24, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Clouaire, T.; Stancheva, I. Methyl-CpG binding proteins: Specialized transcriptional repressors or structural components of chromatin? Cell Mol. Life Sci. 2008, 65, 1509–1522. [Google Scholar] [CrossRef]
- Mo, K.; Wu, S.; Gu, X.; Xiong, M.; Cai, W.; Atianjoh, F.E.; Jobe, E.E.; Zhao, X.; Tu, W.F.; Tao, Y.X. MBD1 Contributes to the Genesis of Acute Pain and Neuropathic Pain by Epigenetic Silencing of Oprm1 and Kcna2 Genes in Primary Sensory Neurons. J. Neurosci. 2018, 38, 9883–9899. [Google Scholar] [CrossRef]
- Manners, M.T.; Tian, Y.; Zhou, Z.; Ajit, S.K. MicroRNAs downregulated in neuropathic pain regulate MeCP2 and BDNF related to pain sensitivity. FEBS Open Bio 2015, 5, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Samaco, R.C.; Neul, J.L. Complexities of Rett syndrome and MeCP2. J. Neurosci. 2011, 31, 7951–7959. [Google Scholar] [CrossRef] [PubMed]
- Downs, J.; Geranton, S.M.; Bebbington, A.; Jacoby, P.; Bahi-Buisson, N.; Ravine, D.; Leonard, H. Linking MECP2 and pain sensitivity: The example of Rett syndrome. Am. J. Med. Genet. A 2010, 152A, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.U.; Hundley, R.J.; Wilson, A.K.; Warren, Z.; Vehorn, A.; Carvalho, C.M.; Lupski, J.R.; Ramocki, M.B. The behavioral phenotype in MECP2 duplication syndrome: A comparison with idiopathic autism. Autism Res. 2013, 6, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, C.; Guo, Q.L.; Yan, J.Q.; Zhu, X.Y.; Huang, C.S.; Zou, W.Y. Intrathecal 5-azacytidine inhibits global DNA methylation and methyl- CpG-binding protein 2 expression and alleviates neuropathic pain in rats following chronic constriction injury. Brain Res. 2011, 1418, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, D.; Dai, Z.; You, Y.; Chen, Y.; Lei, C.; Lv, Y.; Wang, Y. Intraperitoneal 5-Azacytidine Alleviates Nerve Injury-Induced Pain in Rats by Modulating DNA Methylation. Mol. Neurobiol. 2023, 60, 2186–2199. [Google Scholar] [CrossRef] [PubMed]
- Orefice, L.L.; Zimmerman, A.L.; Chirila, A.M.; Sleboda, S.J.; Head, J.P.; Ginty, D.D. Peripheral Mechanosensory Neuron Dysfunction Underlies Tactile and Behavioral Deficits in Mouse Models of ASDs. Cell 2016, 166, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Tochiki, K.K.; Cunningham, J.; Hunt, S.P.; Geranton, S.M. The expression of spinal methyl-CpG-binding protein 2, DNA methyltransferases and histone deacetylases is modulated in persistent pain states. Mol. Pain. 2012, 8, 14. [Google Scholar] [CrossRef]
- Ou, M.; Chen, Y.; Liu, J.; Zhang, D.; Yang, Y.; Shen, J.; Miao, C.; Tang, S.J.; Liu, X.; Mulkey, D.K.; et al. Spinal astrocytic MeCP2 regulates Kir4.1 for the maintenance of chronic hyperalgesia in neuropathic pain. Prog. Neurobiol. 2023, 224, 102436. [Google Scholar] [CrossRef]
- Bhattacherjee, A.; Mu, Y.; Winter, M.K.; Knapp, J.R.; Eggimann, L.S.; Gunewardena, S.S.; Kobayashi, K.; Kato, S.; Krizsan-Agbas, D.; Smith, P.G. Neuronal cytoskeletal gene dysregulation and mechanical hypersensitivity in a rat model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, E6952–E6961. [Google Scholar] [CrossRef]
- Zhang, R.; Huang, M.; Cao, Z.; Qi, J.; Qiu, Z.; Chiang, L.Y. MeCP2 plays an analgesic role in pain transmission through regulating CREB / miR-132 pathway. Mol. Pain. 2015, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Franchini, D.M.; Schmitz, K.M.; Petersen-Mahrt, S.K. 5-Methylcytosine DNA demethylation: More than losing a methyl group. Annu. Rev. Genet. 2012, 46, 419–441. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Y. Reversing DNA methylation: Mechanisms, genomics, and biological functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.K.; Bestor, T.H. The colorful history of active DNA demethylation. Cell 2008, 133, 1145–1148. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Kaas, G.A.; Zhong, C.; Eason, D.E.; Ross, D.L.; Vachhani, R.V.; Ming, G.L.; King, J.R.; Song, H.; Sweatt, J.D. TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron 2013, 79, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Pastor, W.A.; Aravind, L.; Rao, A. TETonic shift: Biological roles of TET proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed]
- Smeriglio, P.; Grandi, F.C.; Davala, S.; Masarapu, V.; Indelli, P.F.; Goodman, S.B.; Bhutani, N. Inhibition of TET1 prevents the development of osteoarthritis and reveals the 5hmC landscape that orchestrates pathogenesis. Sci. Transl. Med. 2020, 12, eaax2332. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Zhang, H.L.; Lu, X.; Du, H.; Li, Y.C.; Zhang, P.A.; Xu, G.Y. Targeting GATA1 and p2x7r Locus Binding in Spinal Astrocytes Suppresses Chronic Visceral Pain by Promoting DNA Demethylation. Neurosci. Bull. 2022, 38, 359–372. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, L.; Xu, Z.H.; Zhu, J.; Ma, J.L.; Gao, Y.P.; Xu, G.Y. Increased ten-eleven translocation methylcytosine dioxygenase one in dorsal root ganglion contributes to inflammatory pain in CFA rats. Mol. Pain. 2022, 18. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Xue, Z.Y.; Li, G.F.; Sun, M.L.; Zhang, M.; Hao, L.Y.; Tang, Q.Q.; Zhu, L.J.; Cao, J.L. DNA Hydroxymethylation by Ten-eleven Translocation Methylcytosine Dioxygenase 1 and 3 Regulates Nociceptive Sensitization in a Chronic Inflammatory Pain Model. Anesthesiology 2017, 127, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Zhang, M.; Ma, T.; Xue, Z.Y.; Li, G.F.; Hao, L.Y.; Zhu, L.J.; Li, Y.Q.; Ding, H.L.; Cao, J.L. Hydroxymethylation of microRNA-365-3p Regulates Nociceptive Behaviors via Kcnh2. J. Neurosci. 2016, 36, 2769–2781. [Google Scholar] [CrossRef] [PubMed]
- Chamessian, A.G.; Qadri, Y.J.; Cummins, M.; Hendrickson, M.; Berta, T.; Buchheit, T.; Van de Ven, T. 5-Hydroxymethylcytosine (5hmC) and Ten-eleven translocation 1-3 (TET1-3) proteins in the dorsal root ganglia of mouse: Expression and dynamic regulation in neuropathic pain. Somatosens. Mot. Res. 2017, 34, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, X.; Sun, Q.; Zhang, Y.; Liu, J.; Hu, T.; Wu, W.; Wei, C.; Liu, M.; Ding, Y.; et al. The upregulation of NLRP3 inflammasome in dorsal root ganglion by ten-eleven translocation methylcytosine dioxygenase 2 (TET2) contributed to diabetic neuropathic pain in mice. J. Neuroinflammation 2022, 19, 302. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.C.; Ho, Y.C.; Lai, C.Y.; Chou, D.; Wang, H.H.; Chen, G.D.; Lin, T.B.; Peng, H.Y. Melatonin impedes Tet1-dependent mGluR5 promoter demethylation to relieve pain. J. Pineal Res. 2017, 63, e12436. [Google Scholar] [CrossRef]
- Hsieh, M.C.; Lai, C.Y.; Ho, Y.C.; Wang, H.H.; Cheng, J.K.; Chau, Y.P.; Peng, H.Y. Tet1-dependent epigenetic modification of BDNF expression in dorsal horn neurons mediates neuropathic pain in rats. Sci. Rep. 2016, 6, 37411. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Wei, G.; Ji, F.; Jia, S.; Wu, S.; Guo, X.; He, L.; Pan, Z.; Miao, X.; Mao, Q.; et al. TET1 Overexpression Mitigates Neuropathic Pain through Rescuing the Expression of mu-Opioid Receptor and Kv1.2 in the Primary Sensory Neurons. Neurotherapeutics 2019, 16, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Stenz, L.; Zewdie, S.; Laforge-Escarra, T.; Prados, J.; La Harpe, R.; Dayer, A.; Paoloni-Giacobino, A.; Perroud, N.; Aubry, J.M. BDNF promoter I methylation correlates between post-mortem human peripheral and brain tissues. Neurosci. Res. 2015, 91, 1–7. [Google Scholar] [CrossRef]
- Nohesara, S.; Ghadirivasfi, M.; Mostafavi, S.; Eskandari, M.R.; Ahmadkhaniha, H.; Thiagalingam, S.; Abdolmaleky, H.M. DNA hypomethylation of MB-COMT promoter in the DNA derived from saliva in schizophrenia and bipolar disorder. J. Psychiatr. Res. 2011, 45, 1432–1438. [Google Scholar] [CrossRef]
- Davies, M.N.; Volta, M.; Pidsley, R.; Lunnon, K.; Dixit, A.; Lovestone, S.; Coarfa, C.; Harris, R.A.; Milosavljevic, A.; Troakes, C.; et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012, 13, R43. [Google Scholar] [CrossRef] [PubMed]
- Shumay, E.; Logan, J.; Volkow, N.D.; Fowler, J.S. Evidence that the methylation state of the monoamine oxidase A (MAOA) gene predicts brain activity of MAO A enzyme in healthy men. Epigenetics 2012, 7, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Massart, R.; Dymov, S.; Millecamps, M.; Suderman, M.; Gregoire, S.; Koenigs, K.; Alvarado, S.; Tajerian, M.; Stone, L.S.; Szyf, M. Overlapping signatures of chronic pain in the DNA methylation landscape of prefrontal cortex and peripheral T cells. Sci. Rep. 2016, 6, 19615. [Google Scholar] [CrossRef] [PubMed]
- Gerra, M.C.; Carnevali, D.; Pedersen, I.S.; Donnini, C.; Manfredini, M.; Gonzalez-Villar, A.; Trinanes, Y.; Pidal-Miranda, M.; Arendt-Nielsen, L.; Carrillo-de-la-Pena, M.T. DNA methylation changes in genes involved in inflammation and depression in fibromyalgia: A pilot study. Scand. J. Pain. 2021, 21, 372–383. [Google Scholar] [CrossRef]
- Gerra, M.C.; Carnevali, D.; Ossola, P.; Gonzalez-Villar, A.; Pedersen, I.S.; Trinanes, Y.; Donnini, C.; Manfredini, M.; Arendt-Nielsen, L.; Carrillo-de-la-Pena, M.T. DNA Methylation Changes in Fibromyalgia Suggest the Role of the Immune-Inflammatory Response and Central Sensitization. J. Clin. Med. 2021, 10, 4992. [Google Scholar] [CrossRef] [PubMed]
- Eller, O.C.; Glidden, N.; Knight, B.; McKearney, N.; Perry, M.; Bernier Carney, K.M.; Starkweather, A.; Young, E.E.; Baumbauer, K.M. A Role for Global DNA Methylation Level and IL2 Expression in the Transition from Acute to Chronic Low Back Pain. Front. Pain Res. 2021, 2, 744148. [Google Scholar] [CrossRef] [PubMed]
- Ikuno, A.; Akeda, K.; Takebayashi, S.I.; Shimaoka, M.; Okumura, K.; Sudo, A. Genome-wide analysis of DNA methylation profile identifies differentially methylated loci associated with human intervertebral disc degeneration. PLoS ONE 2019, 14, e0222188. [Google Scholar] [CrossRef] [PubMed]
- Bruehl, S.; Gamazon, E.R.; Van de Ven, T.; Buchheit, T.; Walsh, C.G.; Mishra, P.; Ramanujan, K.; Shaw, A. DNA methylation profiles are associated with complex regional pain syndrome after traumatic injury. Pain 2019, 160, 2328–2337. [Google Scholar] [CrossRef] [PubMed]
- Muller, D.; Grevet, E.H.; da Silva, B.S.; Charao, M.F.; Rovaris, D.L.; Bau, C.H.D. The neuroendocrine modulation of global DNA methylation in neuropsychiatric disorders. Mol. Psychiatry 2021, 26, 66–69. [Google Scholar] [CrossRef]
- Stefanska, B.; Huang, J.; Bhattacharyya, B.; Suderman, M.; Hallett, M.; Han, Z.G.; Szyf, M. Definition of the landscape of promoter DNA hypomethylation in liver cancer. Cancer Res. 2011, 71, 5891–5903. [Google Scholar] [CrossRef]
- Hansen, K.D.; Timp, W.; Bravo, H.C.; Sabunciyan, S.; Langmead, B.; McDonald, O.G.; Wen, B.; Wu, H.; Liu, Y.; Diep, D.; et al. Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011, 43, 768–775. [Google Scholar] [CrossRef]
- Gombert, S.; Rhein, M.; Winterpacht, A.; Munster, T.; Hillemacher, T.; Leffler, A.; Frieling, H. Transient receptor potential ankyrin 1 promoter methylation and peripheral pain sensitivity in Crohn’s disease. Clin. Epigenetics 2019, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, S.; Sukenaga, N.; Ohmuraya, M.; Matsuki, Y.; Maeda, L.; Takao, Y.; Hirose, M. Association between neuropathic pain characteristics and DNA methylation of transient receptor potential ankyrin 1 in human peripheral blood. Medicine 2020, 99, e19325. [Google Scholar] [CrossRef]
- Bautista, D.M.; Jordt, S.E.; Nikai, T.; Tsuruda, P.R.; Read, A.J.; Poblete, J.; Yamoah, E.N.; Basbaum, A.I.; Julius, D. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 2006, 124, 1269–1282. [Google Scholar] [CrossRef]
- Khan, S.; Patra, P.H.; Somerfield, H.; Benya-Aphikul, H.; Upadhya, M.; Zhang, X. IQGAP1 promotes chronic pain by regulating the trafficking and sensitization of TRPA1 channels. Brain 2023, 146, 2595–2611. [Google Scholar] [CrossRef]
- Tajerian, M.; Alvarado, S.; Millecamps, M.; Dashwood, T.; Anderson, K.M.; Haglund, L.; Ouellet, J.; Szyf, M.; Stone, L.S. DNA methylation of SPARC and chronic low back pain. Mol. Pain. 2011, 7, 65. [Google Scholar] [CrossRef]
- Bradshaw, A.D.; Sage, E.H. SPARC, a matricellular protein that functions in cellular differentiation and tissue response to injury. J. Clin. Investig. 2001, 107, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Stenz, L.; Carre, J.L.; Luthi, F.; Vuistiner, P.; Burrus, C.; Paoloni-Giacobino, A.; Leger, B. Genome-Wide Epigenomic Analyses in Patients with Nociceptive and Neuropathic Chronic Pain Subtypes Reveals Alterations in Methylation of Genes Involved in the Neuro-Musculoskeletal System. J. Pain. 2022, 23, 326–336. [Google Scholar] [CrossRef]
- Montesino-Goicolea, S.; Meng, L.; Rani, A.; Huo, Z.; Foster, T.C.; Fillingim, R.B.; Cruz-Almeida, Y. Enrichment of genomic pathways based on differential DNA methylation profiles associated with knee osteoarthritis pain. Neurobiol. Pain. 2022, 12, 100107. [Google Scholar] [CrossRef] [PubMed]
- Montesino-Goicolea, S.; Sinha, P.; Huo, Z.; Rani, A.; Foster, T.C.; Cruz-Almeida, Y. Enrichment of genomic pathways based on differential DNA methylation profiles associated with chronic musculoskeletal pain in older adults: An exploratory study. Mol. Pain. 2020, 16. [Google Scholar] [CrossRef]
- Achenbach, J.; Rhein, M.; Glahn, A.; Frieling, H.; Karst, M. Leptin promoter methylation in female patients with painful multisomatoform disorder and chronic widespread pain. Clin. Epigenetics 2022, 14, 13. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, S.; Millecamps, M.; Naso, L.; Do Carmo, S.; Cuello, A.C.; Szyf, M.; Stone, L.S. Therapeutic benefits of the methyl donor S-adenosylmethionine on nerve injury-induced mechanical hypersensitivity and cognitive impairment in mice. Pain 2017, 158, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Tajerian, M.; Alvarado, S.; Millecamps, M.; Vachon, P.; Crosby, C.; Bushnell, M.C.; Szyf, M.; Stone, L.S. Peripheral nerve injury is associated with chronic, reversible changes in global DNA methylation in the mouse prefrontal cortex. PLoS ONE 2013, 8, e55259. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Sun, H.; Fu, Y.; Li, Z.; Pais-Vieira, M.; Galhardo, V.; Neugebauer, V. Cognitive impairment in pain through amygdala-driven prefrontal cortical deactivation. J. Neurosci. 2010, 30, 5451–5464. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, S.; Tajerian, M.; Millecamps, M.; Suderman, M.; Stone, L.S.; Szyf, M. Peripheral nerve injury is accompanied by chronic transcriptome-wide changes in the mouse prefrontal cortex. Mol. Pain. 2013, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Descalzi, G.; Mitsi, V.; Purushothaman, I.; Gaspari, S.; Avrampou, K.; Loh, Y.E.; Shen, L.; Zachariou, V. Neuropathic pain promotes adaptive changes in gene expression in brain networks involved in stress and depression. Sci. Signal 2017, 10, eaaj1549. [Google Scholar] [CrossRef] [PubMed]
- Metz, A.E.; Yau, H.J.; Centeno, M.V.; Apkarian, A.V.; Martina, M. Morphological and functional reorganization of rat medial prefrontal cortex in neuropathic pain. Proc. Natl. Acad. Sci. USA 2009, 106, 2423–2428. [Google Scholar] [CrossRef] [PubMed]
- Shiers, S.; Price, T.J. Molecular, circuit, and anatomical changes in the prefrontal cortex in chronic pain. Pain 2020, 161, 1726–1729. [Google Scholar] [CrossRef] [PubMed]
- Parisien, M.; Samoshkin, A.; Tansley, S.N.; Piltonen, M.H.; Martin, L.J.; El-Hachem, N.; Dagostino, C.; Allegri, M.; Mogil, J.S.; Khoutorsky, A.; et al. Genetic pathway analysis reveals a major role for extracellular matrix organization in inflammatory and neuropathic pain. Pain 2019, 160, 932–944. [Google Scholar] [CrossRef]
- Baliki, M.N.; Chialvo, D.R.; Geha, P.Y.; Levy, R.M.; Harden, R.N.; Parrish, T.B.; Apkarian, A.V. Chronic pain and the emotional brain: Specific brain activity associated with spontaneous fluctuations of intensity of chronic back pain. J. Neurosci. 2006, 26, 12165–12173. [Google Scholar] [CrossRef]
- Alvarado, S.; Tajerian, M.; Suderman, M.; Machnes, Z.; Pierfelice, S.; Millecamps, M.; Stone, L.S.; Szyf, M. An epigenetic hypothesis for the genomic memory of pain. Front. Cell. Neurosci. 2015, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Monteggia, L.M. Role of DNA methylation and the DNA methyltransferases in learning and memory. Dialogues Clin. Neurosci. 2014, 16, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.H.; Niu, Z.; Liu, Y.; Liu, P.L.; Lin, X.L.; Zhang, L.; Chen, L.; Song, Y.; Sun, R.; Zhang, H.L. TET1-TRPV4 Signaling Contributes to Bone Cancer Pain in Rats. Brain Sci. 2023, 13, 644. [Google Scholar] [CrossRef] [PubMed]
- Viet, C.T.; Dang, D.; Ye, Y.; Ono, K.; Campbell, R.R.; Schmidt, B.L. Demethylating drugs as novel analgesics for cancer pain. Clin. Cancer Res. 2014, 20, 4882–4893. [Google Scholar] [CrossRef] [PubMed]
- Kuner, R.; Flor, H. Structural plasticity and reorganisation in chronic pain. Nat. Rev. Neurosci. 2016, 18, 20–30. [Google Scholar] [CrossRef]
- Xiong, H.Y.; Hendrix, J.; Schabrun, S.; Wyns, A.; Campenhout, J.V.; Nijs, J.; Polli, A. The Role of the Brain-Derived Neurotrophic Factor in Chronic Pain: Links to Central Sensitization and Neuroinflammation. Biomolecules 2024, 14, 71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, X.; Wang, W.; Lu, Y.G.; Pan, Z.Z. Brain-derived neurotrophic factor-mediated downregulation of brainstem K+-Cl- cotransporter and cell-type-specific GABA impairment for activation of descending pain facilitation. Mol. Pharmacol. 2013, 84, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Franco-Enzastiga, U.; Garcia, G.; Murbartian, J.; Gonzalez-Barrios, R.; Salinas-Abarca, A.B.; Sanchez-Hernandez, B.; Tavares-Ferreira, D.; Herrera, L.A.; Barragan-Iglesias, P.; Delgado-Lezama, R.; et al. Sex-dependent pronociceptive role of spinal α5-GABAA receptor and its epigenetic regulation in neuropathic rodents. J. Neurochem. 2021, 156, 897–916. [Google Scholar] [CrossRef] [PubMed]
- Sikandar, S.; Minett, M.S.; Millet, Q.; Santana-Varela, S.; Lau, J.; Wood, J.N.; Zhao, J. Brain-derived neurotrophic factor derived from sensory neurons plays a critical role in chronic pain. Brain 2018, 141, 1028–1039. [Google Scholar] [CrossRef]
- Sarchielli, P.; Mancini, M.L.; Floridi, A.; Coppola, F.; Rossi, C.; Nardi, K.; Acciarresi, M.; Pini, L.A.; Calabresi, P. Increased levels of neurotrophins are not specific for chronic migraine: Evidence from primary fibromyalgia syndrome. J. Pain. 2007, 8, 737–745. [Google Scholar] [CrossRef]
- Jablochkova, A.; Backryd, E.; Kosek, E.; Mannerkorpi, K.; Ernberg, M.; Gerdle, B.; Ghafouri, B. Unaltered low nerve growth factor and high brain-derived neurotrophic factor levels in plasma from patients with fibromyalgia after a 15-week progressive resistance exercise. J. Rehabil. Med. 2019, 51, 779–787. [Google Scholar] [CrossRef]
- Haas, L.; Portela, L.V.; Bohmer, A.E.; Oses, J.P.; Lara, D.R. Increased plasma levels of brain derived neurotrophic factor (BDNF) in patients with fibromyalgia. Neurochem. Res. 2010, 35, 830–834. [Google Scholar] [CrossRef]
- Simao, A.P.; Mendonca, V.A.; de Oliveira Almeida, T.M.; Santos, S.A.; Gomes, W.F.; Coimbra, C.C.; Lacerda, A.C. Involvement of BDNF in knee osteoarthritis: The relationship with inflammation and clinical parameters. Rheumatol. Int. 2014, 34, 1153–1157. [Google Scholar] [CrossRef]
- Laske, C.; Stransky, E.; Eschweiler, G.W.; Klein, R.; Wittorf, A.; Leyhe, T.; Richartz, E.; Kohler, N.; Bartels, M.; Buchkremer, G.; et al. Increased BDNF serum concentration in fibromyalgia with or without depression or antidepressants. J. Psychiatr. Res. 2007, 41, 600–605. [Google Scholar] [CrossRef]
- Stefani, L.C.; Leite, F.M.; da Graca, L.T.M.; Zanette, S.A.; de Souza, A.; Castro, S.M.; Caumo, W. BDNF and serum S100B levels according the spectrum of structural pathology in chronic pain patients. Neurosci. Lett. 2019, 706, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Nugraha, B.; Korallus, C.; Gutenbrunner, C. Serum level of brain-derived neurotrophic factor in fibromyalgia syndrome correlates with depression but not anxiety. Neurochem. Int. 2013, 62, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Zanette, S.A.; Dussan-Sarria, J.A.; Souza, A.; Deitos, A.; Torres, I.L.; Caumo, W. Higher serum S100B and BDNF levels are correlated with a lower pressure-pain threshold in fibromyalgia. Mol. Pain. 2014, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Kaminskas, E.; Farrell, A.T.; Wang, Y.C.; Sridhara, R.; Pazdur, R. FDA drug approval summary: Azacitidine (5-azacytidine, Vidaza) for injectable suspension. Oncologist 2005, 10, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Gore, S.D.; Jones, C.; Kirkpatrick, P. Decitabine. Nat. Rev. Drug Discov. 2006, 5, 891–892. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, F.; Lu, K.; Wei, A.; Jiang, Q.; Cao, W. PPARgamma preservation via promoter demethylation alleviates osteoarthritis in mice. Ann. Rheum. Dis. 2019, 78, 1420–1429. [Google Scholar] [CrossRef]
- Appel, C.K.; Scheff, N.N.; Viet, C.T.; Schmidt, B.L.; Heegaard, A.M. Decitabine attenuates nociceptive behavior in a murine model of bone cancer pain. Pain 2019, 160, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sahbaie, P.; Liang, D.; Li, W.; Shi, X.; Kingery, P.; Clark, J.D. DNA Methylation Modulates Nociceptive Sensitization after Incision. PLoS ONE 2015, 10, e0142046. [Google Scholar] [CrossRef]
- Liu, R.; Wu, X.M.; He, X.; Wang, R.Z.; Yin, X.Y.; Zhou, F.; Ji, M.H.; Shen, J.C. Contribution of DNA methyltransferases to spared nerve injury induced depression partially through epigenetically repressing Bdnf in hippocampus: Reversal by ketamine. Pharmacol. Biochem. Behav. 2021, 200, 173079. [Google Scholar] [CrossRef] [PubMed]
- Topham, L.; Gregoire, S.; Kang, H.; Salmon-Divon, M.; Lax, E.; Millecamps, M.; Szyf, M.; Stone, L. The methyl donor S-adenosyl methionine reverses the DNA methylation signature of chronic neuropathic pain in mouse frontal cortex. Pain Rep. 2021, 6, e944. [Google Scholar] [CrossRef]
- Rutjes, A.W.; Nuesch, E.; Reichenbach, S.; Juni, P. S-Adenosylmethionine for osteoarthritis of the knee or hip. Cochrane Database Syst. Rev. 2009, 2009, CD007321. [Google Scholar] [CrossRef] [PubMed]
- Velickovic, Z.; Pavlov Dolijanovic, S.; Stojanovic, N.; Janjic, S.; Kovacevic, L.; Soldatovic, I.; Radunovic, G. The short-term effect of glucosamine-sulfate, nonanimal chondroitin-sulfate, and S-adenosylmethionine combination on ultrasonography findings, inflammation, pain, and functionality in patients with knee osteoarthritis: A pilot, double-blind, randomized, placebo-controlled clinical trial. Arch. Rheumatol. 2023, 38, 521–541. [Google Scholar] [CrossRef] [PubMed]
- Choi, L.J.; Huang, J.S. A pilot study of S-adenosylmethionine in treatment of functional abdominal pain in children. Altern. Ther. Health Med. 2013, 19, 61–64. [Google Scholar] [PubMed]
- Vinkers, C.H.; Geuze, E.; van Rooij, S.J.H.; Kennis, M.; Schur, R.R.; Nispeling, D.M.; Smith, A.K.; Nievergelt, C.M.; Uddin, M.; Rutten, B.P.F.; et al. Successful treatment of post-traumatic stress disorder reverses DNA methylation marks. Mol. Psychiatry 2021, 26, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Weaver, I.C.; Champagne, F.A.; Brown, S.E.; Dymov, S.; Sharma, S.; Meaney, M.J.; Szyf, M. Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: Altering epigenetic marking later in life. J. Neurosci. 2005, 25, 11045–11054. [Google Scholar] [CrossRef]
- Lax, E.; Warhaftig, G.; Ohana, D.; Maayan, R.; Delayahu, Y.; Roska, P.; Ponizovsky, A.M.; Weizman, A.; Yadid, G.; Szyf, M. A DNA Methylation Signature of Addiction in T Cells and Its Reversal with DHEA Intervention. Front. Mol. Neurosci. 2018, 11, 322. [Google Scholar] [CrossRef]
- Brueckner, B.; Lyko, F. DNA methyltransferase inhibitors: Old and new drugs for an epigenetic cancer therapy. Trends Pharmacol. Sci. 2004, 25, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Sweatt, J.D. The emerging field of neuroepigenetics. Neuron 2013, 80, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.M.; Hemstedt, T.J.; Bading, H. Rescue of aging-associated decline in Dnmt3a2 expression restores cognitive abilities. Nat. Neurosci. 2012, 15, 1111–1113. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, K.; Pan, H.L. Epigenetic Mechanisms of Neural Plasticity in Chronic Neuropathic Pain. ACS Chem. Neurosci. 2022, 13, 432–441. [Google Scholar] [CrossRef]
- Halder, R.; Hennion, M.; Vidal, R.O.; Shomroni, O.; Rahman, R.U.; Rajput, A.; Centeno, T.P.; van Bebber, F.; Capece, V.; Garcia Vizcaino, J.C.; et al. DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat. Neurosci. 2016, 19, 102–110. [Google Scholar] [CrossRef]
- Santiago, M.; Antunes, C.; Guedes, M.; Sousa, N.; Marques, C.J. TET enzymes and DNA hydroxymethylation in neural development and function—How critical are they? Genomics 2014, 104, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Ma, D.K.; Mo, H.; Ball, M.P.; Jang, M.H.; Bonaguidi, M.A.; Balazer, J.A.; Eaves, H.L.; Xie, B.; Ford, E.; et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 2011, 14, 1345–1351. [Google Scholar] [CrossRef]
- Golzenleuchter, M.; Kanwar, R.; Zaibak, M.; Al Saiegh, F.; Hartung, T.; Klukas, J.; Smalley, R.L.; Cunningham, J.M.; Figueroa, M.E.; Schroth, G.P.; et al. Plasticity of DNA methylation in a nerve injury model of pain. Epigenetics 2015, 10, 200–212. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).