
The Role of Magnesium in Parkinson’s Disease: Status Quo and Implications for Future Research
 ,
,
Abstract
:1. Introduction
2. Methodology
3. Magnesium, an “Eminence Grise” of Cellular Physiology
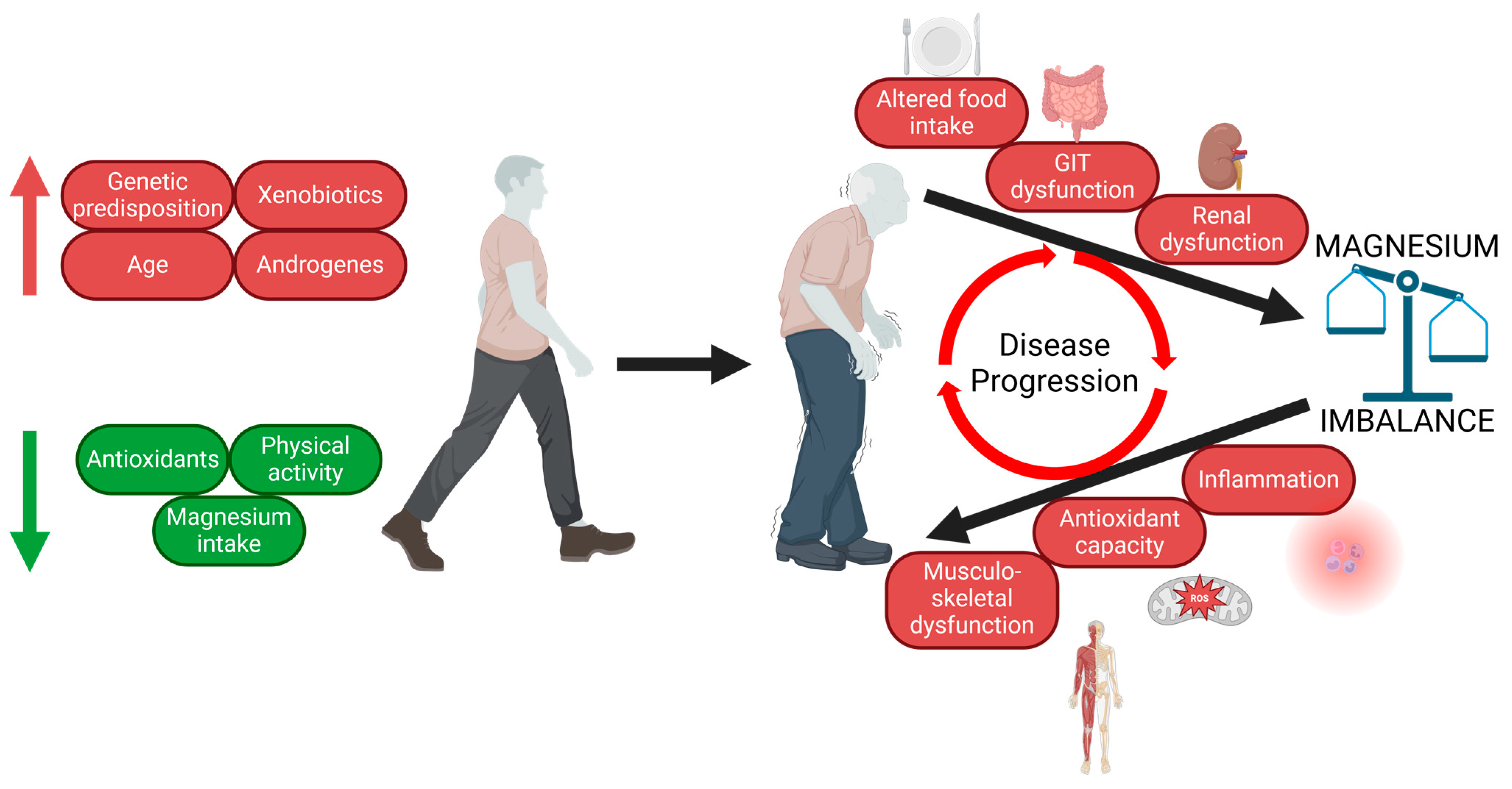
4. Pathomechanisms Involved in PD Onset and Progression
4.1. Oxidative Stress and Mitochondrial Dysfunction in PD
4.2. Neuroinflammation in PD
4.3. Autophagy and α-Synuclein Accumulation in PD
4.4. Dysmicrobiosis in PD
5. The Role of Mg2+ in PD Pathomechanisms
5.1. Magnesium and α-Synuclein
5.2. Magnesium and Guamanian Amyotrophic Lateral Sclerosis and Parkinsonism–Dementia Complex
5.3. Magnesium, Caffeine Intake, and Cigarette Smoking
5.4. Magnesium and Depression
5.5. Magnesium and Oxidative Stress
5.6. Magnesium and Gut Microbiota
5.7. Magnesium and Sleep Disturbances
5.8. Magnesium and Neuroinflammation
5.9. Magnesium and Ageing
6. Mg Intake and Balance in PD
6.1. Serum Magnesium Concentration in PD
6.2. Brain, Hair, and Cerebrospinal Fluid Magnesium Concentration in PD
6.3. Magnesium Intake Assessment
6.4. Magnesium Status Assessment
7. Animal and Cellular Models of PD and Mg Homeostasis
7.1. Cellular Models of PD and Magnesium Homeostasis
7.2. Animal Models of PD and Magnesium Homeostasis
8. Genetic Markers in Mg-Related Genes and PD
9. Future Perspectives
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of the Parkinson Pandemic. J. Park. Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef]
- Yang, W.; Hamilton, J.L.; Kopil, C.; Beck, J.C.; Tanner, C.M.; Albin, R.L.; Dorsey, E.R.; Dahodwala, N.; Cintina, I.; Hogan, P.; et al. Current and Projected Future Economic Burden of Parkinson’s Disease in the U.S. NPJ Park. Dis. 2020, 6, 15. [Google Scholar] [CrossRef]
- Murakami, H.; Shiraishi, T.; Umehara, T.; Omoto, S.; Iguchi, Y. Recent Advances in Drug Therapy for Parkinson’s Disease. Intern. Med. 2023, 62, 33–42. [Google Scholar] [CrossRef]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef]
- De Miranda, B.R.; Goldman, S.M.; Miller, G.W.; Greenamyre, J.T.; Dorsey, E.R. Preventing Parkinson’s Disease: An Environmental Agenda. J. Park. Dis. 2022, 12, 45–68. [Google Scholar] [CrossRef]
- Koziorowski, D.; Figura, M.; Milanowski, M.; Szlufik, S.; Alster, P.; Madetko, N.; Friedman, A. Mechanisms of Neurodegeneration in Various Forms of Parkinsonism—Similarities and Differences. Cells 2021, 10, 656. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A Meta-Analysis of Genome-Wide Association Studies Identifies 17 New Parkinson’s Disease Risk Loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Huang, Y.; Wei, J.; Cooper, A.; Morris, M.J. Parkinson’s Disease: From Genetics to Molecular Dysfunction and Targeted Therapeutic Approaches. Genes. Dis. 2023, 10, 786–798. [Google Scholar] [CrossRef]
- Balestrino, R.; Schapira, A.H.V. Parkinson Disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef]
- Burnashev, N.; Schoepfer, R.; Monyer, H.; Ruppersberg, J.P.; Günther, W.; Seeburg, P.H.; Sakmann, B. Control by Asparagine Residues of Calcium Permeability and Magnesium Blockade in the NMDA Receptor. Science 1992, 257, 1415–1419. [Google Scholar] [CrossRef]
- Ruppersberg, J.P.; Kitzing, E.V.; Schoepfer, R. The Mechanism of Magnesium Block of NMDA Receptors. Semin. Neurosci. 1994, 6, 87–96. [Google Scholar] [CrossRef]
- Feeney, K.A.; Hansen, L.L.; Putker, M.; Olivares-Yañez, C.; Day, J.; Eades, L.J.; Larrondo, L.F.; Hoyle, N.P.; O’Neill, J.S.; van Ooijen, G. Daily Magnesium Fluxes Regulate Cellular Timekeeping and Energy Balance. Nature 2016, 532, 375–379. [Google Scholar] [CrossRef]
- Yamanaka, R.; Shindo, Y.; Hotta, K.; Suzuki, K.; Oka, K. GABA-Induced Intracellular Mg2+ Mobilization Integrates and Coordinates Cellular Information Processing for the Maturation of Neural Networks. Curr. Biol. 2018, 28, 3984–3991. [Google Scholar] [CrossRef]
- Stangherlin, A.; O’Neill, J.S. Signal Transduction: Magnesium Manifests as a Second Messenger. Curr. Biol. 2018, 28, R1403–R1405. [Google Scholar] [CrossRef]
- Olza, J.; Aranceta-Bartrina, J.; González-Gross, M.; Ortega, R.; Serra-Majem, L.; Varela-Moreiras, G.; Gil, Á. Reported Dietary Intake, Disparity between the Reported Consumption and the Level Needed for Adequacy and Food Sources of Calcium, Phosphorus, Magnesium and Vitamin D in the Spanish Population: Findings from the ANIBES Study. Nutrients 2017, 9, 168. [Google Scholar] [CrossRef]
- Rosanoff, A.; Weaver, C.M.; Rude, R.K. Suboptimal Magnesium Status in the United States: Are the Health Consequences Underestimated? Nutr. Rev. 2012, 70, 153–164. [Google Scholar] [CrossRef]
- Belvisi, D.; Pellicciari, R.; Fabbrini, A.; Costanzo, M.; Pietracupa, S.; De Lucia, M.; Modugno, N.; Magrinelli, F.; Dallocchio, C.; Ercoli, T.; et al. Risk Factors of Parkinson Disease: Simultaneous Assessment, Interactions, and Etiologic Subtypes. Neurology 2020, 95, e2500–e2508. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Park. Dis. 2013, 3, 461. [Google Scholar] [CrossRef]
- Pissadaki, E.K.; Bolam, J.P. The Energy Cost of Action Potential Propagation in Dopamine Neurons: Clues to Susceptibility in Parkinson’s Disease. Front. Comput. Neurosci. 2013, 7, 13. [Google Scholar] [CrossRef]
- Liang, C.-L.; Wang, T.T.; Luby-Phelps, K.; German, D.C. Mitochondria Mass Is Low in Mouse Substantia Nigra Dopamine Neurons: Implications for Parkinson’s Disease. Exp. Neurol. 2007, 203, 370–380. [Google Scholar] [CrossRef]
- Venkateshappa, C.; Harish, G.; Mythri, R.B.; Mahadevan, A.; Srinivas Bharath, M.M.; Shankar, S.K. Increased Oxidative Damage and Decreased Antioxidant Function in Aging Human Substantia Nigra Compared to Striatum: Implications for Parkinson’s Disease. Neurochem. Res. 2012, 37, 358–369. [Google Scholar] [CrossRef]
- Tretter, L.; Sipos, I.; Adam-Vizi, V. Initiation of Neuronal Damage by Complex I Deficiency and Oxidative Stress in Parkinson’s Disease. Neurochem. Res. 2004, 29, 569–577. [Google Scholar] [CrossRef]
- Guo, J.-D.; Zhao, X.; Li, Y.; Li, G.-R.; Liu, X.-L. Damage to Dopaminergic Neurons by Oxidative Stress in Parkinson’s Disease (Review). Int. J. Mol. Med. 2018, 41, 1817–1825. [Google Scholar] [CrossRef]
- Noyce, A.J.; Bestwick, J.P.; Silveira-Moriyama, L.; Hawkes, C.H.; Giovannoni, G.; Lees, A.J.; Schrag, A. Meta-Analysis of Early Nonmotor Features and Risk Factors for Parkinson Disease. Ann. Neurol. 2012, 72, 893–901. [Google Scholar] [CrossRef]
- Seifar, F.; Dinasarapu, A.R.; Jinnah, H.A. Uric Acid in Parkinson′s Disease: What Is the Connection? Mov. Disord. 2022, 37, 2173–2183. [Google Scholar] [CrossRef]
- Roe, K. An Inflammation Classification System Using Cytokine Parameters. Scand. J. Immunol. 2021, 93, e12970. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and Immune Dysfunction in Parkinson Disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef]
- Cervellati, C.; Trentini, A.; Pecorelli, A.; Valacchi, G. Inflammation in Neurological Disorders: The Thin Boundary Between Brain and Periphery. Antioxid. Redox Signal. 2020, 33, 191–210. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of Mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Bendor, J.; Logan, T.; Edwards, R.H. The Function of α-Synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired Degradation of Mutant α-Synuclein by Chaperone-Mediated Autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Song, J.-X.; Lu, J.-H.; Liu, L.-F.; Chen, L.-L.; Durairajan, S.S.K.; Yue, Z.; Zhang, H.-Q.; Li, M. HMGB1 Is Involved in Autophagy Inhibition Caused by SNCA/a-Synuclein Overexpression: A Process Modulated by the Natural Autophagy Inducer Corynoxine B. Autophagy 2014, 10, 144–154. [Google Scholar] [CrossRef]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s Disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef]
- Ward, R.J.; Dexter, D.T.; Crichton, R.R. Ageing, Neuroinflammation and Neurodegeneration. FBS 2015, 7, 189–204. [Google Scholar] [CrossRef]
- Lowe, R.; Pountney, D.L.; Jensen, P.H.; Gai, W.P.; Voelcker, N.H. Calcium(II) Selectively Induces α-Synuclein Annular Oligomers via Interaction with the C-Terminal Domain. Protein Sci. 2004, 13, 3245–3252. [Google Scholar] [CrossRef]
- Golts, N.; Snyder, H.; Frasier, M.; Theisler, C.; Choi, P.; Wolozin, B. Magnesium Inhibits Spontaneous and Iron-Induced Aggregation of α-Synuclein. J. Biol. Chem. 2002, 277, 16116–16123. [Google Scholar] [CrossRef]
- Abeyawardhane, D.L.; Lucas, H.R. Iron Redox Chemistry and Implications in the Parkinson’s Disease Brain. Oxid. Med. Cell. Longev. 2019, 2019, 4609702. [Google Scholar] [CrossRef]
- Mochizuki, H.; Choong, C.-J.; Baba, K. Parkinson’s Disease and Iron. J. Neural Transm. 2020, 127, 181–187. [Google Scholar] [CrossRef]
- Hoyer, W.; Antony, T.; Cherny, D.; Heim, G.; Jovin, T.M.; Subramaniam, V. Dependence of Alpha-Synuclein Aggregate Morphology on Solution Conditions. J. Mol. Biol. 2002, 322, 383–393. [Google Scholar] [CrossRef]
- Koichi, I.; Zhi-Gang, X.; Takatoshi, U. The TRPM7 Channel in the Nervous and Cardiovascular Systems. Curr. Protein Pept. Sci. 2020, 21, 985–992. [Google Scholar]
- Abumaria, N.; Li, W.; Clarkson, A.N. Role of the Chanzyme TRPM7 in the Nervous System in Health and Disease. Cell. Mol. Life Sci. 2019, 76, 3301–3310. [Google Scholar] [CrossRef]
- Decker, A.R.; McNeill, M.S.; Lambert, A.M.; Overton, J.D.; Chen, Y.-C.; Lorca, R.A.; Johnson, N.A.; Brockerhoff, S.E.; Mohapatra, D.P.; MacArthur, H.; et al. Abnormal Differentiation of Dopaminergic Neurons in Zebrafish Trpm7 Mutant Larvae Impairs Development of the Motor Pattern. Dev. Biol. 2014, 386, 428–439. [Google Scholar] [CrossRef]
- Vink, R.; Cook, N.L.; Heuvel, C. van den Magnesium in Acute and Chronic Brain Injury: An Update. Magnes. Res. 2009, 22, 158–162. [Google Scholar] [CrossRef]
- Hermosura, M.C.; Nayakanti, H.; Dorovkov, M.V.; Calderon, F.R.; Ryazanov, A.G.; Haymer, D.S.; Garruto, R.M. A TRPM7 Variant Shows Altered Sensitivity to Magnesium That May Contribute to the Pathogenesis of Two Guamanian Neurodegenerative Disorders. Proc. Natl. Acad. Sci. USA 2005, 102, 11510–11515. [Google Scholar] [CrossRef]
- Friedland, R.P.; Armon, C. Tales of Pacific Tangles: Cycad Exposure and Guamanian Neurodegenerative Diseases. Neurology 2007, 68, 1759–1761. [Google Scholar] [CrossRef]
- Yasui, M.; Ota, K.; Yoshida, M. Effects of Low Calcium and Magnesium Dietary Intake on the Central Nervous System Tissues of Rats and Calcium-Magnesium Related Disorders in the Amyotrophic Lateral Sclerosis Focus in the Kii Peninsula of Japan. Magnes. Res. 1997, 10, 39–50. [Google Scholar]
- Hara, K.; Kokubo, Y.; Ishiura, H.; Fukuda, Y.; Miyashita, A.; Kuwano, R.; Sasaki, R.; Goto, J.; Nishizawa, M.; Kuzuhara, S.; et al. TRPM7 Is Not Associated with Amyotrophic Lateral Sclerosis-Parkinsonism Dementia Complex in the Kii Peninsula of Japan. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 153B, 310–313. [Google Scholar] [CrossRef]
- Durlach, J.; Bac, P.; Durlach, V.; Durlach, A.; Bara, M.; Guiet-Bara, A. Are Age-Related Neurodegenerative Diseases Linked with Various Types of Magnesium Depletion? Magnes. Res. 1997, 10, 339–353. [Google Scholar]
- Yasui, M.; Ota, K. Aluminum Decreases the Magnesium Concentration of Spinal Cord and Trabecular Bone in Rats Fed a Low Calcium, High Aluminum Diet. J. Neurol. Sci. 1998, 157, 37–41. [Google Scholar] [CrossRef]
- Purdey, M. Elevated Levels of Ferrimagnetic Metals in Foodchains Supporting the Guam Cluster of Neurodegeneration: Do Metal Nucleated Crystal Contaminants Evoke Magnetic Fields That Initiate the Progressive Pathogenesis of Neurodegeneration? Med. Hypotheses 2004, 63, 793–809. [Google Scholar] [CrossRef]
- Oyanagi, K.; Kawakami, E.; Kikuchi-Horie, K.; Ohara, K.; Ogata, K.; Takahama, S.; Wada, M.; Kihira, T.; Yasui, M. Magnesium Deficiency over Generations in Rats with Special References to the Pathogenesis of the Parkinsonism–Dementia Complex and Amyotrophic Lateral Sclerosis of Guam. Neuropathology 2006, 26, 115–128. [Google Scholar] [CrossRef]
- Taniguchi, R.; Nakagawasai, O.; Tan-No, K.; Yamadera, F.; Nemoto, W.; Sato, S.; Yaoita, F.; Tadano, T. Combined Low Calcium and Lack Magnesium Is a Risk Factor for Motor Deficit in Mice. Biosci. Biotechnol. Biochem. 2013, 77, 266–270. [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M.A. The Epidemiology of Parkinson’s Disease: Risk Factors and Prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Nechifor, M.; Chelarescu, D.; Mândreci, I.; Cartas, N. Magnesium Influence on Nicotine Pharmacodependence and Smoking. Magnes. Res. 2004, 17, 176–181. [Google Scholar]
- Bergman, E.A.; Massey, L.K.; Wise, K.J.; Sherrard, D.J. Effects of Dietary Caffeine on Renal Handling of Minerals in Adult Women. Life Sci. 1990, 47, 557–564. [Google Scholar] [CrossRef]
- Kynast-Gales, S.A.; Massey, L.K. Effect of Caffeine on Circadian Excretion of Urinary Calcium and Magnesium. J. Am. Coll. Nutr. 1994, 13, 467–472. [Google Scholar] [CrossRef]
- Nechifor, M. Magnesium in Addiction—A General View. Magnes. Res. 2018, 31, 90–98. [Google Scholar] [CrossRef]
- Cummings, J.L. Depression and Parkinson’s Disease: A Review. AJP 1992, 149, 443–454. [Google Scholar] [CrossRef]
- Reijnders, J.S.; Ehrt, U.; Weber, W.E.; Aarsland, D.; Leentjens, A.F. A Systematic Review of Prevalence Studies of Depression in Parkinson’s Disease. Mov. Disord. 2008, 23, 183–189. [Google Scholar] [CrossRef]
- Chikatimalla, R.; Dasaradhan, T.; Koneti, J.; Cherukuri, S.P.; Kalluru, R.; Gadde, S. Depression in Parkinson’s Disease: A Narrative Review. Cureus 2022, 14, e27750. [Google Scholar] [CrossRef]
- Mayberg, H.S.; Solomon, D.H. Depression in Parkinson’s Disease: A Biochemical and Organic Viewpoint. Adv. Neurol. 1995, 65, 49–60. [Google Scholar]
- Yamamoto, M. Depression in Parkinson’s Disease: Its Prevalence, Diagnosis, and Neurochemical Background. J. Neurol. 2001, 248, III5–III11. [Google Scholar] [CrossRef]
- Tarleton, E.K.; Kennedy, A.G.; Rose, G.L.; Crocker, A.; Littenberg, B. The Association between Serum Magnesium Levels and Depression in an Adult Primary Care Population. Nutrients 2019, 11, 1475. [Google Scholar] [CrossRef]
- Sun, C.; Wang, R.; Li, Z.; Zhang, D. Dietary Magnesium Intake and Risk of Depression. J. Affect. Disord. 2019, 246, 627–632. [Google Scholar] [CrossRef]
- Tarleton, E.K.; Littenberg, B.; MacLean, C.D.; Kennedy, A.G.; Daley, C. Role of Magnesium Supplementation in the Treatment of Depression: A Randomized Clinical Trial. PLoS ONE 2017, 12, e0180067. [Google Scholar] [CrossRef]
- Iseri, L.T.; French, J.H. Magnesium: Nature’s Physiologic Calcium Blocker. Am. Heart J. 1984, 108, 188–193. [Google Scholar] [CrossRef]
- Górska, N.; Słupski, J.P.; Szałach, Ł.; Włodarczyk, A.; Szarmach, J.; Jakuszkowiak-Wojten, K.; Gałuszko-Węgielnik, M.; Wilkowska, A.; Wiglusz, M.S.; Cubała, W.J. Magnesium and Ketamine in the Treatment of Depression. Psychiatr. Danub. 2019, 31, 549–553. [Google Scholar]
- Hartwig, A. Role of Magnesium in Genomic Stability. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2001, 475, 113–121. [Google Scholar] [CrossRef]
- Maguire, D.; Neytchev, O.; Talwar, D.; McMillan, D.; Shiels, P.G. Telomere Homeostasis: Interplay with Magnesium. Int. J. Mol. Sci. 2018, 19, 157. [Google Scholar] [CrossRef]
- Petrović, J.; Stanić, D.; Dmitrašinović, G.; Plećaš-Solarović, B.; Ignjatović, S.; Batinić, B.; Popović, D.; Pešić, V. Magnesium Supplementation Diminishes Peripheral Blood Lymphocyte DNA Oxidative Damage in Athletes and Sedentary Young Man. Oxid. Med. Cell. Longev. 2016, 2016, 2019643. [Google Scholar] [CrossRef]
- Regan, R.F.; Guo, Y. Magnesium Deprivation Decreases Cellular Reduced Glutathione and Causes Oxidative Neuronal Death in Murine Cortical Cultures. Brain Res. 2001, 890, 177–183. [Google Scholar] [CrossRef]
- Hans, C.P.; Chaudhary, D.P.; Bansal, D.D. Effect of Magnesium Supplementation on Oxidative Stress in Alloxanic Diabetic Rats. Magnes. Res. 2003, 16, 13–19. [Google Scholar]
- Kolisek, M.; Montezano, A.C.; Sponder, G.; Anagnostopoulou, A.; Vormann, J.; Touyz, R.M.; Aschenbach, J.R. PARK7/DJ-1 Dysregulation by Oxidative Stress Leads to Magnesium Deficiency: Implications in Degenerative and Chronic Diseases. Clin. Sci. 2015, 129, 1143–1150. [Google Scholar] [CrossRef]
- Winther, G.; Pyndt Jørgensen, B.M.; Elfving, B.; Nielsen, D.S.; Kihl, P.; Lund, S.; Sørensen, D.B.; Wegener, G. Dietary Magnesium Deficiency Alters Gut Microbiota and Leads to Depressive-like Behaviour. Acta Neuropsychiatr. 2015, 27, 168–176. [Google Scholar] [CrossRef]
- Pachikian, B.D.; Neyrinck, A.M.; Deldicque, L.; De Backer, F.C.; Catry, E.; Dewulf, E.M.; Sohet, F.M.; Bindels, L.B.; Everard, A.; Francaux, M.; et al. Changes in Intestinal Bifidobacteria Levels Are Associated with the Inflammatory Response in Magnesium-Deficient Mice. J. Nutr. 2010, 140, 509–514. [Google Scholar] [CrossRef]
- Wang, L.-J.; Yang, C.-Y.; Chou, W.-J.; Lee, M.-J.; Chou, M.-C.; Kuo, H.-C.; Yeh, Y.-M.; Lee, S.-Y.; Huang, L.-H.; Li, S.-C. Gut Microbiota and Dietary Patterns in Children with Attention-Deficit/Hyperactivity Disorder. Eur. Child Adolesc. Psychiatry 2020, 29, 287–297. [Google Scholar] [CrossRef]
- Del Chierico, F.; Trapani, V.; Petito, V.; Reddel, S.; Pietropaolo, G.; Graziani, C.; Masi, L.; Gasbarrini, A.; Putignani, L.; Scaldaferri, F.; et al. Dietary Magnesium Alleviates Experimental Murine Colitis through Modulation of Gut Microbiota. Nutrients 2021, 13, 4188. [Google Scholar] [CrossRef]
- Schiopu, C.; Ștefănescu, G.; Diaconescu, S.; Bălan, G.G.; Gimiga, N.; Rusu, E.; Moldovan, C.A.; Popa, B.; Tataranu, E.; Olteanu, A.V.; et al. Magnesium Orotate and the Microbiome-Gut-Brain Axis Modulation: New Approaches in Psychological Comorbidities of Gastrointestinal Functional Disorders. Nutrients 2022, 14, 1567. [Google Scholar] [CrossRef]
- Dodet, P.; Houot, M.; Leu-Semenescu, S.; Corvol, J.-C.; Lehéricy, S.; Mangone, G.; Vidailhet, M.; Roze, E.; Arnulf, I. Sleep Disorders in Parkinson’s Disease, an Early and Multiple Problem. npj Park. Dis. 2024, 10, 46. [Google Scholar] [CrossRef]
- Arab, A.; Rafie, N.; Amani, R.; Shirani, F. The Role of Magnesium in Sleep Health: A Systematic Review of Available Literature. Biol. Trace Elem. Res. 2023, 201, 121–128. [Google Scholar] [CrossRef]
- Abbasi, B.; Kimiagar, M.; Sadeghniiat, K.; Shirazi, M.M.; Hedayati, M.; Rashidkhani, B. The Effect of Magnesium Supplementation on Primary Insomnia in Elderly: A Double-Blind Placebo-Controlled Clinical Trial. J. Res. Med. Sci 2012, 17, 1161–1169. [Google Scholar]
- Hou, H.; Wang, L.; Fu, T.; Papasergi, M.; Yule, D.I.; Xia, H. Magnesium Acts as a Second Messenger in the Regulation of NMDA Receptor-Mediated CREB Signaling in Neurons. Mol. Neurobiol. 2020, 57, 2539–2550. [Google Scholar] [CrossRef]
- Papadopol, V.; Nechifor, M. Magnesium in Neuroses and Neuroticism. In Magnesium in the Central Nervous System; Vink, R., Nechifor, M., Eds.; University of Adelaide Press: Adelaide, Australia, 2011; ISBN 978-0-9870730-5-1. [Google Scholar]
- Siegel, J.M. The Neurotransmitters of Sleep. J. Clin. Psychiatry 2004, 65, 4–7. [Google Scholar]
- Irwin, M.R.; Olmstead, R.; Carroll, J.E. Sleep Disturbance, Sleep Duration, and Inflammation: A Systematic Review and Meta-Analysis of Cohort Studies and Experimental Sleep Deprivation. Biol. Psychiatry 2016, 80, 40–52. [Google Scholar] [CrossRef]
- Song, Y.; Li, T.Y.; van Dam, R.M.; Manson, J.E.; Hu, F.B. Magnesium Intake and Plasma Concentrations of Markers of Systemic Inflammation and Endothelial Dysfunction in Women. Am. J. Clin. Nutr. 2007, 85, 1068–1074. [Google Scholar] [CrossRef]
- Guerrero-Romero, F.; Bermudez-Peña, C.; Rodríguez-Morán, M. Severe Hypomagnesemia and Low-Grade Inflammation in Metabolic Syndrome. Magnes. Res. 2011, 24, 45–53. [Google Scholar] [CrossRef]
- Song, Y.; Ridker, P.M.; Manson, J.E.; Cook, N.R.; Buring, J.E.; Liu, S. Magnesium Intake, C-Reactive Protein, and the Prevalence of Metabolic Syndrome in Middle-Aged and Older U.S. Women. Diabetes Care 2005, 28, 1438–1444. [Google Scholar] [CrossRef]
- Shahi, A.; Aslani, S.; Ataollahi, M.; Mahmoudi, M. The Role of Magnesium in Different Inflammatory Diseases. Inflammopharmacol 2019, 27, 649–661. [Google Scholar] [CrossRef]
- Maier, J.A.M.; Locatelli, L.; Fedele, G.; Cazzaniga, A.; Mazur, A. Magnesium and the Brain: A Focus on Neuroinflammation and Neurodegeneration. Int. J. Mol. Sci. 2022, 24, 223. [Google Scholar] [CrossRef]
- Weglicki, W.B.; Chmielinska, J.J.; Tejero-Taldo, M.I.; Kramer, J.H.; Spurney, C.; Viswalingham, K.; Lu, B.; Mak, I.T. Neutral Endopeptidase Inhibition Enhances Substance P Mediated Inflammation Due to Hypomagnesemia. Magnes. Res. 2009, 22, 167S–173S. [Google Scholar] [CrossRef]
- Zhu, J.; Qu, C.; Lu, X.; Zhang, S. Activation of Microglia by Histamine and Substance, P. Cell. Physiol. Biochem. 2014, 34, 768–780. [Google Scholar] [CrossRef]
- Gao, F.; Ding, B.; Zhou, L.; Gao, X.; Guo, H.; Xu, H. Magnesium Sulfate Provides Neuroprotection in Lipopolysaccharide-Activated Primary Microglia by Inhibiting NF-κB Pathway. J. Surg. Res. 2013, 184, 944–950. [Google Scholar] [CrossRef]
- Minnich, V.; Smith, M.B.; Brauner, M.J.; Majerus, P.W. Glutathione Biosynthesis in Human Erythrocytes. J. Clin. Investig. 1971, 50, 507–513. [Google Scholar] [CrossRef]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s Disease: Why Is Advancing Age the Biggest Risk Factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef]
- Barbagallo, M.; Dominguez, L.J. Magnesium and Aging. Curr. Pharm Des. 2010, 16, 832–839. [Google Scholar] [CrossRef]
- Rubin, H. Magnesium: The Missing Element in Molecular Views of Cell Proliferation Control. Bioessays 2005, 27, 311–320. [Google Scholar] [CrossRef]
- Tao, M.-H.; Liu, J.; Cervantes, D. Association between Magnesium Intake and Cognition in US Older Adults: National Health and Nutrition Examination Survey (NHANES) 2011 to 2014. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2022, 8, e12250. [Google Scholar] [CrossRef]
- Eaton, S.B.; Eaton III, S.B. Paleolithic vs. Modern Diets—Slected Pathophysiological Implications. Eur J Nutr 2000, 39, 67–70. [Google Scholar] [CrossRef]
- EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA). Scientific Opinion on Dietary Reference Values for Magnesium. EFSA J. 2015, 13, 4186. [Google Scholar] [CrossRef]
- Rosanoff, A.; Kumssa, D.B. Impact of Rising Body Weight and Cereal Grain Food Processing on Human Magnesium Nutrition. Plant Soil 2020, 457, 5–23. [Google Scholar] [CrossRef]
- Guo, W.; Hussain, N.; Liang, Z.; Yang, D. Magnesium Deficiency in Plants: An Urgent Problem. Crop J. 2016, 4, 83–91. [Google Scholar] [CrossRef]
- Sun, H. Association of Soil Selenium, Strontium, and Magnesium Concentrations with Parkinson’s Disease Mortality Rates in the USA. Environ. Geochem. Health 2018, 9, 349–357. [Google Scholar] [CrossRef]
- Sengupta, P. Potential Health Impacts of Hard Water. Int. J. Prev. Med. 2013, 4, 866–875. [Google Scholar]
- Breckenridge, C.B.; Berry, C.; Chang, E.T.; Sielken, R.L.; Mandel, J.S. Association between Parkinson’s Disease and Cigarette Smoking, Rural Living, Well-Water Consumption, Farming and Pesticide Use: Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0151841. [Google Scholar] [CrossRef]
- Schwab, R.S.; Poryali, A.; Ames, A. Normal Serum Magnesium Levels in Parkinson’s Disease. Neurology 1964, 14, 855. [Google Scholar] [CrossRef]
- Jin, X.; Liu, M.-Y.; Zhang, D.-F.; Gao, H.; Wei, M.-J. Elevated Circulating Magnesium Levels in Patients with Parkinson’s Disease: A Meta-Analysis. Neuropsychiatr. Dis. Treat. 2018, 14, 3159–3168. [Google Scholar] [CrossRef]
- Anirudhan, A.; Angulo-Bejarano, P.I.; Paramasivam, P.; Manokaran, K.; Kamath, S.M.; Murugesan, R.; Sharma, A.; Ahmed, S.S.S.J. RPL6: A Key Molecule Regulating Zinc- and Magnesium-Bound Metalloproteins of Parkinson’s Disease. Front Neurosci 2021, 15, 631892. [Google Scholar] [CrossRef]
- Yasui, M.; Kihira, T.; Ota, K. Calcium, Magnesium and Aluminum Concentrations in Parkinson’s Disease. Neurotoxicology 1992, 13, 593–600. [Google Scholar]
- Barbiroli, B.; Martinelli, P.; Patuelli, A.; Lodi, R.; Iotti, S.; Cortelli, P.; Montagna, P. Phosphorus Magnetic Resonance Spectroscopy in Multiple System Atrophy and Parkinson’s Disease. Mov. Disord. 1999, 14, 430–435. [Google Scholar] [CrossRef]
- Uitti, R.J.; Rajput, A.H.; Rozdilsky, B.; Bickis, M.; Wollin, T.; Yuen, W.K. Regional Metal Concentrations in Parkinson’s Disease, Other Chronic Neurological Diseases, and Control Brains. Can. J. Neurol. Sci. 1989, 16, 310–314. [Google Scholar] [CrossRef]
- Riederer, P.; Sofic, E.; Rausch, W.D.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B. Transition Metals, Ferritin, Glutathione, and Ascorbic Acid in Parkinsonian Brains. J. Neurochem. 1989, 52, 515–520. [Google Scholar] [CrossRef]
- Forte, G.; Alimonti, A.; Violante, N.; Di Gregorio, M.; Senofonte, O.; Petrucci, F.; Sancesario, G.; Bocca, B. Calcium, Copper, Iron, Magnesium, Silicon and Zinc Content of Hair in Parkinson’s Disease. J. Trace Elem. Med. Biol. 2005, 19, 195–201. [Google Scholar] [CrossRef]
- Rajput, K.; Afridi, H.I.; Kazi, T.G.; Talpur, F.N.; Baig, J.A. Sodium, Potassium, Calcium, and Magnesium in the Scalp Hair and Blood Samples Related to the Clinical Stages of the Parkinson’s Disease. Biol. Trace Elem. Res. 2020, 199, 2582–2589. [Google Scholar] [CrossRef]
- Bocca, B.; Alimonti, A.; Senofonte, O.; Pino, A.; Violante, N.; Petrucci, F.; Sancesario, G.; Forte, G. Metal Changes in CSF and Peripheral Compartments of Parkinsonian Patients. J. Neurol. Sci. 2006, 248, 23–30. [Google Scholar] [CrossRef]
- Hozumi, I.; Hasegawa, T.; Honda, A.; Ozawa, K.; Hayashi, Y.; Hashimoto, K.; Yamada, M.; Koumura, A.; Sakurai, T.; Kimura, A.; et al. Patterns of Levels of Biological Metals in CSF Differ among Neurodegenerative Diseases. J. Neurol. Sci. 2011, 303, 95–99. [Google Scholar] [CrossRef]
- Alimonti, A.; Bocca, B.; Pino, A.; Ruggieri, F.; Forte, G.; Sancesario, G. Elemental Profile of Cerebrospinal Fluid in Patients with Parkinson’s Disease. J. Trace Elem. Med. Biol. 2007, 21, 234–241. [Google Scholar] [CrossRef]
- Sanyal, J.; Ahmed, S.S.S.J.; Ng, H.K.T.; Naiya, T.; Ghosh, E.; Banerjee, T.K.; Lakshmi, J.; Guha, G.; Rao, V.R. Metallomic Biomarkers in Cerebrospinal Fluid and Serum in Patients with Parkinson’s Disease in Indian Population. Sci. Rep. 2016, 6, 35097. [Google Scholar] [CrossRef]
- Shi, M.T. [Determination of multiple chemical elements in CSF in Parkinson disease after intracerebral autotransplantation of the adrenal medulla]. Zhonghua Wai Ke Za Zhi Chin. J. Surg. 1991, 29, 129–132, 144. [Google Scholar]
- Maass, F.; Michalke, B.; Leha, A.; Boerger, M.; Zerr, I.; Koch, J.-C.; Tönges, L.; Bähr, M.; Lingor, P. Elemental Fingerprint as a Cerebrospinal Fluid Biomarker for the Diagnosis of Parkinson’s Disease. J. Neurochem. 2018, 145, 342–351. [Google Scholar] [CrossRef]
- Miyake, Y.; Tanaka, K.; Fukushima, W.; Sasaki, S.; Kiyohara, C.; Tsuboi, Y.; Yamada, T.; Oeda, T.; Miki, T.; Kawamura, N.; et al. Dietary Intake of Metals and Risk of Parkinson’s Disease: A Case-Control Study in Japan. J. Neurol. Sci. 2011, 306, 98–102. [Google Scholar] [CrossRef]
- Powers, K.M.; Smith-Weller, T.; Franklin, G.M.; Longstreth, W.T.; Swanson, P.D.; Checkoway, H. Parkinson’s Disease Risks Associated with Dietary Iron, Manganese, and Other Nutrient Intakes. Neurology 2003, 60, 1761–1766. [Google Scholar] [CrossRef]
- Sukumar, D.; DeLuccia, R.; Cheung, M.; Ramadoss, R.; Ng, T.; Lamoureux, A. Validation of a Newly Developed Food Frequency Questionnaire to Assess Dietary Intakes of Magnesium. Nutrients 2019, 11, 2789. [Google Scholar] [CrossRef]
- Rosanoff, A.; West, C.; Elin, R.J.; Micke, O.; Baniasadi, S.; Barbagallo, M.; Campbell, E.; Cheng, F.-C.; Costello, R.B.; Gamboa-Gomez, C.; et al. Recommendation on an Updated Standardization of Serum Magnesium Reference Ranges. Eur. J. Nutr. 2022, 61, 3697–3706. [Google Scholar] [CrossRef]
- Chia, S.J.; Tan, E.-K.; Chao, Y.-X. Historical Perspective: Models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 2464. [Google Scholar] [CrossRef]
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.-Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson’s Disease. New Engl. J. Med. 2020, 382, 1926–1932. [Google Scholar] [CrossRef]
- Smits, L.M.; Schwamborn, J.C. Midbrain Organoids: A New Tool to Investigate Parkinson’s Disease. Front. Cell Dev. Biol. 2020, 8, 359. [Google Scholar] [CrossRef]
- Shindo, Y.; Yamanaka, R.; Suzuki, K.; Hotta, K.; Oka, K. Intracellular Magnesium Level Determines Cell Viability in the MPP+ Model of Parkinson’s Disease. Biochim. Et Biophys. Acta (BBA) Mol. Cell Res. 2015, 1853, 3182–3191. [Google Scholar] [CrossRef]
- Kolisek, M.; Sponder, G.; Pilchova, I.; Cibulka, M.; Tatarkova, Z.; Werner, T.; Racay, P. Magnesium Extravaganza: A Critical Compendium of Current Research into Cellular Mg2+ Transporters Other than TRPM6/7. Rev. Physiol. Biochem. Pharmacol. 2019, 176, 65–105. [Google Scholar] [CrossRef]
- Shindo, Y.; Yamanaka, R.; Suzuki, K.; Hotta, K.; Oka, K. Altered Expression of Mg(2+) Transport Proteins during Parkinson’s Disease-like Dopaminergic Cell Degeneration in PC12 Cells. Biochim. Biophys. Acta 2016, 1863, 1979–1984. [Google Scholar] [CrossRef]
- Lin, L.; Ke, Z.; Lv, M.; Lin, R.; Wu, B.; Zheng, Z. Effects of MgSO4 and Magnesium Transporters on 6-Hydroxydopamine-Induced SH-SY5Y Cells. Life Sci. 2017, 172, 48–54. [Google Scholar] [CrossRef]
- Lin, L.; Yan, M.; Wu, B.; Lin, R.; Zheng, Z. Expression of Magnesium Transporter SLC41A1 in the Striatum of 6-Hydroxydopamine-Induced Parkinsonian Rats. Brain Res. Bull. 2018, 142, 338–343. [Google Scholar] [CrossRef]
- Wu, C.; Xue, L.-D.; Su, L.-W.; Xie, J.-L.; Jiang, H.; Yu, X.-J.; Liu, H.-M. Magnesium Promotes the Viability and Induces Differentiation of Neural Stem Cells Both in Vitro and in Vivo. Neurol. Res. 2019, 41, 208–215. [Google Scholar] [CrossRef]
- Lupp, A.; Lücking, C.H.; Koch, R.; Jackisch, R.; Feuerstein, T.J. Inhibitory Effects of the Antiparkinsonian Drugs Memantine and Amantadine on N-Methyl-D-Aspartate-Evoked Acetylcholine Release in the Rabbit Caudate Nucleus in Vitro. J. Pharmacol. Exp. Ther. 1992, 263, 717–724. [Google Scholar]
- Tariq, M.; Khan, H.A.; al Moutaery, K.; al Deeb, S.M. Effect of Chronic Administration of Magnesium Sulfate on 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Neurotoxicity in Mice. Pharmacol. Toxicol. 1998, 82, 218–222. [Google Scholar] [CrossRef]
- Shindo, Y.; Yamanaka, R.; Hotta, K.; Oka, K. Inhibition of Mg2+ Extrusion Attenuates Glutamate Excitotoxicity in Cultured Rat Hippocampal Neurons. Nutrients 2020, 12, 2768. [Google Scholar] [CrossRef]
- Nowak, L.; Bregestovski, P.; Ascher, P.; Herbet, A.; Prochiantz, A. Magnesium Gates Glutamate-Activated Channels in Mouse Central Neurones. Nature 1984, 307, 462–465. [Google Scholar] [CrossRef]
- Lan, A.; Chen, J.; Zhao, Y.; Chai, Z.; Hu, Y. mTOR Signaling in Parkinson’s Disease. Neuromol Med 2017, 19, 1–10. [Google Scholar] [CrossRef]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR Kinase Structure, Mechanism and Regulation by the Rapamycin-Binding Domain. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef]
- Xie, J.; Cheng, C.; Zhu, X.Y.; Shen, Y.H.; Song, L.B.; Chen, H.; Chen, Z.; Liu, L.M.; Meng, Z.Q. Magnesium Transporter Protein Solute Carrier Family 41 Member 1 Suppresses Human Pancreatic Ductal Adenocarcinoma through Magnesium-Dependent Akt/mTOR Inhibition and Bax-Associated Mitochondrial Apoptosis. Aging 2019, 11, 2681–2698. [Google Scholar] [CrossRef]
- Chassain, C.; Eschalier, A.; Durif, F. Antidyskinetic Effect of Magnesium Sulfate in MPTP-Lesioned Monkeys. Exp. Neurol. 2003, 182, 490–496. [Google Scholar] [CrossRef]
- Morgenroth, V.H.; Hegstrand, L.R.; Roth, R.H.; Greengard, P. Evidence for Involvement of Protein Kinase in the Activation by Adenosine 3’:5’-Monophosphate of Brain Tyrosine 3-Monooxygenase. J. Biol. Chem. 1975, 250, 1946–1948. [Google Scholar] [CrossRef]
- Hashimoto, T.; Nishi, K.; Nagasao, J.; Tsuji, S.; Oyanagi, K. Magnesium Exerts Both Preventive and Ameliorating Effects in an in Vitro Rat Parkinson Disease Model Involving 1-Methyl-4-Phenylpyridinium (MPP+) Toxicity in Dopaminergic Neurons. Brain Res. 2008, 1197, 143–151. [Google Scholar] [CrossRef]
- Muroyama, A.; Inaka, M.; Matsushima, H.; Sugino, H.; Marunaka, Y.; Mitsumoto, Y. Enhanced Susceptibility to MPTP Neurotoxicity in Magnesium-Deficient C57BL/6N Mice. Neurosci. Res. 2009, 63, 72–75. [Google Scholar] [CrossRef]
- Shen, Y.; Dai, L.; Tian, H.; Xu, R.; Li, F.; Li, Z.; Zhou, J.; Wang, L.; Dong, J.; Sun, L. Treatment of Magnesium-L-Threonate Elevates The Magnesium Level in the Cerebrospinal Fluid and Attenuates Motor Deficits and Dopamine Neuron Loss in a Mouse Model of Parkinson’s Disease. Neuropsychiatr. Dis. Treat. 2019, 15, 3143–3153. [Google Scholar] [CrossRef]
- Kronbauer, M.; Metz, V.G.; Roversi, K.; Dias, V.T.; de David Antoniazzi, C.T.; da Silva Barcelos, R.C.; Burger, M.E. Influence of Magnesium Supplementation on Movement Side Effects Related to Typical Antipsychotic Treatment in Rats. Behav. Brain Res. 2017, 320, 400–411. [Google Scholar] [CrossRef]
- Sienaert, P.; van Harten, P.; Rhebergen, D. The Psychopharmacology of Catatonia, Neuroleptic Malignant Syndrome, Akathisia, Tardive Dyskinesia, and Dystonia. Handb. Clin. Neurol. 2019, 165, 415–428. [Google Scholar] [CrossRef]
- Kolisek, M.; Nestler, A.; Vormann, J.; Schweigel-Röntgen, M. Human Gene SLC41A1 Encodes for the Na+/Mg2+ Exchanger. Am. J. Physiol. Cell Physiol. 2012, 302, C318–C326. [Google Scholar] [CrossRef]
- Kolisek, M.; Launay, P.; Beck, A.; Sponder, G.; Serafini, N.; Brenkus, M.; Froschauer, E.M.; Martens, H.; Fleig, A.; Schweigel, M. SLC41A1 Is a Novel Mammalian Mg2+ Carrier. J. Biol. Chem. 2008, 283, 16235–16247. [Google Scholar] [CrossRef] [PubMed]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-Wide Association Study Identifies Common Variants at Four Loci as Genetic Risk Factors for Parkinson’s Disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.-K.; Kwok, H.-K.; Tan, L.C.; Zhao, W.-T.; Prakash, K.M.; Au, W.-L.; Pavanni, R.; Ng, Y.-Y.; Satake, W.; Zhao, Y.; et al. Analysis of GWAS-Linked Loci in Parkinson Disease Reaffirms PARK16 as a Susceptibility Locus. Neurology 2010, 75, 508–512. [Google Scholar] [CrossRef]
- Chang, X.-L.; Mao, X.-Y.; Li, H.-H.; Zhang, J.-H.; Li, N.-N.; Burgunder, J.-M.; Peng, R.; Tan, E.-K. Association of GWAS Loci with PD in China. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2011, 156, 334–339. [Google Scholar] [CrossRef]
- Chang, K.-H.; Chen, C.-M.; Chen, Y.-C.; Lyu, R.-K.; Chang, H.-S.; Ro, L.-S.; Lee-Chen, G.-J.; Wu, Y.-R. Association between PARK16 and Parkinson’s Disease in the Han Chinese Population: A Meta-Analysis. Neurobiol. Aging 2013, 34, 2442.e5–2442.e9. [Google Scholar] [CrossRef]
- Wang, L.; Cheng, L.; Li, N.-N.; Yu, W.-J.; Sun, X.-Y.; Peng, R. Genetic Analysis of SLC41A1 in Chinese Parkinson’s Disease Patients. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2015, 168, 706–711. [Google Scholar] [CrossRef]
- Madadi, F.; Khaniani, M.S.; Shandiz, E.E.; Ayromlou, H.; Najmi, S.; Emamalizadeh, B.; Taghavi, S.; Jamshidi, J.; Tafakhori, A.; Shahidi, G.-A.; et al. Genetic Analysis of the ZNF512B, SLC41A1, and ALDH2 Polymorphisms in Parkinson’s Disease in the Iranian Population. Genet. Test. Mol. Biomark. 2016, 20, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Tucci, A.; Nalls, M.A.; Houlden, H.; Revesz, T.; Singleton, A.B.; Wood, N.W.; Hardy, J.; Paisán-Ruiz, C. Genetic Variability at the PARK16 Locus. Eur. J. Hum. Genet. 2010, 18, 1356–1359. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Tian, J.; Mo, X.; Zhao, G.; Yin, X.; Pu, J.; Zhang, B. Genetic Variants in the RAB7L1 and SLC41A1 Genes of the PARK16 Locus in Chinese Parkinson’s Disease Patients. Int. J. Neurosci. 2011, 121, 632–636. [Google Scholar] [CrossRef]
- Cibulka, M.; Brodnanova, M.; Grendar, M.; Grofik, M.; Kurca, E.; Pilchova, I.; Osina, O.; Tatarkova, Z.; Dobrota, D.; Kolisek, M. SNPs Rs11240569, Rs708727, and Rs823156 in SLC41A1 Do Not Discriminate Between Slovak Patients with Idiopathic Parkinson’s Disease and Healthy Controls: Statistics and Machine-Learning Evidence. Int. J. Mol. Sci. 2019, 20, 4688. [Google Scholar] [CrossRef]
- Cibulka, M.; Brodnanova, M.; Grendar, M.; Necpal, J.; Benetin, J.; Han, V.; Kurca, E.; Nosal, V.; Skorvanek, M.; Vesely, B.; et al. Alzheimer’s Disease-Associated SNP Rs708727 in SLC41A1 May Increase Risk for Parkinson’s Disease: Report from Enlarged Slovak Study. Int. J. Mol. Sci. 2022, 23, 1604. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mut, J.V.; Heyn, H.; Silva, B.A.; Dixsaut, L.; Garcia-Esparcia, P.; Vidal, E.; Sayols, S.; Glauser, L.; Monteagudo-Sánchez, A.; Perez-Tur, J.; et al. PM20D1 Is a Quantitative Trait Locus Associated with Alzheimer’s Disease. Nat. Med. 2018, 24, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, Y.; Readhead, B.; Chen, K.; Su, Y.; Reiman, E.M.; Dudley, J.T. Longitudinal Data in Peripheral Blood Confirm That PM20D1 Is a Quantitative Trait Locus (QTL) for Alzheimer’s Disease and Implicate Its Dynamic Role in Disease Progression. Clin. Epigenetics 2020, 12, 189. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Tanaka, K.; Fukushima, W.; Kiyohara, C.; Sasaki, S.; Tsuboi, Y.; Oeda, T.; Shimada, H.; Kawamura, N.; Sakae, N.; et al. PARK16 Polymorphisms, Interaction with Smoking, and Sporadic Parkinson’s Disease in Japan. J. Neurol. Sci. 2016, 362, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.J.; Jung, Y.; Hong, M.; Kim, M.J.; You, S.; Kim, Y.J.; Kim, J.; Song, K. Alzheimer’s Disease and Parkinson’s Disease Genome-Wide Association Study Top Hits and Risk of Parkinson’s Disease in Korean Population. Neurobiol. Aging 2013, 34, 2695.e1–2695.e7. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.-P.; Mo, X.-Y.; Tian, J.; Zhao, G.-H.; Yin, X.-Z.; Jin, F.-Y.; Zhang, B.-R. An Association between the PARK16 Locus and Parkinson’s Disease in a Cohort from Eastern China. Park. Relat. Disord. 2011, 17, 737–739. [Google Scholar] [CrossRef] [PubMed]
- Mata, I.F.; Yearout, D.; Alvarez, V.; Coto, E.; de Mena, L.; Ribacoba, R.; Lorenzo-Betancor, O.; Samaranch, L.; Pastor, P.; Cervantes, S.; et al. Replication of MAPT and SNCA, but Not PARK16-18, as Susceptibility Genes for Parkinson’s Disease. Mov. Disord. 2011, 26, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Gopalai, A.A.; Ahmad-Annuar, A.; Li, H.-H.; Zhao, Y.; Lim, S.-Y.; Tan, A.H.; Lim, T.T.; Eow, G.B.; Santhi, P.; Shanthi, V.; et al. PARK16 Is Associated with PD in the Malaysian Population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2016, 171, 839–847. [Google Scholar] [CrossRef]
- Pihlstrøm, L.; Rengmark, A.; Bjørnarå, K.A.; Dizdar, N.; Fardell, C.; Forsgren, L.; Holmberg, B.; Larsen, J.P.; Linder, J.; Nissbrandt, H.; et al. Fine Mapping and Resequencing of the PARK16 Locus in Parkinson’s Disease. J. Hum. Genet. 2015, 60, 357–362. [Google Scholar] [CrossRef]
- Bai, Y.; Dong, L.; Huang, X.; Zheng, S.; Qiu, P.; Lan, F. Associations of Rs823128, Rs1572931, and Rs823156 Polymorphisms with Reduced Parkinson’s Disease Risks. Neuroreport 2017, 28, 936–941. [Google Scholar] [CrossRef]
- Kolisek, M.; Sponder, G.; Mastrototaro, L.; Smorodchenko, A.; Launay, P.; Vormann, J.; Schweigel-Röntgen, M. Substitution p.A350V in Na+/Mg2+ Exchanger SLC41A1, Potentially Associated with Parkinson’s Disease, Is a Gain-of-Function Mutation. PLoS ONE 2013, 8, e71096. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Wu, Y.-R.; Chen, W.-L.; Wang, H.-C.; Lee, C.-M.; Lee-Chen, G.-J.; Chen, C.-M. Variant R244H in Na+/Mg2+ Exchanger SLC41A1 in Taiwanese Parkinson’s Disease Is Associated with Loss of Mg2+ Efflux Function. Park. Relat. Disord. 2014, 20, 600–603. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Mg2+ Transporting System | Mode of Action Δ | Cellular Localisation Δ | Detected in Brain * |
|---|---|---|---|
| NIPA1 | Non-specific cation channel | Plasma membrane | + |
| NIPA2 | Non-specific cation channel | Plasma membrane | + |
| NIPAL1 | Non-specific cation channel | Plasma membrane | − |
| NIPAL4 | Non-specific cation channel | Plasma membrane | + |
| TRPM6 | Chanzyme | Plasma membrane | − |
| TRPM7 | Chanzyme | Plasma membrane | + |
| SLC41A1 | Na+/Mg2+ exchanger | Plasma membrane | + |
| SLC41A2 | Putative Mg2+ transporter | Undetermined | + |
| SLC41A3 | Na+-coupled Mg2+ transporter | IMM | + |
| MMGT1 | Mg2+ transporter | Golgi apparatus | + |
| MRS2 | Mg2+ channel | IMM | + |
| APC (SLC25A23, SLC25A24 SLC25A25, SLC25A41) | MgATP2−/HPO42− exchanger | IMM | + + + + |
| ATP13A2 | Mg2+ pump | Lysosomal membrane | + |
| ATP13A4 | Mg2+ pump | Endoplasmic reticulum | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cibulka, M.; Brodnanova, M.; Halasova, E.; Kurca, E.; Kolisek, M.; Grofik, M. The Role of Magnesium in Parkinson’s Disease: Status Quo and Implications for Future Research. Int. J. Mol. Sci. 2024, 25, 8425. https://doi.org/10.3390/ijms25158425
Cibulka M, Brodnanova M, Halasova E, Kurca E, Kolisek M, Grofik M. The Role of Magnesium in Parkinson’s Disease: Status Quo and Implications for Future Research. International Journal of Molecular Sciences. 2024; 25(15):8425. https://doi.org/10.3390/ijms25158425
Chicago/Turabian StyleCibulka, Michal, Maria Brodnanova, Erika Halasova, Egon Kurca, Martin Kolisek, and Milan Grofik. 2024. "The Role of Magnesium in Parkinson’s Disease: Status Quo and Implications for Future Research" International Journal of Molecular Sciences 25, no. 15: 8425. https://doi.org/10.3390/ijms25158425