Abstract
The transmembrane protein β-amyloid precursor protein (APP) is central to the pathophysiology of Alzheimer’s disease (AD). The β-amyloid hypothesis posits that aberrant processing of APP forms neurotoxic β-amyloid aggregates, which lead to the cognitive impairments observed in AD. Although numerous additional factors contribute to AD, there is a need to better understand the synaptic function of APP. We have found that Drosophila APP-like (APPL) has both shared and non-shared roles at the synapse with Kismet (Kis), a chromatin helicase binding domain (CHD) protein. Kis is the homolog of CHD7 and CHD8, both of which are implicated in neurodevelopmental disorders including CHARGE Syndrome and autism spectrum disorders, respectively. Loss of function mutations in kis and animals expressing human APP and BACE in their central nervous system show reductions in the glutamate receptor subunit, GluRIIC, the GTPase Rab11, and the bone morphogenetic protein (BMP), pMad, at the Drosophila larval neuromuscular junction (NMJ). Similarly, processes like endocytosis, larval locomotion, and neurotransmission are deficient in these animals. Our pharmacological and epistasis experiments indicate that there is a functional relationship between Kis and APPL, but Kis does not regulate appl expression at the larval NMJ. Instead, Kis likely influences the synaptic localization of APPL, possibly by promoting rab11 transcription. These data identify a potential mechanistic connection between chromatin remodeling proteins and aberrant synaptic function in AD.
1. Introduction
Alzheimer’s disease (AD) is characterized by the loss of synapses and neurons in the cortex and hippocampus leading to cognitive impairments [1]. Neuronal dysfunction in AD is correlated with the accumulation of synaptic amyloid beta (Aβ) oligomers [2], which are generated by cleavage of the transmembrane protein, amyloid precursor protein (APP) [3]. APP is composed of a large extracellular domain, which may function in cell adhesion, ligand binding, and dimerization, a transmembrane domain, and a short C-terminus [3]. Cleavage by α- or β-secretase produces the soluble ectodomains, APPα or APPβ, respectively. Subsequent cleavage of αAPP by γ-secretase releases the P3 peptide in the non-amyloidogenic pathway while βAPP cleavage by γ-secretase yields Aβ in the amyloidogenic pathway. Both pathways produce an APP intracellular domain (AICD), which has putative roles in intracellular signaling [4]. Both the synaptic levels of APP [5] and the localization of APP to endosomes versus plasma membrane [6,7] influence Aβ production.
APP, along with the related proteins APP-like (APPL) 1 and APPL2, play partly functionally redundant roles in cell adhesion, proliferation, differentiation, neurite outgrowth, and synaptogenesis [3]. In the developing nervous system, the initial expression of APP and APPL correlates with neurogenesis [8], neurite outgrowth, and synaptogenesis [9]. APP is expressed in both pre- and postsynaptic neurons and loss-of-function mutations reduce hippocampal CA1 and cortical dendritic complexity in older animals [10]. Similarly, fly appld loss-of-function mutants exhibit a significant reduction in the number of boutons at the neuromuscular junction (NMJ) [11]. APP is important for synaptic plasticity and memory in mice [12,13] and flies [14] suggesting critical synaptic functions. Decades of research, however, have failed to yield a clear understanding of the non-pathological roles of APP. Further, focusing on APP and Aβ production has demonstrated little therapeutic benefit for AD patients [15]. AD is multi-faceted and includes perturbations in cytoskeletal dynamics, endolysosomal trafficking, lipid homeostasis, mitochondrial function, RNA splicing, microglial immune activation, and blood–brain barrier integrity [16].
To better understand the pathology of AD, next-generation sequencing and transcriptomic analyses have identified disrupted genomic loci in affected patients. Hundreds of mutations are correlated with late-onset AD including genes that regulate synaptic transmission (downregulated), biological adhesion (upregulated) [17,18,19], and the synaptic vesicle pathway [20]. The altered transcriptional programs associated with AD are caused by structural modifications to chromatin, which were first documented four decades ago [21,22]. Chromatin structure is established and maintained by a combination of epigenetic regulators, including chromatin modifying and chromatin remodeling enzymes, that collectively determine the accessibility of RNA polymerase II to proximal promoter sequences [23]. Expression of genes encoding enzymes that influence chromatin assembly and disassembly and nucleosome organization is altered in AD mouse models [24]. As such, epigenetic changes including DNA methylation and histone acetylation and methylation are found in AD patients and mouse models of AD [25].
Chromatin remodeling enzymes orchestrate epigenetic modifications by using ATP to slide nucleosomes relative to DNA, interchange histone protein subunits, and remove histones from DNA [23]. The chromodomain helicase DNA (CHD) binding protein family of chromatin remodelers plays critical roles in neurodevelopment and adult neurogenesis [26]. Although CHD proteins are better known for their roles in neurodevelopment including neural migration and differentiation [27,28], much less is known about their functions in mature neurons where they continue to be expressed [29,30].
CHD proteins and APP possess overlapping neuronal functions. Both CHD7 [31] and APP [32] are expressed in neural crest cells and are important for neural migration [31,33]. AD patients exhibit hyperacetylation of histone 3 lysine residue 27 (H3K27) loci in the entorhinal cortex that extensively overlap with loci affected in patients with Kallman Syndrome [17,34], a neurodevelopmental disorder correlated with Chd7 mutations [35]. The link between AD phenotypes and aberrant histone acetylation has led to the use of histone deacetylase (HDAC) inhibitors to improve cognitive function [36]. Class I and II HDAC inhibitors alleviate the production of Aβ [37] and improve spatial memory in APP-overexpressing mice [38]. Notably, Class I and II HDAC inhibitors suppress morphological defects associated with zebrafish Chd7 mutations [39], and Class I HDAC inhibitors suppress deficits in evoked neurotransmission, movement [40], and endocytosis [41] in kis mutants. Kis is the Drosophila ortholog of CHD7 [42] and CHD8. Collectively, these data suggest that there may be a functional relationship between CHD proteins and APP.
We sought to investigate whether CHD proteins and APP mediate similar synaptic processes in mature neurons using the Drosophila NMJ, which is a glutamatergic synapse similar to mammalian central nervous system synapses [43]. Drosophila is widely used to examine disease processes and synaptic pathology because of its non-redundant genome and conservation of molecular processes [44]. Drosophila expresses a single APP, APPL, which includes the conserved extracellular domains and the intracellular C-terminal domain but lacks the Aβ sequence [45]. Human APP can be cleaved by the fly γ-secretase [46,47] and APPL processing by secretases produces secreted fragments, an Aβ-like neurotoxic peptide, and an AICD [48]. We found that kis loss-of-function mutants and animals expressing human APP and BACE in their central nervous system share some, but not all, synaptic phenotypes examined. Although Kis does not regulate appl expression, Kis and APPL regulate the synaptic localization of Rab11 with mutations in both producing a similar loss of Rab11 as that of the single mutants. In contrast, simultaneous mutations in kis and appl produce additive deficits in larval locomotion suggesting that while Kis and APPL may cooperatively regulate some synaptic functions, they also possess unique synaptic functions.
2. Results
2.1. Expression of APP and BACE in Neurons Partly Phenocopies Loss of Function Mutations in Kis
CHD proteins affect the expression of genes and gene families identified as risk factors for neurodegenerative diseases [28,49]. We investigated the relationship between Kis and APPL by first examining synaptic characteristics regulated by Kis. Kis promotes the postsynaptic localization of glutamate receptors [50], the expression and synaptic localization of the GTPase Rab11, and endocytosis [41]. We examined these phenotypes in a fly model of AD, which was generated by expressing human APP [46] and its cleaving enzyme, β-secretase/β-site APP cleaving enzyme (BACE) [47], in the Drosophila central nervous system (CNS) using the elav-Gal4 driver. Expression of these transgenes in the CNS leads to the deposition of amyloid plaques and degeneration of photoreceptor cells [47] and neurons in the mushroom bodies [51,52] of adult flies. Similar to kisLM27/kisk13416 loss of function mutants [41,50], expression of APP and BACE in the CNS produced significant reductions compared to controls in synaptic levels of the glutamate receptor subunit, GluRIIC (Figure 1A), and in endocytosis as indicated by internalization of the lipophilic dye, FM 1-43FX (Figure 1B), at the third instar larval 6/7 NMJ. Similarly, synaptic levels of Rab11, the GTPase that regulates trafficking between recycling endosomes and the plasma membrane [53], was also significantly reduced in larvae expressing APP and BACE in the CNS (Figure 1C).
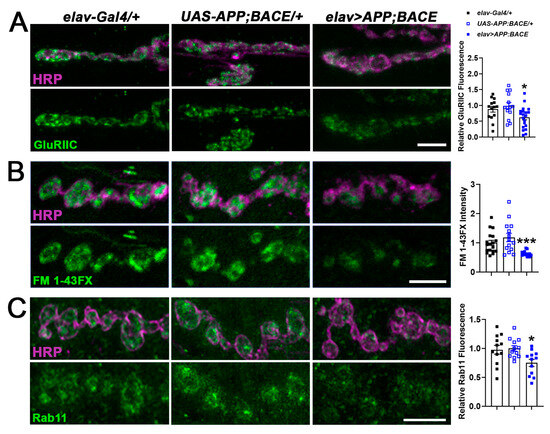
Figure 1.
Expression of APP and BACE in the larval CNS mimics kis loss of function mutant phenotypes. High-resolution confocal micrographs showing the glutamate receptor subunit, GluRIIC (A, green), endocytosis as indicated by FM 1-43FX (B, green), and the GTPase Rab11 (C, green) at the 6/7 NMJ in the listed genotypes. Fluorescence intensities, relative to controls, are depicted in right histograms. HRP (magenta) denotes the presynaptic motor neuron. Scale bars = 5 µm. * = p < 0.05, *** = p < 0.001.
Transforming growth factor (TGF)-β signaling is prevalent across metazoans and regulates diverse processes involved in embryogenesis, development, and adult tissue maintenance [54]. Expression of the downstream TGF-β component, SMAD4, is increased in AD patient brains and is a proposed biomarker for the disease [55]. Kis binds near or within the coding region of several components of the TGF-β/BMP signaling pathway including mothers against dpp (mad), thick veins (tkv), and wishful thinking (wit) in Drosophila intestinal stem cells [56]. Therefore, we examined levels of the phosphorylated R-SMAD, Mad (pMad), which is phosphorylated after TGF-β receptor binding to a ligand [43]. Synaptic levels of pMad were increased both in kisLM27/kisk13416 mutants compared with controls (Figure 2A) and in animals expressing APP and BACE in the CNS compared with outcrossed controls (Figure 2B). Collectively, these data indicate that expression of APP and BACE in the CNS phenocopies kis loss of function mutants.
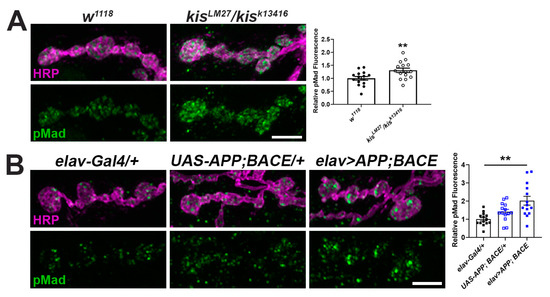
Figure 2.
There is an increase in the TGF-β/BMP signaling component pMad at kis mutant and APP and BACE expressing NMJs. (A,B) High-resolution confocal micrographs of the 6/7 NMJ of genotypes listed showing synaptic pMad (green) and HRP (magenta), which labels the presynaptic motor neuron. Fluorescence intensities, relative to controls, are depicted in right histograms. Scale bars = 5 µm. ** = p < 0.01, F = 9.24.
We next used two-electrode voltage clamp electrophysiology to more broadly assess the synaptic function of kis mutants and APP;BACE-expressing larvae. Neurotransmission was compromised in kis mutants as previously reported [41,50]. Similar to kisLM27/kisk13416 mutants, larvae expressing APP and BACE in the CNS showed reduced amplitudes of spontaneous miniature endplate junctional currents (mEJCs) (Figure 3B,D), consistent with the loss of synaptic GluRIIC (Figure 1A). APP- and BACE-expressing animals also showed a decrease in mEJC frequency, indicating a reduction in the number of active zones, but this was unchanged in kis mutants (Figure 3D, right). The latter, however, exhibited decreased evoked EJC (eEJC) amplitudes, which were unchanged in APP;BACE-expressing larvae compared to UAS-APP;BACE outcrossed controls (Figure 3A,C).
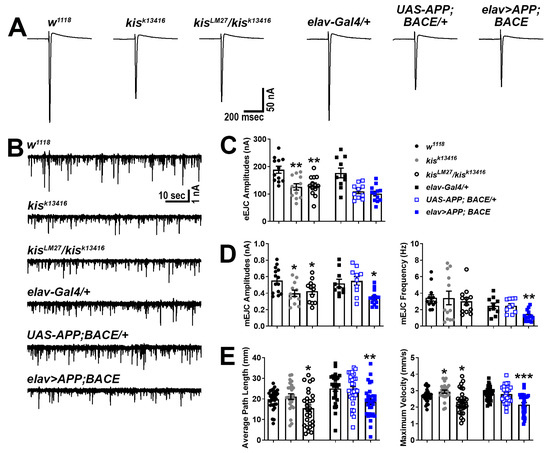
Figure 3.
Kis mutants and APP;BACE-expressing larvae show deficits in neurotransmission and locomotion. (A) Representative eEJC traces recorded from muscle 6 in response to administration of a suprathreshold stimulus to the presynaptic motor neuron. (B) Representative mEJC traces from larval body wall muscle 6. (C) Quantification of eEJC amplitudes. ** = p < 0.01 (D) Quantification of mEJC amplitudes (left) and frequency (right). * = p < 0.05, F = 8.50; ** = p < 0.01, F = 9.24 (E) wrMTrck quantification of larval crawling behavior on an agar arena for 30 s. Distance traveled or average path length (left) and maximum crawling velocity (right) were normalized to body size (body lengths per second). * = p < 0.05; ** = p < 0.01, F = 6.89; *** = p < 0.001, F = 15.14.
To determine whether the deficits in neurotransmission were sufficient to produce behavioral deficits, we examined the locomotor activity of kis mutants and APP;BACE-expressing larvae. Larval locomotor behavior is produced by CNS central pattern generators that send cholinergic inputs to presynaptic motor neurons [57]. The frequency and duration of presynaptic motor neuron activity positively correlate with postsynaptic muscle contractile force [58]. Both kisLM27/kisk13416 mutants and larvae expressing APP and BACE in the CNS exhibited reductions in the distance traveled/average path length and maximum velocity of movement compared with controls (Figure 3E). Thus, APP;BACE-expressing larvae phenocopy some, but not all, characteristics of kis loss of function mutant synapses. In support of this, we noted that synaptic levels of the endocytic proteins Dynamin (Dyn) and Endophilin A (EndoA), which are enriched at kisLM27/kisk13416 NMJs [41], showed no differences in APP;BACE-expressing larvae (Dyn: elav-Gal4/+ = 1.00 ± 0.06 AU, n = 14; UAS-APP;BACE/+ = 1.06 ± 0.03 AU, n = 14; elav>APP;BACE = 0.94 ± 0.07 AU, n = 14, p = 0.39. EndoA: elav-Gal4/+ = 1.00 ± 0.06 AU, n = 14; UAS-APP;BACE/+ = 1.05 ± 0.07 AU, n = 14; elav>APP;BACE = 1.10 ± 0.10 AU, n = 14, p = 0.66). These collective findings indicate that Kis, APP, and BACE likely have shared and non-shared roles at the synapse.
2.2. Kis Promotes the Synaptic Localization of APPL
Based on our data, we hypothesized that Kis could promote synaptic function and the expression and localization of synaptic proteins including Rab11 by restricting the expression of appl (Figure 4A). Kis binds near the appl transcription start site in Drosophila intestinal stem cells [56] and APP is differentially expressed in Chd8 heterozygous loss of function mutations in adult mice [59]. Alternatively, it is possible that AICD, generated by APP/APPL processing, promotes kis expression (Figure 4B). AICD is stabilized by the cytoplasmic adapter protein Fe65, transported to the nucleus, and associates with the histone acetylase Tip60 to form the AFT complex in HEK293 and neuroblastoma cells [60]. In addition, AICD and Kis may function in a complex (Figure 4C) to cooperatively regulate some synaptic functions by regulating the expression of synaptic gene products like rab11. Both AICD [61] and CHD7 regulate the expression of TP53 [62], which encodes the transcription factor, p53.
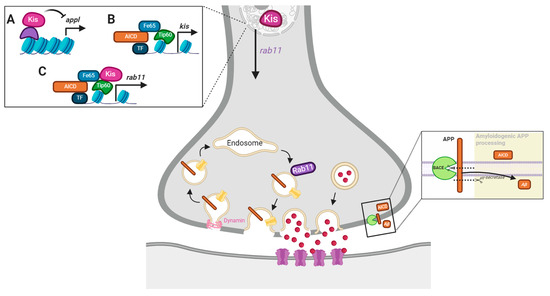
Figure 4.
Model of potential Kis and APPL interactions. Both Kis and AICD are transcriptional regulators. Therefore, Kis may influence synaptic function by repressing neuronal APPL (A), being a target of AICD-mediated transcription (B), or being recruited by the AFT complex to chromatin to promote rab11 transcription (C).
To begin to distinguish between these models, we examined APPL expression in kisLM27/kisk13416 mutants. We found that kisLM27/kisk13416 mutants exhibited a significant decrease in APPL protein at the NMJ (Figure 5A) but a 1.8-fold increase in appl transcripts in the CNS (Figure 5B). These data support a role for Kis in promoting APPL protein but not mRNA expression. Next, we examined synaptic function in appld mutants, a null mutant [63], and tip60e02395, a hypomorph generated by the insertion of a piggybac element in the coding region of tip60 [64]. If mutations in appl and tip60 produce similar phenotypes as kis mutants, then Kis, APPL, and Tip60 may function in a complex to regulate the expression of genes important for synaptic function. Both appld and tip60e02395 mutants showed reductions in eEJC amplitudes (Figure 5C,E) and these were not significantly different from the reduction in eEJC amplitudes in kisLM27/kisk13416 mutants. Similarly, kisLM27/kisk13416 and tip60e02395 mutants showed reduced mEJC amplitudes (Figure 5D,E), and kisLM27/kisk13416 and appld mutants showed a decrease in quantal content, which is an estimation of the number of vesicles released by the presynaptic motor neuron [65]. There was no change in mEJC frequency in any of the genotypes examined (w1118 = 2.40 Hz, n = 12; kisLM27/kisk13416 = 2.53 Hz, n = 12, p = 0.66; appld = 3.00 Hz, n = 13, p = 0.24; tip60e02395 = 2.43 Hz, n = 17, p = 0.93). Although these data support the hypothesis that Kis, APPL, and Tip60 cooperate to ensure proper neurotransmission, it is notable that kis mutants show a decrease in APPL protein at the synapse but an increase in appl mRNA. Thus, Kis may not regulate appl transcription but instead may influence the localization of APPL to the synapse.
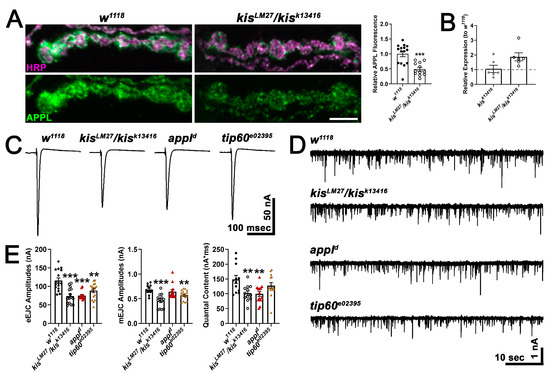
Figure 5.
Kis regulates APPL localization and may act in a transcriptional complex with AICD and Tip60. (A) High-resolution confocal micrographs of terminal 6/7 boutons immunolabeled for APPL (green). HRP (magenta) was used to delineate presynaptic motor neurons. Scale bar = 5 µm. Right histogram shows the quantification of relative APPL fluorescence intensity. *** = p < 0.001 (B) Histogram showing appl transcript levels in kis mutants relative to w1118 controls. (C) Representative eEJC traces recorded from muscle 6 of third instar larvae in response to administration of a suprathreshold stimulus to the presynaptic motor neuron. (D) Representative mEJC traces from larval body wall muscle 6. (E) Quantification of eEJC amplitudes (left), mEJC amplitudes (middle), and quantal content (right). ** = p < 0.01, *** = p < 0.001.
2.3. Disrupting APPL Processing Impairs Neurotransmission and Locomotion in Controls but Not Kis Mutants
If Kis regulates the synaptic localization of APPL, then interfering with APPL processing would not exacerbate the deficits in neurotransmission and locomotion observed in kis mutants. To examine this possibility, we used L-685,458, which binds to γ-secretase thereby proportionally decreasing Aβ production in adult rat hippocampal slices [66] and preventing the formation of the AFT complex in HEK293 cells [60]. Raising larvae on food containing 100 nM L-685,458 partially suppresses deficits in movement and significantly increases synaptic mitochondria and localization of the postsynaptic scaffold, Discs Large, to NMJs in third instar larvae expressing APP;BACE in the CNS [67]. Exposing larvae to L-685,458 throughout development resulted in decreased eEJC amplitudes for w1118 controls but not for kisk13416 or kisLM27/kisk13416 mutants (Figure 6A,B). These data support previous work demonstrating that APP processing is important for evoked neurotransmission and synaptic plasticity [68,69]. There were no differences, however, in mEJC amplitudes, mEJC frequency, or quantal content of controls or kisLM27/kisk13416 mutants raised in food containing the γ-secretase inhibitor, L-685,458.
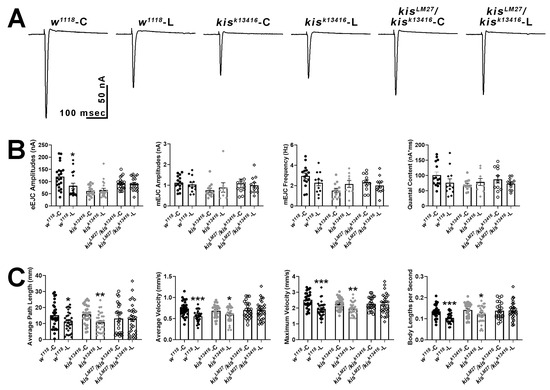
Figure 6.
Pharmacological inhibition of γ-secretase activity with L-685,458 impairs evoked neurotransmission and locomotion in controls but not in kisLM27/kisk13416 mutants. (A) Representative eEJC traces recorded from muscle 6 of third instar larvae in response to administration of a suprathreshold stimulus to the presynaptic motor neuron. C represents animals exposed to the vehicle, DMSO, while L represents animals exposed to 100 nM L-685,458 throughout development. (B) Quantification of eEJC amplitudes, mEJC amplitudes and frequency, and quantal content. * = p < 0.05 (C) wrMTrck quantification of larval crawling behavior on an agar arena for 30 s. C represents animals exposed to the vehicle, DMSO, while L represents animals exposed to 100 nM L-685,458 for 24 h. Histograms show average path length, average and maximum velocities, and body lengths per second. * = p < 0.05, ** = p < 0.01, *** = p < 0.001.
To distinguish the developmental effects of inhibiting APPL processing from those in mature neurons, we exposed third-instar larvae to 100 nM L-685,485 for 24 h. Further, inhibiting γ-secretase for at least two days induces lysosomal dysfunction followed by endosomal dysfunction in mouse embryonic fibroblasts [70]. Exposing larvae to L-685,485 for 24 h was sufficient to produce locomotor deficits including reductions in average path length, average and maximum velocity, and body lengths per second in treated w1118 larvae compared with those exposed to an equal volume of DMSO (Figure 6C). The same deficits were not observed in kisLM27/kisk13416 mutants but were retained in kisk13416 mutants. Thus, both evoked neurotransmission and inhibition of γ-secretase activity resulted in locomotor deficits in controls but did not affect these phenotypes in kisLM27/kisk13416 mutants. Collectively, these data indicate that Kis acts upstream of APPL to promote neurotransmission and larval locomotor behaviors.
2.4. Kis and APPL Promote the Synaptic Localization of Rab11
Synaptic Rab11 is decreased in kis mutants [41] and in larvae expressing APP;BACE in the CNS (Figure 1C). Therefore, it is possible that both APPL and Kis regulate rab11 expression. To test this possibility, we assessed synaptic Rab11 levels in appld mutants and in animals heterozygous for mutations in both appl and kis. Appld mutants exhibit a reduction in Rab11 at the synapse similar to that of kisLM27/kisk13416 mutants (Figure 7A,B). If APPL and Kis function in a transcriptional complex to regulate synaptic Rab11 levels, we would expect that the loss of Rab11 in animals containing mutations in both appl and kis would be similar as that observed in animals with mutations in either appl or kis. Consistent with this, larvae with heterozygous loss of function mutations in both appl and kis show a decrease in synaptic Rab11 that is comparable to that of appld/+ and kisLM27/+ mutants. In addition, knockdown of APPL in kisLM27/kisk13416 mutants did not exacerbate the loss of Rab11 observed at kis mutant NMJs (Figure 7C,D).
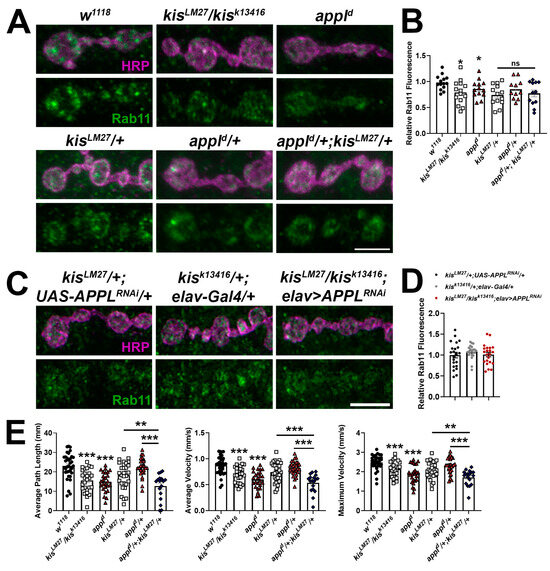
Figure 7.
APPL and Kis may cooperatively regulate the synaptic localization of Rab11. (A) High-resolution confocal micrographs of terminal 6/7 boutons immunolabeled for Rab11 (green). HRP (magenta) was used to label presynaptic motor neurons. Scale bar = 5 µm. (B) Histogram showing relative Rab11 fluorescence intensities. * = p < 0.05 (C) High-resolution confocal micrographs of terminal 6/7 boutons immunolabeled for Rab11 (green). HRP (magenta) was used to denote presynaptic motor neurons. Scale bar = 5 µm. (D) Histogram showing relative Rab11 fluorescence intensities. (E) wrMTrck quantification of larval crawling behavior on an agar arena for 30 s. Histograms show average path length and average and maximum velocities normalized to body lengths per second. * = p < 0.05, ** = p < 0.01, *** = p < 0.001.
To more broadly assess whether APPL and Kis work cooperatively to regulate synaptic function, we examined locomotor behavior in animals containing mutations in both appl and kis. Both kisLM27/kisk13416 and appld mutants demonstrated impaired locomotion with reductions in average path length and average and maximum velocities (Figure 7E). Larvae with mutations in both kisLM27 and appld mutants showed additive effects compared with both appld/+ and kisLM27/+ mutants. Collectively, these data indicate that while Kis and APPL regulate common synaptic targets like Rab11, they also regulate non-overlapping synaptic targets that contribute to the generation and execution of locomotor behaviors.
Chromatin-remodeling enzymes influence the expression of thousands of genes [56,59,71]. Thus, the reduction in APPL at kisLM27/kisk13416 mutant synapses (Figure 5A) likely results from the collective action of many differentially expressed gene products important for synaptic function. Therefore, to test the relationship between Rab11 and APPL, we manipulated Rab11 expression and activity in a control genetic background. If APPL promotes rab11 transcription, we would expect that changing the expression or activity of Rab11 would not affect synaptic levels of APPL. If, however, Rab11 regulates the localization of APPL by determining the recycling of APPL, then changing the expression or activity of Rab11 would affect synaptic levels of APPL. In support of the latter, knockdown of Rab11 in presynaptic motor neurons using the C380-Gal4 driver resulted in a reduction in synaptic APPL while knockdown of Rab11 in postsynaptic muscle using the 24B-Gal4 driver did not affect APPL (Figure 8A). Expression of wild-type Rab11 tagged with enhanced yellow fluorescent protein (Rab11eYFP) in either presynaptic motor neurons or postsynaptic muscle increased APPL at the NMJ. Similarly, expression of a constitutively active Rab11, which does not hydrolyze its bound GTP (Rab11Q70L), also increased synaptic APPL when expressed in presynaptic motor neurons or postsynaptic muscle (UAS-Rab11Q70L/+ = 1.00 ± 0.07 AU, n = 15; C380-Gal4/+ = 0.80 ± 0.07 AU, n = 14; C380> Rab11Q70L = 1.28 ± 0.19 AU, n = 13, p = 0.03; 24B-Gal4/+ = 0.99 ± 0.09 AU, n = 13; 24B> Rab11Q70L = 2.08 ± 0.18 AU, n = 13, p < 0.0001). These data indicate that the reduction in synaptic APPL in kis mutants may occur due to reduced levels of Rab11.
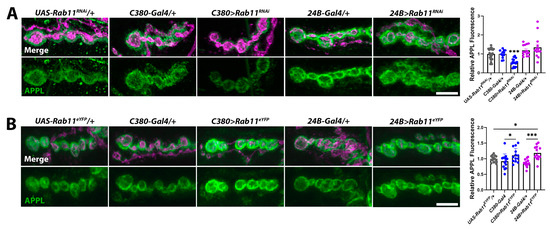
Figure 8.
Rab11 regulates synaptic levels of APPL. (A) High-resolution confocal micrographs of terminal 6/7 boutons immunolabeled for APPL (green). HRP (magenta) was used to label presynaptic motor neurons. (A) Rab11 was knocked down in presynaptic motor neurons using the C380-Gal4 driver and in postsynaptic muscle using the 24B-Gal4 driver. Scale bar = 5 µm. Right histogram shows APPL fluorescence intensities. *** = p < 0.001, F = 6.51 (B) Rab11 was overexpressed in presynaptic motor neurons by expressing UAS-Rab11eYFP using the C380-Gal4 driver and in postsynaptic muscle using the 24B-Gal4 driver. Scale bar = 5 µm. Right histogram showing relative APPL fluorescence intensities. * = p < 0.05, F = 4.61; *** = p < 0.001, F = 14.05.
3. Discussion
Chromatin-regulatory proteins establish and maintain the epigenome, the collective set of chemical modifications that occur on DNA and histones. The epigenome integrates both cell-intrinsic programs and environmental cues [72] to regulate gene expression by determining chromatin structure. The CHD proteins, CHD7 and CHD8, are best known for their roles in neurodifferentiation and neurodevelopment [27,28] but are expressed in mature neurons [29,30]. Epigenetic changes in gene expression underlie persistent synaptic remodeling that occurs in learning and memory [73] and cognitive decline [74]. The transcriptional profiles from individuals with autism spectrum disorders or AD illustrate shared and distinct sets of affected neuronal genes [75] suggesting that these processes share a subset of altered molecular pathways.
3.1. Kis and APPL Functionally Interact and Localize Rab11 to the Synapse
We investigated the link between chromatin remodeling and AD by examining synaptic phenotypes in Drosophila larvae expressing human APP and BACE in their CNS and comparing them with kis mutants. Loss of function kis mutants and animals expressing human APP and BACE in their CNS exhibit reduced synaptic levels of the glutamate receptor subunit, GluRIIC, the GTPase Rab11 (Figure 1A,C), and the BMP component, pMad (Figure 2), at the Drosophila larval NMJ. Collectively, these synaptic abnormalities, along with others not investigated here, likely produced reductions in endocytosis (Figure 1B), neurotransmission, and movement of kisLM27/kisk13416 mutants and APP;BACE-expressing larvae (Figure 3). Some characteristics of neurotransmission and the localization of the endocytic proteins EndoA and Dyn, however, differed between kis mutants and APP;BACE-expressing larvae. Collectively, these data suggest that there may be a functional interaction between Kis and APPL to influence some aspects of synaptic function.
It is possible that the functional interaction between Kis and APPL occurs because Kis restricts appl transcription in the larval CNS (Figure 4A). Several transcriptional programs including those downstream of growth factors [76], androgens [77], cytokines [78], apolipoprotein E, and extracellular regulated kinase (ERK) [79] promote APP transcription in neurons. Transcription of a subset of ERK targets is enhanced by interactions between CHD8 and the transcription factor, ELK1, in mature human neurons [80]. In Drosophila intestinal stem cells, Kis restricts expression of a negative regulator of the epidermal growth factor receptor, which is upstream of ERK, and binds near the appl transcription start site [56]. Alternatively, Kis may be a target of the AFT complex (Figure 4B), formed by APPL’s AICD, which interacts with the adapter protein FE65 in the cytoplasm, translocates to the nucleus, and binds to the histone acetylase TIP60 [81]. Although appl mRNA was increased in the CNS of kisLM27/kisk13416 mutants (Figure 5B), there was a decrease in APPL at the NMJ synapse (Figure 5A). These data do not support a model where Kis restricts appl transcription in the larval CNS. Instead, Kis may indirectly influence APPL localization via direct transcriptional regulation of other synaptic proteins as Kis differentially regulates hundreds of synaptic targets [50]. Kis also promotes trafficking through the endomembrane system [82], a process that is disrupted in AD and other neurodegenerative diseases [83]. The loss of APPL at kis mutant synapses may, therefore, result from aberrant intracellular trafficking and/or a compensatory response that downregulates appl translation [84].
It is also possible that Kis is recruited to the AFT complex (Figure 4C) to regulate the transcription of genes that encode synaptic proteins thereby promoting synaptic function. Chromatin remodeling proteins form unique multisubunit complexes by associating with different combinations of transcription factors and chromatin modifying enzymes in a cell- and developmental-dependent manner [85,86]. Neurotransmission at appld and tip60e02395 mutant synapses shared some features with kisLM27/kisk13416 mutants (Figure 5C–E). Further, kisLM27/kisk13416 mutants do not show exacerbated deficits in evoked neurotransmission and locomotion after inhibition of APPL processing using L-685,458 [60]. Inhibition of APPL processing in controls, however, decreased evoked neurotransmission and locomotion (Figure 6). These data support a role for Kis in AFT complex-mediated transcription of synaptic targets that enable proper locomotion and neurotransmission. These data are also consistent, however, with a model where Kis indirectly influences APPL localization via direct transcriptional regulation of other synaptic and/or trafficking proteins.
Rab11 may be a target of a potential Kis–AFT complex as Kis and APPL promoted the synaptic localization of Rab11 and simultaneous loss of function mutations in each did not produce an additive loss of synaptic Rab11 (Figure 7A). Similarly, the knockdown of APPL in the kisLM27/kisk13416 mutant background did not produce a further decrease in Rab11 (Figure 7B). Although these data are consistent with a Kis–AFT complex regulating rab11 transcription, it is also important to consider the possibility that Kis regulates synaptic levels of APPL indirectly by promoting rab11 transcription [87]. Rab11 is associated with intracellular vesicles containing APP in mouse forebrain neurons [88] suggesting that Rab11, at least partly, determines the concentration of APPL localized to the plasma membrane. Rab11 also directly interacts with the BACE subunits Presenilin 1 and Presenilin 2 [89], transports BACE1 along hippocampal dendrites and axons [90], and promotes the production of Aβ [91]. Thus, Kis may influence the synaptic localization of APPL by promoting rab11 transcription thereby increasing synaptic levels of Rab11 and, as a result, APPL. In support of this, tknockdown of Rab11 in presynaptic motor neurons reduced synaptic APPL (Figure 8A) while overexpression of Rab11 (Figure 8B) or expression of a constitutively active Rab11 in either presynaptic motor neurons or postsynaptic muscle increased synaptic APPL.
3.2. APPL and Rab11 May Influence the Synaptic Localization of One Another
Our results also illustrate a potentially reciprocal relationship between Rab11 and APPL. Appld mutants exhibited decreased synaptic levels of Rab11 (Figure 7A,B) and Rab11 influenced the localization of APPL. Knockdown of Rab11 decreased, while overexpression of Rab11 increased, synaptic levels of APPL (Figure 8). While Rab11 traffics APPL to and from the plasma membrane [88], the localization of Rab11 may be partly determined by APPL processing. Processing of APP generates Aβ, which hydrolyzes the phosphatidylinositol, PI(4,5)P2 by activating type I metabotropic glutamate receptors and phospholipase C [92]. Although Rab11 is recruited to membranes enriched in PI(3,5)P2 [93], PI(3,5)P2 and PI(4,5)P2 share the precursor, PI4P [94]. Therefore, the membrane concentrations of PI(3,5)P2 and PI(4,5)P2 are partly dependent on one another. Thus, APPL processing may restrict synaptic levels of Rab11.
Collectively, our results support a model where Kis acts upstream of APPL to regulate some aspects of synaptic function. Our conclusions, however, are limited because we cannot exclude the possibility that Kis is recruited to the AFT complex to cooperatively regulate the transcription of synaptic gene products. Indeed, our data are consistent with a combination of the two models where Kis and APPL work cooperatively to promote rab11 expression and Kis acts upstream, through Rab11, to properly localize APPL to the synapse. We are further limited because the phenotypes we observe in kis mutants and animals expressing APP and BACE in their CNS are the sum of hundreds to thousands of differentially expressed gene products. Kis influences the transcription of thousands of potential targets [56] and the expression of hundreds of genes is affected in AD [20]. Despite this, it is important to use models that emulate the complexity of the disease process to gain a better understanding of AD.
3.3. Aberrant Chromatin Remodeling Contributes to AD
Chromatin remodeling contributes to neurodifferentiation and neurodevelopment [95], learning and memory [73], healthy aging [96], and neurodegeneration [25]. Our data add to a growing body of work indicating that aberrant chromatin remodeling is a driving force in AD. Genome-wide changes in histone acetylation influence transcriptional regulation and likely contribute to the differential expression of genes in AD. Genome-wide hypo- and hyperacetylation of H3K27 is observed in AD patient entorhinal cortex genomes compared with non-diseased age-matched controls [17] and increased H3 acetylation precedes cognitive impairments in an AD mouse model. Similarly, AD patients show a net loss of H4K16 acetylation in the temporal lobe [97]. Kis promotes multiple histone modifications including H3K27 [98] and H4K16 acetylation [87]. Thus, the similarities we observed between kis mutants and APP- and BACE-expressing larvae may occur due to the loss of histone acetylation.
Histone acetylation and other chromatin modifications are dynamic processes [23] and AD pathology is characterized by the dysregulation of hundreds of gene products [17,18,19,20]. Thus, targeting gene expression globally by altering neuronal epigenomes offers another potential therapeutic strategy for treating AD patients. Therapeutic approaches like targeting Aβ and inhibiting γ-secretase activity have shown limited success. The Food and Drug Administration (FDA) recently approved the use of lecanemab and donanemab, which are monoclonal Aβ antibodies, for the treatment of early AD. These compounds slightly delayed cognitive decline in patients with early AD but ultimately do not slow the production of Aβ [99] or restore the synapses or neurons that are lost when patients seek treatment [15]. γ-secretase inhibitors, which include L-685,458, limit the production of Aβ [66] but produce side effects as chronic administration inhibits Notch signaling [100]. Chromatin modifications also occur in cancer epigenomes and several different HDAC inhibitors are FDA approved to treat hematological malignancies. With the continued development of and several HDAC inhibitors in clinical trials [101], it is promising to investigate the of potential of repurposing these compounds. However, we need to better understand the specific changes in chromatin structure that occur in AD, when they occur in the disease process, and how they contribute to disease pathology.
4. Materials and Methods
4.1. Drosophila Stocks and Husbandry
Flies were raised at 25 °C in a Percival DR-36NL incubator with a 12 h light:dark cycle in standard vials containing Jazz Mix (Fisher Scientific AS153; Hampton, NH, USA) food. The following fly stocks were obtained from the Bloomington Drosophila Stock Center: 24B-Gal4 (BL 1767), appld (BL 43632), C380-Gal4 (BL 80580), elav-Gal4 on X (BL 458), elav-Gal4 on chromosome II (BL 8765), kisk13416 (BL 10442), tip60e02395 (BL 18052), UAS-applRNAi (BL 28043), UAS-Rab11Q70L (BL 50783), UAS-Rab11eYFP (BL 50782), UAS-Rab11RNAi (BL 27730), and w1118 (BL 3605), which was used as the isogenic control for most experiments. Both kisLM27 [102] and UAS-APP;BACE [67] were obtained from the laboratory of Dr. Dan Marenda. KisLM27/kisk13416 was generated by crossing the respective stocks because kisLM27 is an embryonic lethal mutation. Outcrossed controls for experiments using UAS transgenes and Gal4 drivers were generated by crossing each respective stock with w1118. Both male and female flies and larvae were used for all experiments except for those including appld and tip60e02395. Appl and tip60 are located on the X chromosome, necessitating the use of females for these experiments.
Larvae treated with 100 nM L685,458 (Tocris 2627; Bristol, United Kingdom) were raised on instant food (Genessee Scientific Nutri-Fly 66–117; Morrisville, NC, USA) containing 1.6% of 10% w/v Tegosept (Genessee Scientific 20-258) for 24 h or for their lifetime. Controls were raised on food containing the same components except an equal volume of DMSO was added in place of L685,458. Food was made in small batches, stored at 4 °C, and used within five days of preparation.
4.2. Immunocytochemistry and FM 1-43FX Labeling
Third-instar larvae were dissected at room temperature in Roger’s Ringer (135 mM NaCl, 5 mM KCl, 4 mM MgCl2*6H2O, 1.8 mM CaCl2*2H2O, 5 mM TES, 72 mM Sucrose, 2 mM glutamate, pH 7.15) on Sylgard (World Precision Instruments 501986; Sarasota, FL, USA)-lined petri dishes. Larvae were fixed in either Bouin’s Fixative (Ricca Chemical 112016; Arlington, TX, USA) or 3.7% formaldehyde freshly diluted from a 10× stock (37% Formaldehyde, Fisher Scientific BP531-500). After fixing for 30 min, larvae were placed in 1.5 mL centrifuge tubes containing PTX (1 × PBS + 0.1% Triton X-100, Fisher Scientific AAA16046AP) and washed three times for 10 min. Larvae were then washed two times for 30 min in PBTX (1 × PBS + 0.1% Triton X-100 + 1% Bovine Serum Albumin, Fisher Scientific BP1600-100) and incubated at 4 °C overnight in PBTX containing primary antibodies. Primary antibodies included guinea pig α-APPL (1:250, generated for this study), mouse α-Dynamin (1:300, BD Biosciences 610245; Franklin Lakes, NJ, USA), guinea pig α-Endophilin A (1:5000, Hugo Bellen lab), rabbit α-GluRIIC (1:5000, generated by Genscript using the sequence described in [103]), rabbit α-pMad (1:100, Abcam ab52903; Cambridge, United Kingdom), and mouse α-Rab11 (1:50, BD Biosciences 610657). The following day larvae were washed three times for 10 min and two times for 30 min in PBTX. Then, secondary antibodies including α-mouse FITC (113-095-003), α-mouse TRITC (115-025-003), α-rabbit FITC (106-095-003), or α-guinea pig FITC (111-095-003) were applied at 1:400 along with either Cy3-HRP (123-165-021) or A647-HRP (123-605-021) at 1:125 in PBTX for 2 h. All secondary antibodies and HRP were obtained from Jackson Immunoresearch Laboratories. Larvae were washed three times for 10 min and two times for 30 min in PBTX, placed on slides, and covered with VectaShield mounting medium (Vector Laboratories H1000; Newark, CA, USA).
Polyclonal APPL antibodies were generated by Genscript by optimizing and expressing the full-length appl cDNA sequence in E. coli. Purified APPL protein was injected into guinea pigs with boosters administered at two, four, and six weeks after initial injection. Seven weeks after primary immunization, serum was collected from animals. The serum was affinity purified and the APPL antibody was tested by comparing synapse-specific immunolabeling with total guinea pig IgG in both control (w1118) and appld null larvae.
FM 1-43FX (Fisher Scientific F35355) labeling was performed as previously described [104]. Third-instar larvae were dissected in Ca2+-free HL-3 (100 mM NaCl, 5 mM KCl, 10 mM NaHCO3, 5 mM HEPES, 30 mM sucrose, 5 mM trehalose, 10 mM MgCl2, pH 7.2) in Sylgard-coated petri dishes. Once larvae were fillet dissected, they were washed once with Ca2+-free HL-3, the motor neurons were cut, and 4 μM FM 1–43FX in HL-3 containing 90 mM KCl and 1.5 mM CaCl2 was applied for 1 min. Residual/unbound FM 1-43FX was washed off by replacing the Ca2+-free HL-3 five times over 5–10 min. Larvae were then fixed with 3.7% formaldehyde in Ca2+-free HL-3 for 5 min and subsequently transferred to 1.5 mL centrifuge tubes. Larvae were washed 10 times over 10–15 min with Ca2+-free HL-3 containing 2.5% goat serum and then incubated at room temperature in A647-HRP in Ca2+-free HL-3 containing 2.5% goat serum for 30 min. Finally, larvae were washed with Ca2+-free HL-3 10 times over 10–15 min, placed on slides, covered with Vectashield, and immediately imaged via confocal microscopy.
Larval 6/7 NMJs of segments A3 or A4 were imaged using the 60× oil immersion objective of an Olympus Fluoview 1000 microscope. Image acquisition parameters were determined based on the mean fluorescence intensities used for control larvae. Approximately equal numbers of control and experimental animals were imaged on the same day. Max projected z-stacks were used for image analyses, which were performed using Fiji [105]. Mean pixel intensities were obtained from immunolabeled images by outlining the NMJ and subtracting the background pixel intensity obtained from an area of equal size that did not include the NMJ from the value that represented the NMJ. For FM 1-43FX labeling, relative fluorescence was obtained for each z-stack slice by subtracting background fluorescence from NMJ fluorescence. The mean of all z-stacks was used and reported for FM 1-43FX intensity.
4.3. RNA Isolation and RT-qPCR
Third-instar larvae CNSs were dissected at room temperature in Roger’s Ringer (135 mM NaCl, 5 mM KCl, 4 mM MgCl2*6H2O, 1.8 mM CaCl2*2H2O, 5 mM TES, 72 mM Sucrose, 2 mM glutamate, pH 7.15) on Sylgard (World Precision Instruments 501986)-filled 60 mm petri dishes. Isolated CNSs were placed in nuclease-free 1.5 mL centrifuge tubes containing RNAlater solution (Fisher Scientific AM7020) and stored at −20 °C until RNA was isolated from tissues using an Invitrogen RNAqueous Total RNA Isolation Kit (Fisher Scientific AM1912). RNA concentrations were assessed using an Implen NanoPhotometer N50. RT-qPCR was performed using the iTaq Universal SYBR Green One-Step Kit (BioRad 1725151) and a CFX Connect Real-Time PCR Detection System (Bio-Rad; Hercules, CA, USA). 100 ng of RNA and 50 pmol/µL of cDNA-specific primers were added to each reaction. Each technical replicate included 30 CNSs and two biological replicates were used to calculate relative fold changes. Relative fold changes were calculated by determining the difference between the C(t) value of the target transcript reaction and the C(t) value for GAPDH to obtain ΔC(t) for each transcript. Next, the difference between control and kis mutant ΔC(t)s was calculated and log transformed to obtain the 2−ΔΔC(t) [106].
4.4. Electrophysiology and Behavior
Third-instar larvae were fillet dissected in ice-cold HL-3 + 0.25% CaCl2 by gluing larval pelts to Sylgard-coated 18 mm round coverslips. The CNS was removed by cutting the motor neurons and the HL-3 was replaced with room temperature HL-3 + 1.0% CaCl2. Recordings were acquired from muscle 6 of segment A3 or A4 using glass microelectrodes with 10–20 MΩ of resistance filled with 3 M KCl. Muscles with input resistances < 5 MΩ were voltage-clamped at −60 mV by an Axoclamp 900A amplifier (Molecular Devices; San Jose, CA, USA). Segmental nerves were stimulated by a fire-polished glass electrode filled with room temperature HL-3 + 1.0% CaCl2 and a Grass S88 stimulator with a SIU5 isolation unit (A-M Systems; Sequim, WA, USA). Recordings were digitized with a Digidata 1443 digitizer (Molecular Devices) and data were analyzed in Clampfit (v 11.1, Molecular Devices). Three minutes of mEJCs were analyzed to determine mEJC amplitudes and frequencies. The mean amplitude of 10 eEJCs elicited by suprathreshold stimuli was used to represent eEJC amplitudes. Quantal content was calculated by dividing the integrated area of eEJCs by that of mEJCs [107]. Approximately equal numbers of controls and experimental animals were used daily for recordings.
Larvae used for locomotor assays were transferred from vials to a 100 mm plate containing 1.6% agar. They were allowed to acclimate to the crawling surface and shed home vial debris for 1 min. Next, larvae were placed on a 1.6% agar arena and locomotion was recorded after animals began moving. Locomotor behavior was recorded for five animals simultaneously on a Canon EOS M50 camera at 29.97 frames per second. Video recordings were analyzed in Fiji using the wrMTrck plugin (written by Jesper S. Pedersen).
4.5. Experimental Design and Statistical Analyses
Two to four biological replicates including 3–8 animals were used for each experiment. Approximately equal numbers of controls and experimental animals were used for each biological replicate. The total number of technical replicates is represented by the points (circles, squares, triangles, or diamonds) found associated with histogram bars. Data analyses were performed with GraphPad Prism (v. 10.2.3). Unpaired t-tests were used to analyze data from experiments that included a single control group while one-way ANOVAs followed by Tukey’s or Bonferroni’s post hoc tests were used to analyze data from experiments that included more than one control group. Bartlett’s test for homogeneity of variance was used to assess the variances between data sets. Statistical significance is represented on bar graphs as follows: * = <0.05, ** = <0.01, *** = <0.001 with error bars representing standard error of the mean (SEM).
Author Contributions
Conceptualization, F.L.W.L.; methodology, E.L.H., N.L., and F.L.W.L.; formal analysis, I.R.S., E.L.H., N.L., and F.L.W.L.; investigation, I.R.S., E.L.H., N.L., and F.L.W.L.; resources, F.L.W.L.; writing—original draft preparation, F.L.W.L.; writing—review and editing, F.L.W.L.; supervision, F.L.W.L.; project administration, F.L.W.L.; funding acquisition, F.L.W.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the National Institutes of Health, NINDS 1R15NS101608-01A1 (FL), and NINDS 2R15NS101608-02A1 (FL). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available upon request from the corresponding author. Data are available in the form of spreadsheets documenting data analysis of raw data.
Acknowledgments
We thank Daniel Marenda for generously providing the kisLM27 and UAS-APP;BACE fly stocks used in our experiments. We also thank the Bloomington Drosophila Stock Center for fly stocks, the Hugo Bellen lab for the Endophilin A antibody, and the Developmental Studies Hybridoma Bank (University of Iowa) for additional antibodies. Figure 4 and graphical abstract created with BioRender.com. Finally, we thank Dave Featherstone for his guidance and mentorship.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Forner, S.; Baglietto-Vargas, D.; Martini, A.C.; Trujillo-Estrada, L.; LaFerla, F.M. Synaptic Impairment in Alzheimer’s Disease: A Dysregulated Symphony. Trends Neurosci. 2017, 40, 347–357. [Google Scholar] [CrossRef]
- Puzzo, D.; Piacentini, R.; Fa, M.; Gulisano, W.; Li Puma, D.D.; Staniszewski, A.; Zhang, H.; Tropea, M.R.; Cocco, S.; Palmeri, A.; et al. LTP and memory impairment caused by extracellular Abeta and Tau oligomers is APP-dependent. elife 2017, 6, e26991. [Google Scholar] [CrossRef]
- Dawkins, E.; Small, D.H. Insights into the physiological function of the beta-amyloid precursor protein: Beyond Alzheimer’s disease. J. Neurochem. 2014, 129, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Coronel, R.; Bernabeu-Zornoza, A.; Palmer, C.; Muñiz-Moreno, M.; Zambrano, A.; Cano, E.; Liste, I. Role of Amyloid Precursor Protein (APP) and Its Derivatives in the Biology and Cell Fate Specification of Neural Stem Cells. Mol. Neurobiol. 2018, 55, 7107–7117. [Google Scholar] [CrossRef]
- Hermann, D.; Both, M.; Ebert, U.; Gross, G.; Schoemaker, H.; Draguhn, A.; Wicke, K.; Nimmrich, V. Synaptic transmission is impaired prior to plaque formation in amyloid precursor protein–overexpressing mice without altering behaviorally-correlated sharp wave–ripple complexes. Neuroscience 2009, 162, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Kaden, D.; Voigt, P.; Munter, L.-M.; Bobowski, K.D.; Schaefer, M.; Multhaup, G. Subcellular localization and dimerization of APLP1 are strikingly different from APP and APLP2. J. Cell Sci. 2009, 122, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Sannerud, R.; Declerck, I.; Peric, A.; Raemaekers, T.; Menendez, G.; Zhou, L.; Veerle, B.; Coen, K.; Munck, S.; De Strooper, B.; et al. ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proc. Natl. Acad. Sci. USA 2011, 108, E559–E568. [Google Scholar] [CrossRef]
- Salbaum, J.M.; Ruddle, F.H. Embryonic expression pattern of amyloid protein precursor suggests a role in differentiation of specific subsets of neurons. J. Exp. Zool. 1994, 269, 116–127. [Google Scholar] [CrossRef]
- Clarris, H.J.; Key, B.; Beyreuther, K.; Masters, C.L.; Small, D.H. Expression of the amyloid protein precursor of Alzheimer’s disease in the developing rat olfactory system. Brain Res. Dev. Brain. Res. 1995, 88, 87–95. [Google Scholar] [CrossRef]
- Müller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef]
- Torroja, L.; Chu, H.; Kotovsky, I.; White, K. Neuronal overexpression of APPL, the Drosophila homologue of the amyloid precursor protein (APP), disrupts axonal transport. Curr. Biol. 1999, 9, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Dawson, G.R.; Seabrook, G.R.; Zheng, H.; Smith, D.W.; Graham, S.; O’dowd, G.; Bowery, B.J.; Boyce, S.; Trumbauer, M.E.; Chen, H.Y.; et al. Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor protein. Neuroscience 1999, 90, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Deyts, C.; Clutter, M.; Pierce, N.; Chakrabarty, P.; Ladd, T.B.; Goddi, A.; Rosario, A.M.; Cruz, P.; Vetrivel, K.; Wagner, S.L.; et al. APP-Mediated Signaling Prevents Memory Decline in Alzheimer’s Disease Mouse Model. Cell Rep. 2019, 27, 1345–1355.e6. [Google Scholar] [CrossRef]
- Rieche, F.; Carmine-Simmen, K.; Poeck, B.; Kretzschmar, D.; Strauss, R. Drosophila Full-Length Amyloid Precursor Protein Is Required for Visual Working Memory and Prevents Age-Related Memory Impairment. Curr. Biol. 2018, 28, 817–823.e3. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Young-Pearse, T.; Lee, H.; Hsieh, Y.C.; Chou, V.; Selkoe, D.J. Moving beyond amyloid and tau to capture the biological heterogeneity of Alzheimer’s disease. Trends Neurosci. 2023, 46, 426–444. [Google Scholar] [CrossRef]
- Marzi, S.J.; Leung, S.K.; Ribarska, T.; Hannon, E.; Smith, A.R.; Pishva, E.; Poschmann, J.; Moore, K.; Troakes, C.; Al-Sarraj, S.; et al. A histone acetylome-wide association study of Alzheimer’s disease identifies disease-associated H3K27ac differences in the entorhinal cortex. Nat. Neurosci. 2018, 21, 1618–1627. [Google Scholar] [CrossRef]
- Morabito, S.; Miyoshi, E.; Michael, N.; Swarup, V. Integrative genomics approach identifies conserved transcriptomic networks in Alzheimer’s disease. Hum. Mol. Genet. 2020, 29, 2899–2919. [Google Scholar] [CrossRef]
- Satoh, J.-I.; Yamamoto, Y.; Asahina, N.; Kitano, S.; Kino, Y. RNA-Seq data mining: Downregulation of neuroD6 serves as a possible biomarker for Alzheimer’s disease brains. Dis. Markers 2014, 2014, 1–10. [Google Scholar] [CrossRef]
- Patel, H.; Hodges, A.K.; Curtis, C.; Lee, S.H.; Troakes, C.; Dobson, R.J.; Newhouse, S.J. Transcriptomic analysis of probable asymptomatic and symptomatic alzheimer brains. Brain. Behav. Immun. 2019, 80, 644–656. [Google Scholar] [CrossRef]
- Lewis, P.N.; Lukiw, W.J.; De Boni, U.; Crapper McLachlan, D.R. Changes in chromatin structure associated with Alzheimer’s disease. J. Neurochem. 1981, 37, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, D.C.; Lewis, P. Alzheimer’s disease: Errors in gene expression. Can. J. Neurol. Sci./J. Can. Sci. Neurol. 1985, 12, 1–5. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Petty, E.; Pillus, L. Balancing chromatin remodeling and histone modifications in transcription. Trends Genet. 2013, 29, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Gjoneska, E.; Pfenning, A.R.; Mathys, H.; Quon, G.; Kundaje, A.; Tsai, L.-H.; Kellis, M. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 2015, 518, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Lardenoije, R.; Iatrou, A.; Kenis, G.; Kompotis, K.; Steinbusch, H.W.; Mastroeni, D.; Coleman, P.; Lemere, C.A.; Hof, P.R.; van den Hove, D.L.A.; et al. The epigenetics of aging and neurodegeneration. Prog. Neurobiol. 2015, 131, 21–64. [Google Scholar] [CrossRef] [PubMed]
- Alendar, A.; Berns, A. Sentinels of chromatin: Chromodomain helicase DNA-binding proteins in development and disease. Genes Dev. 2021, 35, 1403–1430. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, R.; Chen, D.A.; Rada-Iglesias, A.; Zhang, J.; Xiong, Y.; Helms, J.; Chang, C.-P.; Zhao, Y.; Swigut, T.; Wysocka, J. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 2010, 463, 958–962. [Google Scholar] [CrossRef] [PubMed]
- Sugathan, A.; Biagioli, M.; Golzio, C.; Erdin, S.; Blumenthal, I.; Manavalan, P.; Ragavendran, A.; Brand, H.; Lucente, D.; Miles, J.; et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl. Acad. Sci. USA 2014, 111, E4468–E4477. [Google Scholar] [CrossRef] [PubMed]
- Maussion, G.; Diallo, A.B.; Gigek, C.O.; Chen, E.S.; Crapper, L.; Théroux, J.-F.; Chen, G.G.; Vasuta, C.; Ernst, C. Investigation of genes important in neurodevelopment disorders in adult human brain. Hum. Genet. 2015, 134, 1037–1053. [Google Scholar] [CrossRef]
- Platt, R.J.; Zhou, Y.; Slaymaker, I.M.; Shetty, A.S.; Weisbach, N.R.; Kim, J.-A.; Sharma, J.; Desai, M.; Sood, S.; Kempton, H.R.; et al. Chd8 Mutation Leads to Autistic-like Behaviors and Impaired Striatal Circuits. Cell Rep. 2017, 19, 335–350. [Google Scholar] [CrossRef]
- Schulz, Y.; Wehner, P.; Opitz, L.; Salinas-Riester, G.; Bongers, E.M.H.F.; van Ravenswaaij-Arts, C.M.A.; Wincent, J.; Schoumans, J.; Kohlhase, J.; Borchers, A.; et al. CHD7, the gene mutated in CHARGE syndrome, regulates genes involved in neural crest cell guidance. Hum. Genet. 2014, 133, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Sarasa, M.; Sorribas, V.; Terrado, J.; Climent, S.; Palacios, J.M.; Mengod, G. Alzheimer beta-amyloid precursor proteins display specific patterns of expression during embryogenesis. Mech. Dev. 2000, 94, 233–236. [Google Scholar] [CrossRef]
- Young-Pearse, T.L.; Bai, J.; Chang, R.; Zheng, J.B.; LoTurco, J.J.; Selkoe, D.J. A critical function for beta-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J. Neurosci. 2007, 27, 14459–14469. [Google Scholar] [CrossRef] [PubMed]
- Jongmans, M.; Van Ravenswaaij-Arts, C.; Pitteloud, N.; Ogata, T.; Sato, N.; der Grinten, H.C.; Van Der Donk, K.; Seminara, S.; Bergman, J.; Brunner, H.; et al. CHD7 mutations in patients initially diagnosed with Kallmann syndrome—The clinical overlap with CHARGE syndrome. Clin. Genet. 2009, 75, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-G.; Kurth, I.; Lan, F.; Meliciani, I.; Wenzel, W.; Eom, S.H.; Kang, G.B.; Rosenberger, G.; Tekin, M.; Ozata, M.; et al. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet. 2008, 83, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-S.; Zhang, R.; Wang, G.; Zhang, Y.-F. The development prospection of HDAC inhibitors as a potential therapeutic direction in Alzheimer’s disease. Transl. Neurodegener. 2017, 6, 1–6. [Google Scholar] [CrossRef]
- Sung, Y.M.; Lee, T.; Yoon, H.; DiBattista, A.M.; Song, J.M.; Sohn, Y.; Moffat, E.I.; Turner, R.S.; Jung, M.; Kim, J.; et al. Mercaptoacetamide-based class II HDAC inhibitor lowers Abeta levels and improves learning and memory in a mouse model of Alzheimer’s disease. Exp. Neurol. 2013, 239, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Janczura, K.J.; Volmar, C.H.; Sartor, G.C.; Rao, S.J.; Ricciardi, N.R.; Lambert, G.; Brothers, S.P.; Wahlestedt, C. Inhibition of HDAC3 reverses Alzheimer’s disease-related pathologies in vitro and in the 3xTg-AD mouse model. Proc. Natl. Acad. Sci. USA 2018, 115, E11148–E11157. [Google Scholar] [CrossRef] [PubMed]
- Asad, Z.; Sachidanandan, C. Chemical screens in a zebrafish model of CHARGE syndrome identifies small molecules that ameliorate disease-like phenotypes in embryo. Eur. J. Med. Genet. 2020, 63, 103661. [Google Scholar] [CrossRef] [PubMed]
- Latcheva, N.K.; Viveiros, J.M.; Waddell, E.A.; Nguyen, P.T.; Liebl, F.L.; Marenda, D.R. Epigenetic crosstalk: Pharmacological inhibition of HDACs can rescue defective synaptic morphology and neurotransmission phenotypes associated with loss of the chromatin reader Kismet. Mol. Cell. Neurosci. 2018, 87, 77–85. [Google Scholar] [CrossRef]
- Latcheva, N.K.; Delaney, T.L.; Viveiros, J.M.; Smith, R.A.; Bernard, K.M.; Harsin, B.; Marenda, D.R.; Liebl, F.L.W. The CHD Protein, Kismet, is Important for the Recycling of Synaptic Vesicles during Endocytosis. Sci. Rep. 2019, 9, 1–13. [Google Scholar]
- Latcheva, N.K.; Ghosh, R.; Marenda, D.R. The epigenetics of CHARGE syndrome. Front. Biol. 2016, 11, 85–95. [Google Scholar] [CrossRef]
- Chou, V.T.; Johnson, S.A.; Van Vactor, D. Synapse development and maturation at the drosophila neuromuscular junction. Neural Dev. 2020, 15, 1–17. [Google Scholar]
- Bouleau, S.; Tricoire, H. Drosophila models of Alzheimer’s disease: Advances, limits, and perspectives. J. Alzheimers Dis. 2015, 45, 1015–1038. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, M.; Hassan, B.A. Amyloid precursor protein and neural development. Development 2014, 141, 2543–2548. [Google Scholar] [CrossRef] [PubMed]
- Fossgreen, A.; Brückner, B.; Czech, C.; Masters, C.L.; Beyreuther, K.; Paro, R. Transgenic Drosophila expressing human amyloid precursor protein show gamma-secretase activity and a blistered-wing phenotype. Proc. Natl. Acad. Sci. USA 1998, 95, 13703–13708. [Google Scholar] [CrossRef]
- Greeve, I.; Kretzschmar, D.; Tschäpe, J.A.; Beyn, A.; Brellinger, C.; Schweizer, M.; Nitsch, R.M.; Reifegerste, R. Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J. Neurosci. 2004, 24, 3899–3906. [Google Scholar] [CrossRef] [PubMed]
- Cassar, M.; Kretzschmar, D. Analysis of Amyloid Precursor Protein Function in Drosophila melanogaster. Front. Mol. Neurosci. 2016, 9, 61. [Google Scholar] [CrossRef]
- Wang, P.; Lin, M.; Pedrosa, E.; Hrabovsky, A.; Zhang, Z.; Guo, W.; Lachman, H.M.; Zheng, D. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in neurodevelopment. Mol. Autism 2015, 6, 1–18. [Google Scholar] [CrossRef]
- Ghosh, R.; Vegesna, S.; Safi, R.; Bao, H.; Zhang, B.; Marenda, D.R.; Liebl, F.L.W. Kismet positively regulates glutamate receptor localization and synaptic transmission at the Drosophila neuromuscular junction. PLoS ONE 2014, 9, e113494. [Google Scholar] [CrossRef]
- Chakraborty, R.; Vepuri, V.; Mhatre, S.D.; Paddock, B.E.; Miller, S.; Michelson, S.J.; Delvadia, R.; Desai, A.; Vinokur, M.; Melicharek, D.J.; et al. Characterization of a Drosophila Alzheimer’s disease model: Pharmacological rescue of cognitive defects. PLoS ONE 2011, 6, e20799. [Google Scholar] [CrossRef] [PubMed]
- Sarantseva, S.V.; Bolshakova, O.I.; Timoshenko, S.I.; Rodin, D.I.; Vitek, M.; Schwarzman, A.L. Studying the pathogenesis of Alzheimer’s disease in a Drosophila melanogaster model: Human APP overexpression in the brain of transgenic flies leads to deficit of the synaptic protein synaptotagmin. Russ. J. Genet. 2009, 45, 105–112. [Google Scholar] [CrossRef]
- Nieuwenhuis, B.; Eva, R. ARF6 and Rab11 as intrinsic regulators of axon regeneration. Small GTPases 2020, 11, 392–401. [Google Scholar] [CrossRef]
- Tzavlaki, K.; Moustakas, A. TGF-beta Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Su, L.; Pan, M.; Chen, L.; Li, H.; Zou, C.; Xie, J.Q.; Huang, X.; Lu, M.; Zou, D. Exploration of novel biomarkers in Alzheimer’s disease based on four diagnostic models. Front. Aging Neurosci. 2023, 15, 1079433. [Google Scholar] [CrossRef] [PubMed]
- Gervais, L.; van den Beek, M.; Josserand, M.; Sallé, J.; Stefanutti, M.; Perdigoto, C.N.; Skorski, P.; Mazouni, K.; Marshall, O.J.; Andrea, H.; et al. Cell Proliferation Is Kept in Check by the Chromatin Regulators Kismet/CHD7/CHD8 and Trr/MLL3/4. Dev. Cell 2019, 49, 556–573.e6. [Google Scholar] [CrossRef] [PubMed]
- Gjorgjieva, J.; Berni, J.; Evers, J.F.; Eglen, S.J. Neural circuits for peristaltic wave propagation in crawling Drosophila larvae: Analysis and modeling. Front. Comput. Neurosci. 2013, 7, 24. [Google Scholar] [CrossRef]
- Ormerod, K.G.; Scibelli, A.E.; Littleton, J.T. Regulation of excitation-contraction coupling at the Drosophila neuromuscular junction. J. Physiol. 2022, 600, 349–372. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Nishiyama, M.; Shoji, H.; Ohkawa, Y.; Kawamura, A.; Sato, T.; Suyama, M.; Takumi, T.; Miyakawa, T.; Nakayama, K.I. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature 2016, 537, 675–679. [Google Scholar] [CrossRef]
- Von Rotz, R.C.; Kohli, B.M.; Bosset, J.; Meier, M.; Suzuki, T.; Nitsch, R.M.; Konietzko, U. The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J. Cell Sci. 2004, 117, 4435–4448. [Google Scholar] [CrossRef]
- Checler, F.; Sunyach, C.; Pardossi-Piquard, R.; Sevalle, J.; Vincent, B.; Kawarai, T.; Girardot, N.; George-Hyslop, P.S.; da Costa, C.A. The gamma/epsilon-secretase-derived APP intracellular domain fragments regulate p53. Curr. Alzheimer. Res. 2007, 4, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Marie, C.; Clavairoly, A.; Frah, M.; Hmidan, H.; Yan, J.; Zhao, C.; Van Steenwinckel, J.; Daveau, R.; Zalc, B.; Hassan, B.; et al. Oligodendrocyte precursor survival and differentiation requires chromatin remodeling by Chd7 and Chd8. Proc. Natl. Acad. Sci. USA 2018, 115, E8246–E8255. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Tully, T.; White, K. Human amyloid precursor protein ameliorates behavioral deficit of flies deleted for appl gene. Neuron 1992, 9, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Bellen, H.J.; Levis, R.W.; He, Y.; Carlson, J.W.; Evans-Holm, M.; Bae, E.; Kim, J.; Metaxakis, A.; Savakis, C.; Schulze, K.L.; et al. The Drosophila gene disruption project: Progress using transposons with distinctive site specificities. Genetics 2011, 188, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.A.; Fetter, R.D.; Noordermeer, J.N.; Goodman, C.S.; DiAntonio, A. Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron 1997, 19, 1237–1248. [Google Scholar] [CrossRef] [PubMed]
- Xiong, K.; Clough, R.W.; Luo, X.G.; Struble, R.G.; Li, Y.M.; Yan, X.X. [3H]-L-685,458 as a radiotracer that maps gamma-secretase complex in the rat brain: Relevance to Abeta genesis and presence of active presenilin-1 components. Brain Res. 2007, 1157, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Mhatre, S.D.; Satyasi, V.; Killen, M.; Paddock, B.E.; Moir, R.D.; Saunders, A.J.; Marenda, D.R. Synaptic abnormalities in a Drosophila model of Alzheimer’s disease. Dis. Model Mech. 2014, 7, 373–385. [Google Scholar] [PubMed]
- Hick, M.; Herrmann, U.; Weyer, S.W.; Mallm, J.P.; Tschäpe, J.A.; Borgers, M.; Mercken, M.; Roth, F.C.; Draguhn, A.; Slomianka, L.; et al. Acute function of secreted amyloid precursor protein fragment APPsalpha in synaptic plasticity. Acta. Neuropathol. 2015, 129, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Klevanski, M.; Herrmann, U.; Weyer, S.W.; Fol, R.; Cartier, N.; Wolfer, D.P.; Caldwell, J.H.; Korte, M.; Müller, U.C. The APP Intracellular Domain Is Required for Normal Synaptic Morphology, Synaptic Plasticity, and Hippocampus-Dependent Behavior. J. Neurosci. 2015, 35, 16018–16033. [Google Scholar] [CrossRef]
- Bretou, M.; Sannerud, R.; Escamilla-Ayala, A.; Leroy, T.; Vrancx, C.; Van Acker, Z.P.; Perdok, A.; Vermeire, W.; Vorsters, I.; Van Keymolen, S.; et al. Accumulation of APP C-terminal fragments causes endolysosomal dysfunction through the dysregulation of late endosome to lysosome-ER contact sites. Dev. Cell 2024, 59, 1571–1592.e9. [Google Scholar] [CrossRef]
- Schnetz, M.P.; Bartels, C.F.; Shastri, K.; Balasubramanian, D.; Zentner, G.E.; Balaji, R.; Zhang, X.; Song, L.; Wang, Z.; LaFramboise, T.; et al. Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Res. 2009, 19, 590–601. [Google Scholar] [CrossRef]
- Feil, R.; Fraga, M.F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kaang, B.-K. Epigenetic regulation and chromatin remodeling in learning and memory. Exp. Mol. Med. 2017, 49, e281. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-Y.; Aromolaran, K.A.; Zukin, R.S. The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat. Rev. Neurosci. 2017, 18, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Parikshak, N.N.; Gandal, M.J.; Geschwind, D.H. Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat. Rev. Genet. 2015, 16, 441–458. [Google Scholar] [CrossRef] [PubMed]
- Ringheim, G.E.; Szczepanik, A.M.; Petko, W.; Burgher, K.L.; Zu Zhu, S.; Chao, C.C. Enhancement of beta-amyloid precursor protein transcription and expression by the soluble interleukin-6 receptor/interleukin-6 complex. Mol. Brain Res. 1998, 55, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.I.; Fujiwara, K.; Inoue, S. Amyloid precursor protein, an androgen-regulated gene, is targeted by RNA-binding protein PSF/SFPQ in neuronal cells. Genes Cells 2019, 24, 719–730. [Google Scholar] [CrossRef]
- Grilli, M.; Goffi, F.; Memo, M.; Spano, P. Interleukin-1beta and glutamate activate the NF-kappaB/Rel binding site from the regulatory region of the amyloid precursor protein gene in primary neuronal cultures. J. Biol. Chem. 1996, 271, 15002–15007. [Google Scholar] [CrossRef]
- Huang, Y.W.A.; Zhou, B.; Nabet, A.M.; Wernig, M.; Südhof, T.C. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell 2017, 168, 427–441.e21. [Google Scholar] [CrossRef]
- Derafshi, B.H.; Danko, T.; Chanda, S.; Batista, P.J.; Litzenburger, U.; Lee, Q.Y.; Ng, Y.H.; Sebin, A.; Chang, H.Y.; Südhof, T.C.; et al. The autism risk factor CHD8 is a chromatin activator in human neurons and functionally dependent on the ERK-MAPK pathway effector ELK1. Sci. Rep. 2022, 12, 1–28. [Google Scholar]
- Bukhari, H.; Glotzbach, A.; Kolbe, K.; Leonhardt, G.; Loosse, C.; Müller, T. Small things matter: Implications of APP intracellular domain AICD nuclear signaling in the progression and pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2017, 156, 189–213. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.R.; Hendricks, E.L.; Latcheva, N.K.; Marenda, D.R.; Liebl, F.L.W. The CHD Protein Kismet Restricts the Synaptic Localization of Cell Adhesion Molecules at the Drosophila Neuromuscular Junction. Int. J. Mol. Sci. 2024, 25, 3074. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.S.M.; Ng, K.Y.; Koh, R.Y.; Chok, K.C.; Chye, S.M. Endosomal-lysosomal dysfunctions in Alzheimer’s disease: Pathogenesis and therapeutic interventions. Metab. Brain Dis. 2021, 36, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Perl, K.; Ushakov, K.; Pozniak, Y.; Yizhar-Barnea, O.; Bhonker, Y.; Shivatzki, S.; Geiger, T.; Avraham, K.B.; Shamir, R. Reduced changes in protein compared to mRNA levels across non-proliferating tissues. BMC Genom. 2017, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, D.C.; Crabtree, G.R. ATP-dependent chromatin remodeling: Genetics, genomics and mechanisms. Cell Res. 2011, 21, 396–420. [Google Scholar] [CrossRef] [PubMed]
- Meier, K.; Brehm, A. Chromatin regulation: How complex does it get? Epigenetics 2014, 9, 1485–1495. [Google Scholar] [CrossRef] [PubMed]
- Latcheva, N.K.; Viveiros, J.M.; Marenda, D.R. The Drosophila Chromodomain Protein Kismet Activates Steroid Hormone Receptor Transcription to Govern Axon Pruning and Memory In Vivo. iScience 2019, 16, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Steuble, M.; Gerrits, B.; Ludwig, A.; Mateos, J.M.; Diep, T.M.; Tagaya, M.; Stephan, A.; Schätzle, P.; Kunz, B.; Streit, P.; et al. Molecular characterization of a trafficking organelle: Dissecting the axonal paths of calsyntenin-1 transport vesicles. Proteomics 2010, 10, 3775–3788. [Google Scholar] [CrossRef]
- Dumanchin, C.; Czech, C.; Campion, D.; Cuif, M.H.; Poyot, T.; Martin, C.; Charbonnier, F.; Goud, B.; Pradier, L.; Frebourg, T. Presenilins interact with Rab11, a small GTPase involved in the regulation of vesicular transport. Hum. Mol. Genet. 1999, 8, 1263–1269. [Google Scholar] [CrossRef]
- Buggia-Prévot, V.; Fernandez, C.G.; Riordan, S.; Vetrivel, K.S.; Roseman, J.; Waters, J.; Bindokas, V.P.; Vassar, R.; Thinakaran, G. Axonal BACE1 dynamics and targeting in hippocampal neurons: A role for Rab11 GTPase. Mol. Neurodegener. 2014, 9, 1–18. [Google Scholar] [CrossRef]
- Udayar, V.; Buggia-Prevot, V.; Guerreiro, R.L.; Siegel, G.; Rambabu, N.; Soohoo, A.L.; Ponnusamy, M.; Siegenthaler, B.; Bali, J.; Guerreiro, R.; et al. A paired RNAi and RabGAP overexpression screen identifies Rab11 as a regulator of beta-amyloid production. Cell Rep. 2013, 5, 1536–1551. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wei, M.; Wu, Y.; Qin, H.; Li, W.; Ma, X.; Cheng, J.; Ren, J.; Shen, Y.; Chen, Z. Amyloid beta oligomers suppress excitatory transmitter release via presynaptic depletion of phosphatidylinositol-4,5-bisphosphate. Nat. Commun. 2019, 10, 1193. [Google Scholar] [CrossRef] [PubMed]
- Seebohm, G.; Neumann, S.; Theiss, C.; Novkovic, T.; Hill, E.V.; Tavaré, J.M.; Lang, F.; Hollmann, M.; Manahan-Vaughan, D.; Strutz-Seebohm, N. Identification of a novel signaling pathway and its relevance for GluA1 recycling. PLoS ONE 2012, 7, e33889. [Google Scholar] [CrossRef] [PubMed]
- Raghu, P.; Joseph, A.; Krishnan, H.; Singh, P.; Saha, S. Phosphoinositides: Regulators of Nervous System Function in Health and Disease. Front. Mol. Neurosci. 2019, 12, 208. [Google Scholar] [CrossRef] [PubMed]
- Salinas, R.; Connolly, D.R.; Song, H. Invited Review: Epigenetics in neurodevelopment. Neuropathol. Appl. Neurobiol. 2020, 46, 6–27. [Google Scholar] [CrossRef] [PubMed]
- Emerson, F.J.; Lee, S.S. Chromatin: The old and young of it. Front. Mol. Biosci. 2023, 10, 1270285. [Google Scholar] [CrossRef] [PubMed]
- Nativio, R.; Donahue, G.; Berson, A.; Lan, Y.; Amlie-Wolf, A.; Tuzer, F.; Toledo, J.B.; Gosai, S.J.; Gregory, B.D.; Torres, C.; et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Dorighi, K.M.; Tamkun, J.W. The trithorax group proteins Kismet and ASH1 promote H3K36 dimethylation to counteract Polycomb group repression in Drosophila. Development 2013, 140, 4182–4192. [Google Scholar] [CrossRef] [PubMed]
- Volloch, V.; Rits-Volloch, S. Effect of Lecanemab in Early Alzheimer’s Disease: Mechanistic Interpretation in the Amyloid Cascade Hypothesis 2.0 Perspective. J. Alzheimers Dis. 2023, 93, 1277–1284. [Google Scholar] [CrossRef]
- Hur, J.Y. gamma-Secretase in Alzheimer’s disease. Exp. Mol. Med. 2022, 54, 433–446. [Google Scholar] [CrossRef]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef] [PubMed]
- Melicharek, D.J.; Ramirez, L.C.; Singh, S.; Thompson, R.; Marenda, D.R. Kismet/CHD7 regulates axon morphology, memory and locomotion in a Drosophila model of CHARGE syndrome. Hum. Mol. Genet. 2010, 19, 4253–4264. [Google Scholar] [CrossRef]
- Marrus, S.B.; Portman, S.L.; Allen, M.J.; Moffat, K.G.; DiAntonio, A. Differential localization of glutamate receptor subunits at the drosophila neuromuscular junction. J. Neurosci. 2004, 24, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Verstreken, P.; Ohyama, T.; Bellen, H.J. FM 1-43 labeling of synaptic vesicle pools at the Drosophila neuromuscular junction. Methods Mol. Biol. 2008, 440, 349–369. [Google Scholar] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bykhovskaia, M. Making quantal analysis more convenient, fast, and accurate: User-friendly software QUANTAN. J. Neurosci. Methods 2008, 168, 500–513. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).