
Comprehensive Transcriptome Analysis Expands lncRNA Functional Profiles in Breast Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
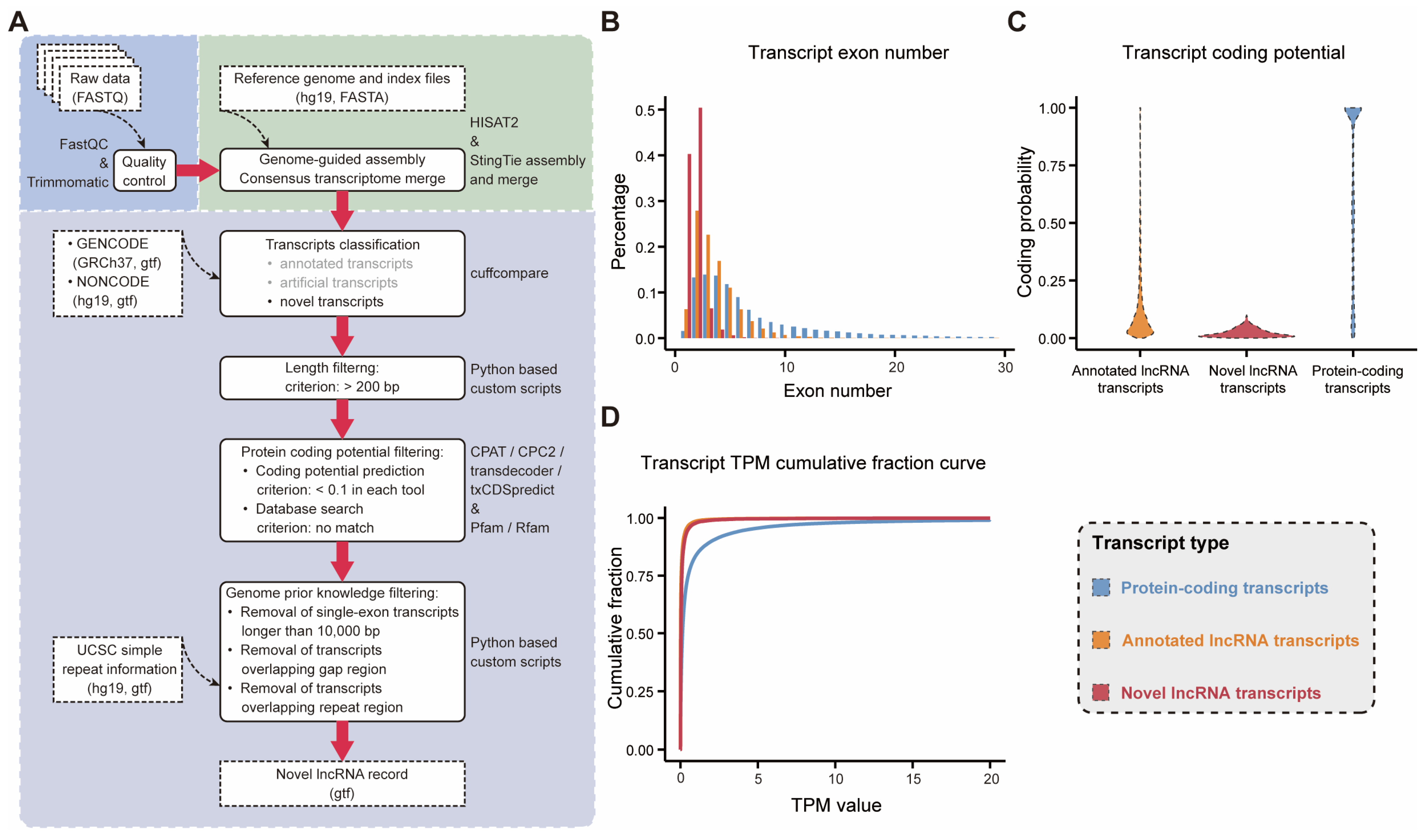
2.1. A Framework for the Identification of Novel lncRNAs in the SEU-BRCA Dataset
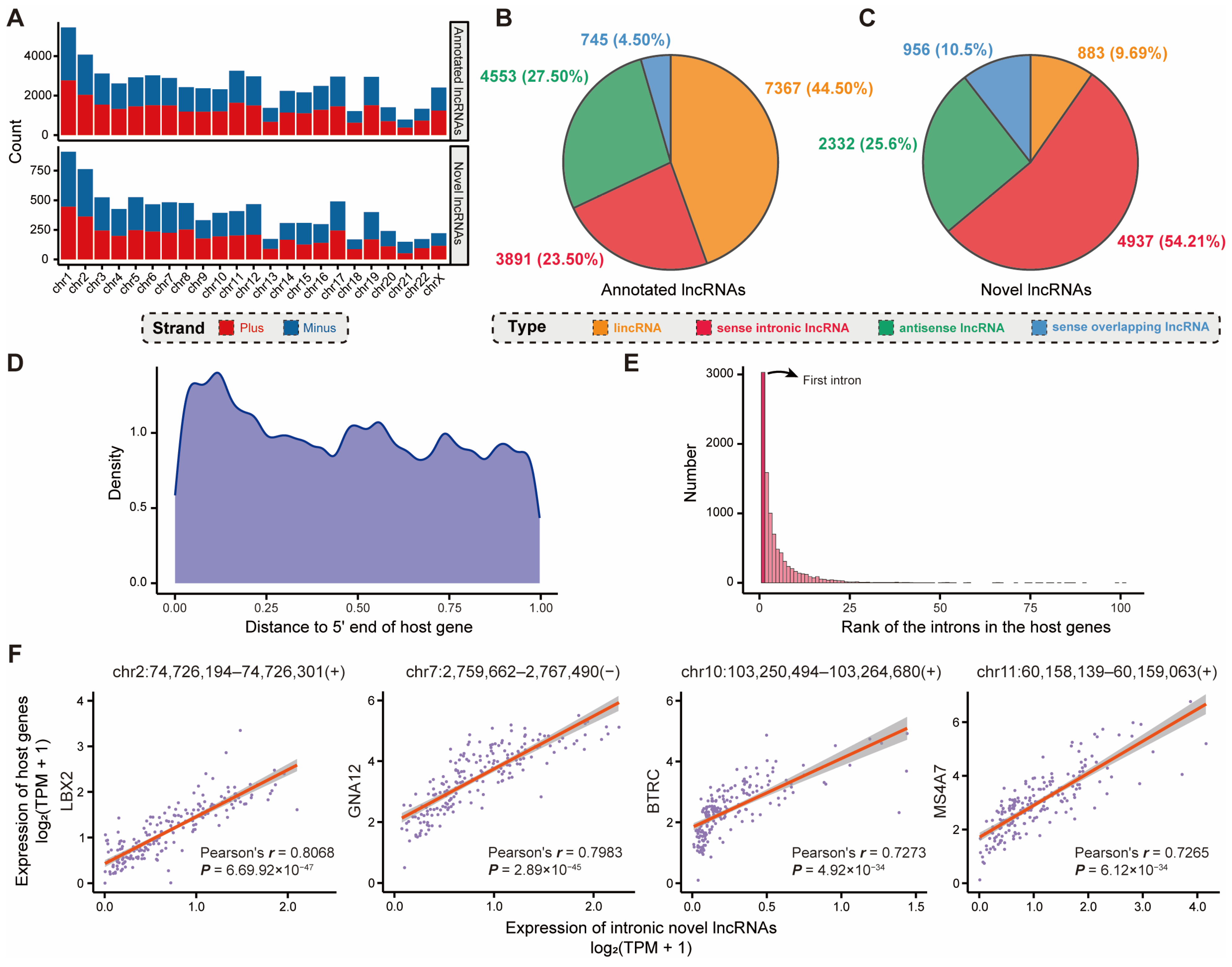
2.2. Novel Intronic lncRNAs Involved in Breast-Cancer-Specific Biological Processes
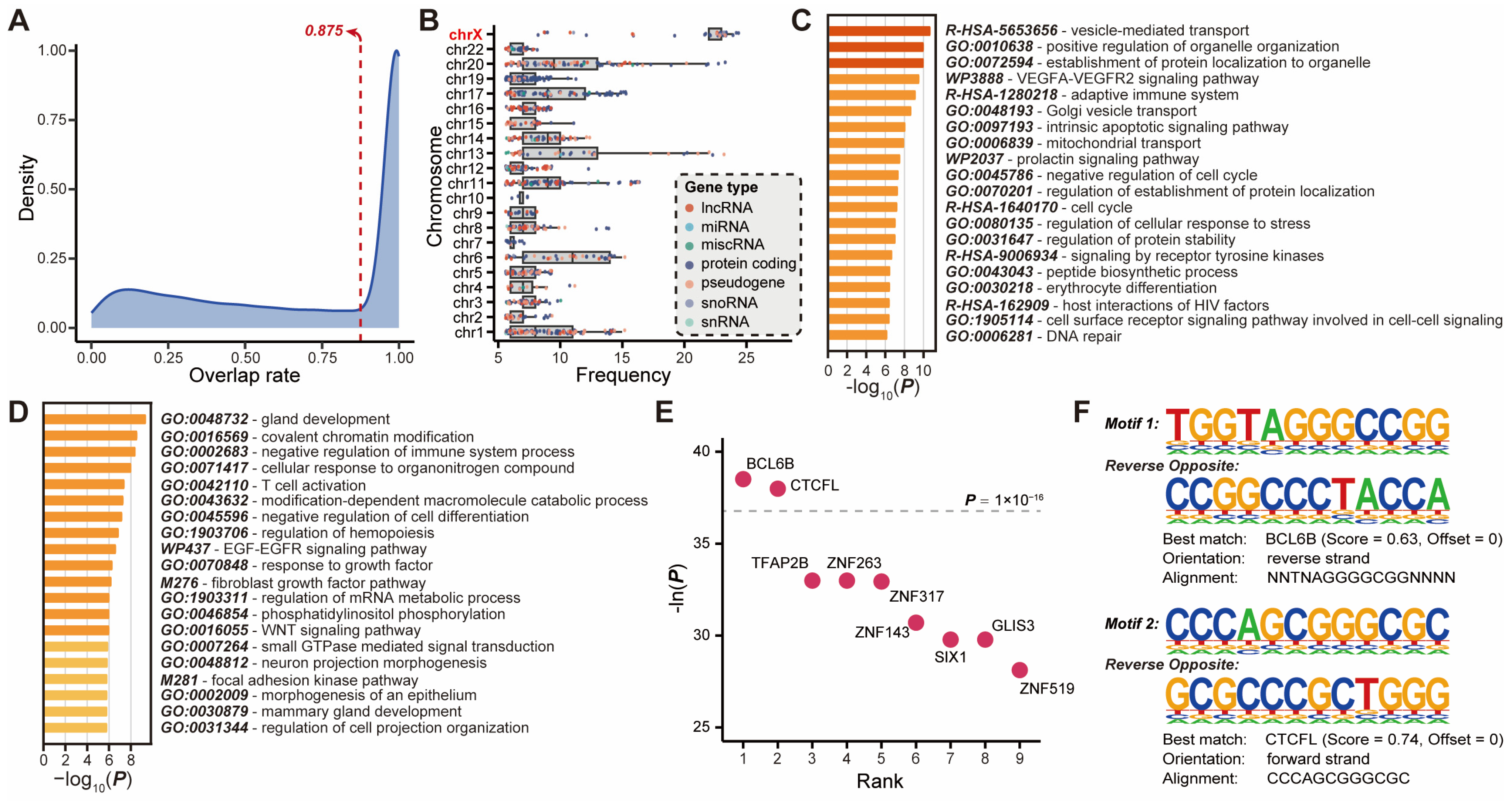
2.3. Novel lncRNAs Associated with Putative Enhancers Were Involved in Breast Cancer Regulatory Network
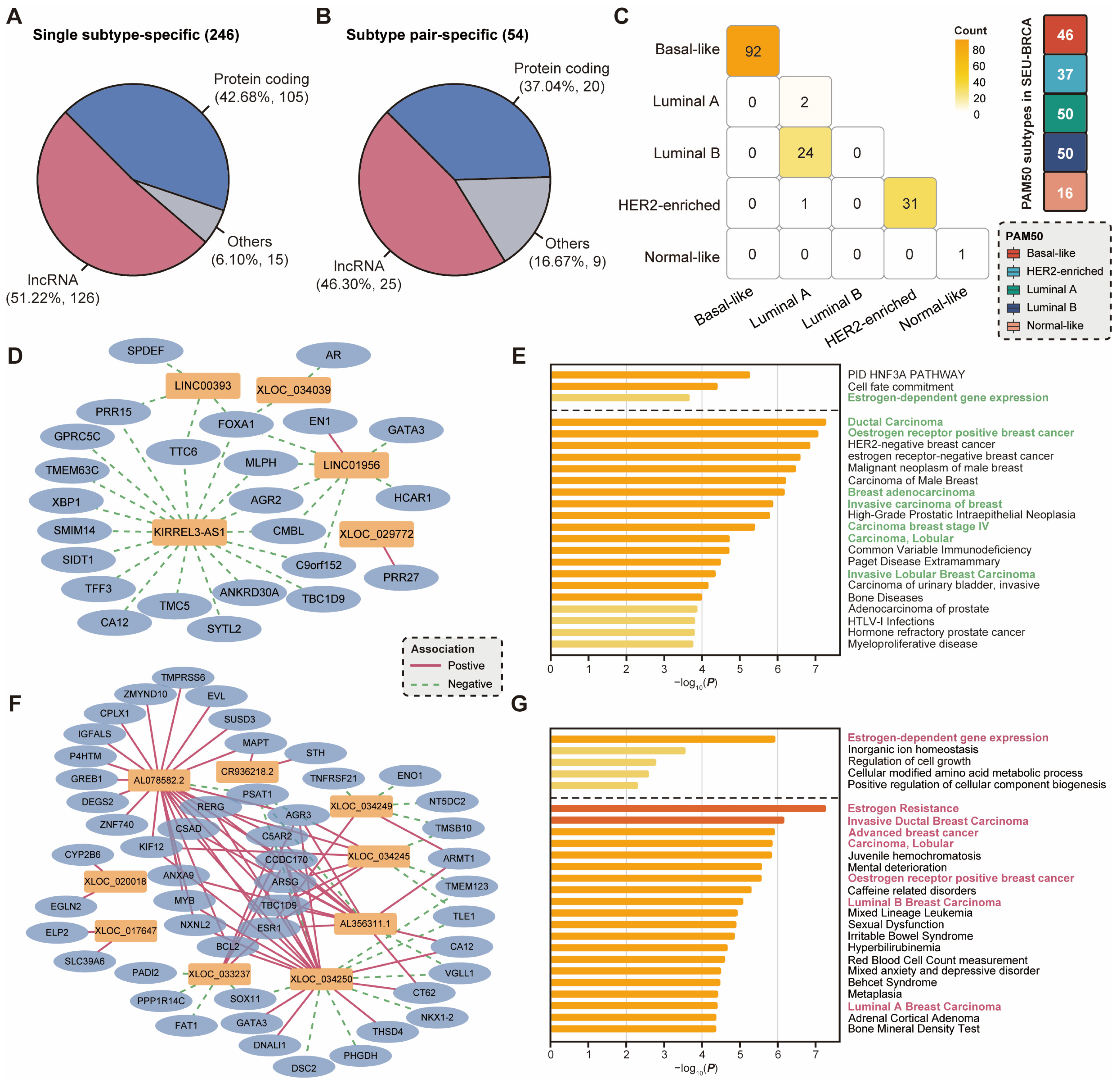
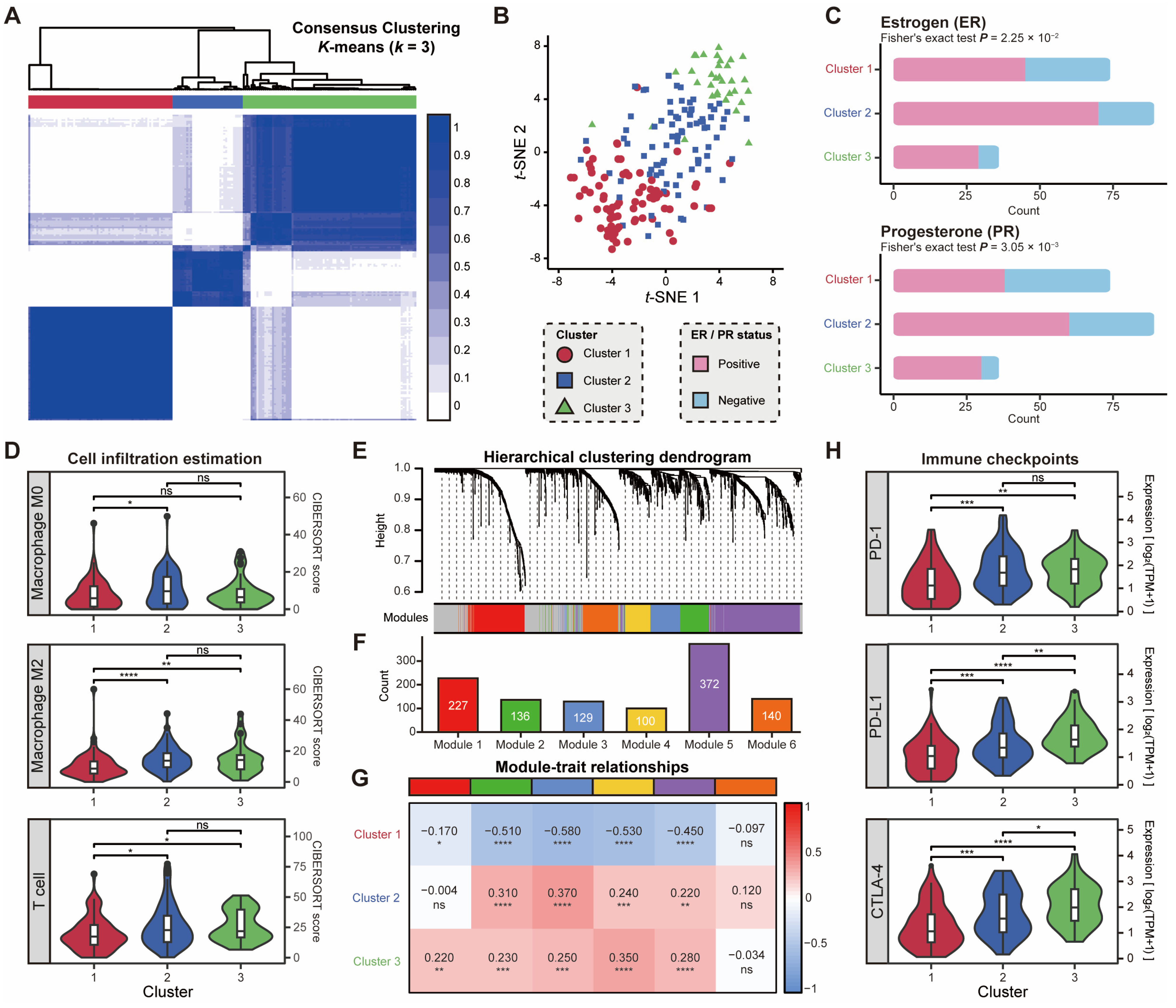
2.4. Subtype-Specific lncRNAs Correlate with Dysregulated Pathways and Hormonal Characteristics
2.5. Expression Patterns of lncRNAs Associated with Hormonal Characteristics and Immune Activation
3. Discussion
4. Materials and Methods
4.1. Raw Data Processing
4.2. Identification of Novel lncRNA Transcripts
4.3. Function Analysis of Novel lncRNAs Associated Protein-Coding Genes
4.4. Extraction of Breast-Cancer-Related Putative Enhancers
4.5. Detection of Subtype-Specific Expressed lncRNAs
4.6. Construction of lncRNA Regulatory Networks
4.7. Consensus Clustering Based on Expression Patterns of lncRNAs
4.8. Construction of Co-Expression Module of Immune-Related Genes
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K. Heterogeneity in breast cancer. J. Clin. Investig. 2011, 121, 3786–3788. [Google Scholar] [CrossRef] [PubMed]
- Dvir, K.; Giordano, S.; Leone, J.P. Immunotherapy in Breast Cancer. Int. J. Mol. Sci. 2024, 25, 7517. [Google Scholar] [CrossRef]
- Blows, F.M.; Driver, K.E.; Schmidt, M.K.; Broeks, A.; Van Leeuwen, F.E.; Wesseling, J.; Cheang, M.C.; Gelmon, K.; Nielsen, T.O.; Blomqvist, C. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: A collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med. 2010, 7, e1000279. [Google Scholar] [CrossRef]
- Zhao, L.; Lee, V.H.; Ng, M.K.; Yan, H.; Bijlsma, M.F. Molecular subtyping of cancer: Current status and moving toward clinical applications. Brief. Bioinform. 2019, 20, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Song, Y.; Zhang, Y.; Wu, H.; Chen, L.; Pang, J.; Zhou, L.; Shen, S.; Liang, Z. Prognostic significance of different molecular typing methods and immune status based on RNA sequencing in HR-positive and HER2-negative early-stage breast cancer. BMC Cancer 2022, 22, 548. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, S.; de Anda-Jáuregui, G.; Hernández-Lemus, E. Multi-omic regulation of the PAM50 gene signature in breast cancer molecular subtypes. Front. Oncol. 2020, 10, 845. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V. A comprehensive pan-cancer molecular study of gynecologic and breast cancers. Cancer Cell 2018, 33, 690–705.e9. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.-W.; Wang, Y.; Chen, L.-L. Cellular functions of long noncoding RNAs. Nat. Cell Biol. 2019, 21, 542–551. [Google Scholar] [CrossRef]
- Jadaliha, M.; Gholamalamdari, O.; Tang, W.; Zhang, Y.; Petracovici, A.; Hao, Q.; Tariq, A.; Kim, T.G.; Holton, S.E.; Singh, D.K. A natural antisense lncRNA controls breast cancer progression by promoting tumor suppressor gene mRNA stability. PLoS Genet. 2018, 14, e1007802. [Google Scholar] [CrossRef]
- Peng, W.-X.; Koirala, P.; Mo, Y.-Y. LncRNA-mediated regulation of cell signaling in cancer. Oncogene 2017, 36, 5661–5667. [Google Scholar] [CrossRef]
- Chandra Gupta, S.; Nandan Tripathi, Y. Potential of long non-coding RNAs in cancer patients: From biomarkers to therapeutic targets. Int. J. Cancer 2017, 140, 1955–1967. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, T.; Zhou, W.; Li, J.; Li, X.; Wang, Q.; Jin, X.; Yin, J.; Chen, L.; Zhang, Y. Pan-cancer characterization of immune-related lncRNAs identifies potential oncogenic biomarkers. Nat. Commun. 2020, 11, 1000. [Google Scholar] [CrossRef]
- Zhu, W.; Huang, H.; Ming, W.; Zhang, R.; Gu, Y.; Bai, Y.; Liu, X.; Liu, H.; Liu, Y.; Gu, W. Delineating highly transcribed noncoding elements landscape in breast cancer. Comput. Struct. Biotechnol. J. 2023, 21, 4432–4445. [Google Scholar] [CrossRef] [PubMed]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Huang, H.-T.; Liang, Y.; Trimarchi, T.; Aifantis, I.; Tsirigos, A. lncRNA-screen: An interactive platform for computationally screening long non-coding RNAs in large genomics datasets. BMC Genom. 2017, 18, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Zhao, Y.; Wang, H.; Sun, H. Sebnif: An integrated bioinformatics pipeline for the identification of novel large intergenic noncoding rnas (lincrnas)-application in human skeletal muscle cells. PLoS ONE 2014, 9, e84500. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Nair, A.; Chen, X.; Prodduturi, N.; Wang, J.; Kocher, J.-P. UClncR: Ultrafast and comprehensive long non-coding RNA detection from RNA-seq. Sci. Rep. 2017, 7, 14196. [Google Scholar] [CrossRef]
- Zhao, Q.; Sun, Y.; Wang, D.; Zhang, H.; Yu, K.; Zheng, J.; Zuo, Z. LncPipe: A Nextflow-based pipeline for identification and analysis of long non-coding RNAs from RNA-Seq data. J. Genet. Genom. 2018, 45, 399–401. [Google Scholar] [CrossRef]
- Baruzzo, G.; Hayer, K.E.; Kim, E.J.; Di Camillo, B.; FitzGerald, G.A.; Grant, G.R. Simulation-based comprehensive benchmarking of RNA-seq aligners. Nat. Methods 2017, 14, 135–139. [Google Scholar] [CrossRef]
- Corchete, L.A.; Rojas, E.A.; Alonso-López, D.; De Las Rivas, J.; Gutiérrez, N.C.; Burguillo, F.J. Systematic comparison and assessment of RNA-seq procedures for gene expression quantitative analysis. Sci. Rep. 2020, 10, 19737. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yin, H.; Li, B.; Yu, C.; Wang, F.; Xu, X.; Cao, J.; Bao, Y.; Wang, L.; Abbasi, A.A. Characterization and identification of long non-coding RNAs based on feature relationship. Bioinformatics 2019, 35, 2949–2956. [Google Scholar] [CrossRef] [PubMed]
- Arunima, A.; van Schaik, E.J.; Samuel, J.E. The emerging roles of long non-coding RNA in host immune response and intracellular bacterial infections. Front. Cell. Infect. Microbiol. 2023, 13, 1160198. [Google Scholar] [CrossRef]
- Szempruch, A.; Guttman, M. Linking protein and RNA function within the same gene. Cell 2017, 168, 753–755. [Google Scholar] [CrossRef]
- Barkovskaya, A.; Buffone Jr, A.; Žídek, M.; Weaver, V.M. Proteoglycans as mediators of cancer tissue mechanics. Front. Cell Dev. Biol. 2020, 8, 569377. [Google Scholar] [CrossRef]
- Ordaz-Ramos, A.; Tellez-Jimenez, O.; Vazquez-Santillan, K. Signaling pathways governing the maintenance of breast cancer stem cells and their therapeutic implications. Front. Cell Dev. Biol. 2023, 11, 1221175. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, M.; Xu, F.; Jiang, S. Wnt signaling in breast cancer: Biological mechanisms, challenges and opportunities. Mol. Cancer 2020, 19, 165. [Google Scholar] [CrossRef]
- Böhmdorfer, G.; Wierzbicki, A.T. Control of chromatin structure by long noncoding RNA. Trends Cell Biol. 2015, 25, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Li, C.; Fang, Y.; Gu, X.; Zheng, Q.; Lu, J.; Li, L. The role of LncRNA LBX2-AS1 in cancers: Functions, mechanisms and potential clinical utility. Clin. Transl. Oncol. 2023, 25, 293–305. [Google Scholar] [CrossRef]
- Fang, J.; Yang, J.; Chen, H.; Sun, W.; Xiang, L.; Feng, J. Long non-coding RNA LBX2-AS1 predicts poor survival of colon cancer patients and promotes its progression via regulating miR-627-5p/RAC1/PI3K/AKT pathway. Hum. Cell 2022, 35, 1521–1534. [Google Scholar] [CrossRef]
- Mattiola, I.; Mantovani, A.; Locati, M. The tetraspan MS4A family in homeostasis, immunity, and disease. Trends Immunol. 2021, 42, 764–781. [Google Scholar] [CrossRef]
- Rasheed, S.A.K.; Subramanyan, L.V.; Lim, W.K.; Udayappan, U.K.; Wang, M.; Casey, P.J. The emerging roles of Gα12/13 proteins on the hallmarks of cancer in solid tumors. Oncogene 2022, 41, 147–158. [Google Scholar] [CrossRef]
- Shen, H.-y.; Xu, J.-l.; Zhang, W.; Chen, Q.-n.; Zhu, Z.; Mao, Y. Exosomal circRHCG promotes breast cancer metastasis via facilitating M2 polarization through TFEB ubiquitination and degradation. NPJ Precis. Oncol. 2024, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Satomi-Tsushita, N.; Shimomura, A.; Matsuzaki, J.; Yamamoto, Y.; Kawauchi, J.; Takizawa, S.; Aoki, Y.; Sakamoto, H.; Kato, K.; Shimizu, C. Serum microRNA-based prediction of responsiveness to eribulin in metastatic breast cancer. PLoS ONE 2019, 14, e0222024. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zhang, R.; Sun, X. Enhancer LncRNAs influence chromatin interactions in different ways. Front. Genet. 2019, 10, 484553. [Google Scholar] [CrossRef] [PubMed]
- Ropri, A.S.; DeVaux, R.S.; Eng, J.; Chittur, S.V.; Herschkowitz, J.I. Cis-acting super-enhancer lncRNAs as biomarkers to early-stage breast cancer. Breast Cancer Res. 2021, 23, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Sartorelli, V.; Lauberth, S.M. Enhancer RNAs are an important regulatory layer of the epigenome. Nat. Struct. Mol. Biol. 2020, 27, 521–528. [Google Scholar] [CrossRef]
- Zawadka, P.; Zielińska, W.; Gagat, M.; Izdebska, M. Role of Filamin A in Growth and Migration of Breast Cancer. Curr. Issues Mol. Biol. 2024, 46, 3408–3423. [Google Scholar] [CrossRef]
- Krøigård, A.B.; Larsen, M.J.; Lænkholm, A.-V.; Knoop, A.S.; Jensen, J.D.; Bak, M.; Mollenhauer, J.; Thomassen, M.; Kruse, T.A. Identification of metastasis driver genes by massive parallel sequencing of successive steps of breast cancer progression. PLoS ONE 2018, 13, e0189887. [Google Scholar] [CrossRef]
- Bergmaier, P.; Weth, O.; Dienstbach, S.; Boettger, T.; Galjart, N.; Mernberger, M.; Bartkuhn, M.; Renkawitz, R. Choice of binding sites for CTCFL compared to CTCF is driven by chromatin and by sequence preference. Nucleic Acids Res. 2018, 46, 7097–7107. [Google Scholar] [CrossRef]
- Nishana, M.; Ha, C.; Rodriguez-Hernaez, J.; Ranjbaran, A.; Chio, E.; Nora, E.P.; Badri, S.B.; Kloetgen, A.; Bruneau, B.G.; Tsirigos, A. Defining the relative and combined contribution of CTCF and CTCFL to genomic regulation. Genome Biol. 2020, 21, 1–34. [Google Scholar] [CrossRef]
- Debaugny, R.E.; Skok, J.A. CTCF and CTCFL in cancer. Curr. Opin. Genet. Dev. 2020, 61, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Lu, H.; Liu, J.; Wu, S.; Kim, P.; Zhou, X. lncRNAfunc: A knowledgebase of lncRNA function in human cancer. Nucleic Acids Res. 2022, 50, D1295–D1306. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Chen, F.; Liu, D.; Gu, F.; Chen, Z.; Wang, Y. Prognostic value of immune checkpoint molecules in breast cancer. Biosci. Rep. 2020, 40, BSR20201054. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wei, D.; Zhang, Y.; Wang, P.; Zhang, W. Long non-coding RNAs as potential diagnostic and prognostic biomarkers in breast cancer: Progress and prospects. Front. Oncol. 2021, 11, 710538. [Google Scholar] [CrossRef] [PubMed]
- Kornienko, A.E.; Dotter, C.P.; Guenzl, P.M.; Gisslinger, H.; Gisslinger, B.; Cleary, C.; Kralovics, R.; Pauler, F.M.; Barlow, D.P. Long non-coding RNAs display higher natural expression variation than protein-coding genes in healthy humans. Genome Biol. 2016, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, S.U.; Grote, P.; Herrmann, B.G. Mechanisms of long noncoding RNA function in development and disease. Cell. Mol. Life Sci. 2016, 73, 2491–2509. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef]
- Wang, W.; Min, L.; Qiu, X.; Wu, X.; Liu, C.; Ma, J.; Zhang, D.; Zhu, L. Biological function of long non-coding RNA (LncRNA) Xist. Front. Cell Dev. Biol. 2021, 9, 645647. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Chen, S.; Hu, Y.; Huang, W. Landscape of immune microenvironment under immune cell infiltration pattern in breast cancer. Front. Immunol. 2021, 12, 711433. [Google Scholar] [CrossRef] [PubMed]
- Quezada, S.; Peggs, K. Exploiting CTLA-4, PD-1 and PD-L1 to reactivate the host immune response against cancer. Br. J. Cancer 2013, 108, 1560–1565. [Google Scholar] [CrossRef] [PubMed]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, W.; Huang, H.; Hu, Z.; Gu, Y.; Zhang, R.; Shu, H.; Liu, H.; Sun, X. Comprehensive Transcriptome Analysis Expands lncRNA Functional Profiles in Breast Cancer. Int. J. Mol. Sci. 2024, 25, 8456. https://doi.org/10.3390/ijms25158456
Zhu W, Huang H, Hu Z, Gu Y, Zhang R, Shu H, Liu H, Sun X. Comprehensive Transcriptome Analysis Expands lncRNA Functional Profiles in Breast Cancer. International Journal of Molecular Sciences. 2024; 25(15):8456. https://doi.org/10.3390/ijms25158456
Chicago/Turabian StyleZhu, Wenyong, Hao Huang, Zixi Hu, Yu Gu, Rongxin Zhang, Huiling Shu, Hongjia Liu, and Xiao Sun. 2024. "Comprehensive Transcriptome Analysis Expands lncRNA Functional Profiles in Breast Cancer" International Journal of Molecular Sciences 25, no. 15: 8456. https://doi.org/10.3390/ijms25158456
APA StyleZhu, W., Huang, H., Hu, Z., Gu, Y., Zhang, R., Shu, H., Liu, H., & Sun, X. (2024). Comprehensive Transcriptome Analysis Expands lncRNA Functional Profiles in Breast Cancer. International Journal of Molecular Sciences, 25(15), 8456. https://doi.org/10.3390/ijms25158456