
Allele-Selective Thiomorpholino Antisense Oligonucleotides as a Therapeutic Approach for Fused-in-Sarcoma Amyotrophic Lateral Sclerosis

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
2.1. FUS Common Variant Analysis
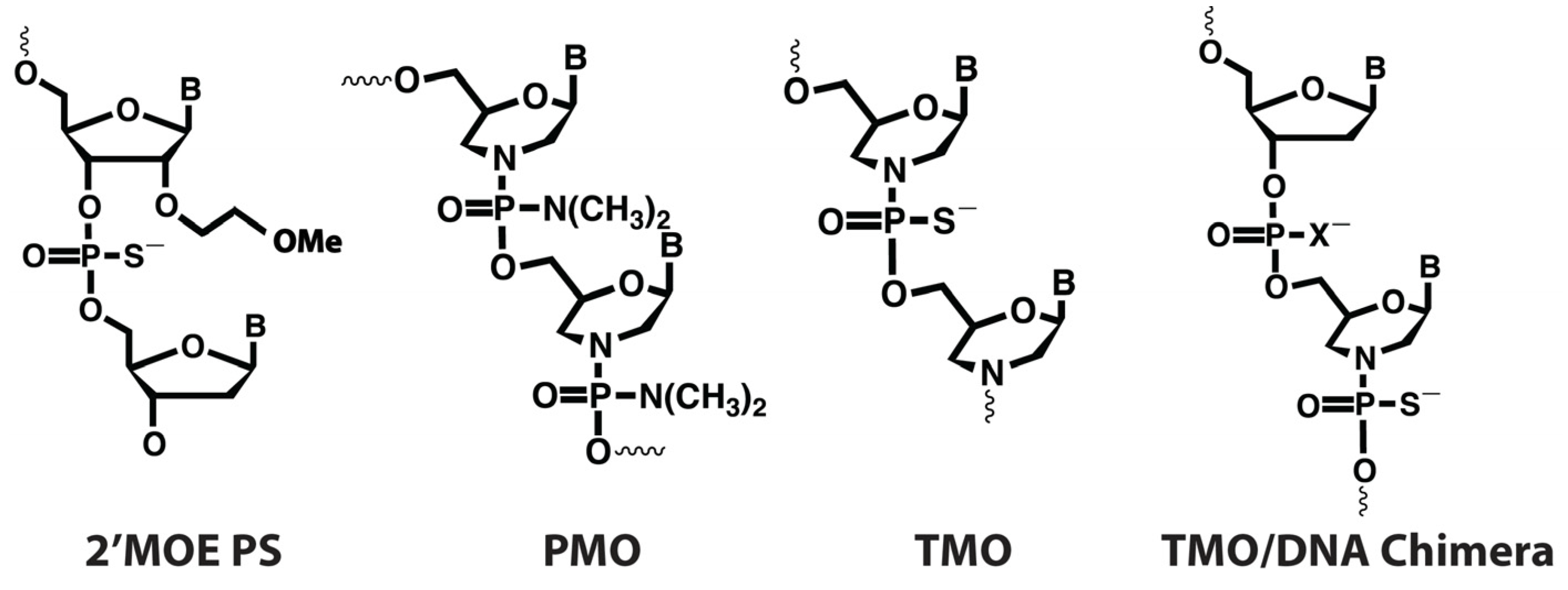
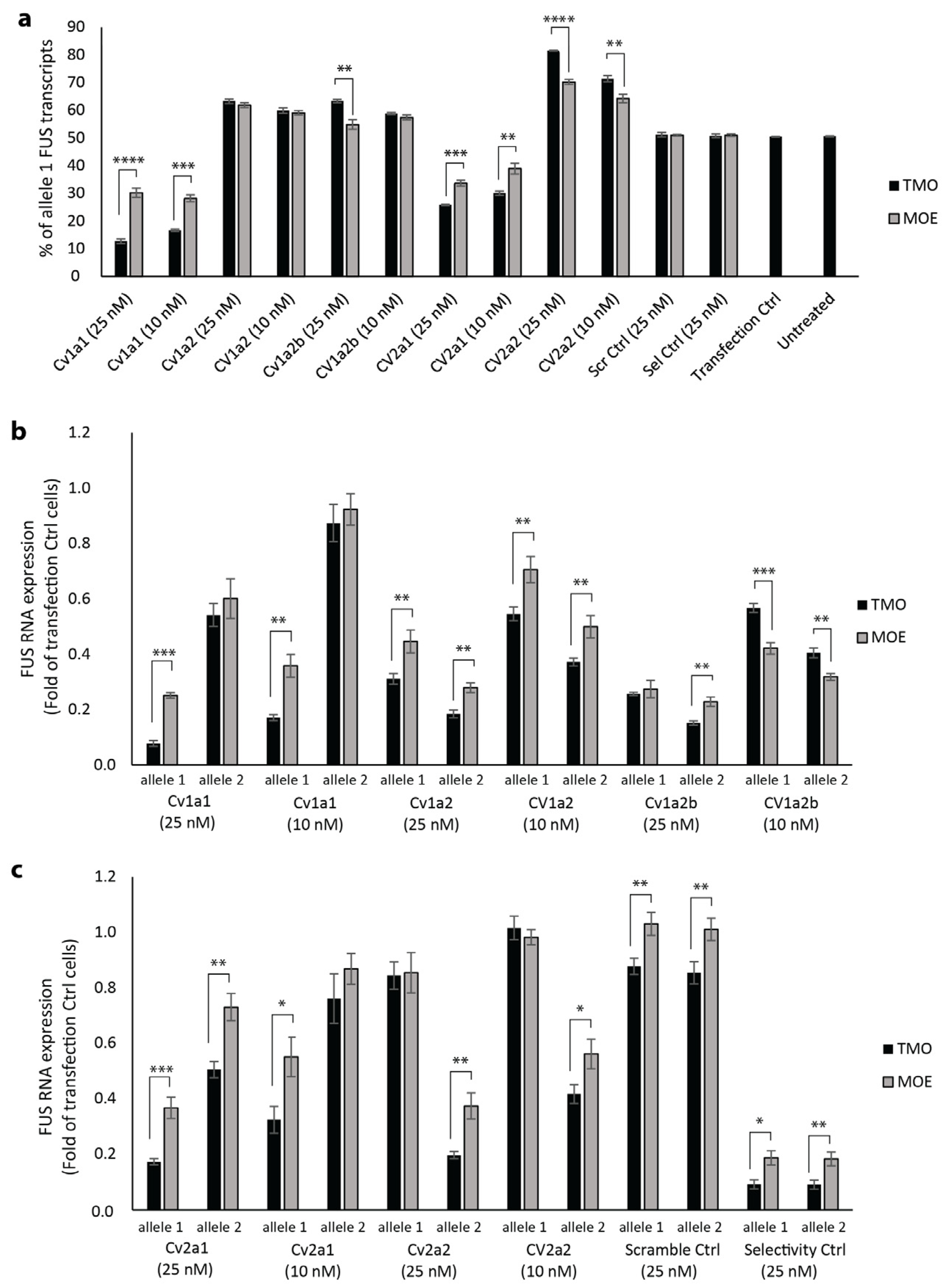
2.2. Allele Selectivity of MOE and TMO-Modified FUS-Targeted AOs
2.3. TMO Reduces SFPQ Positive Protein Aggregation
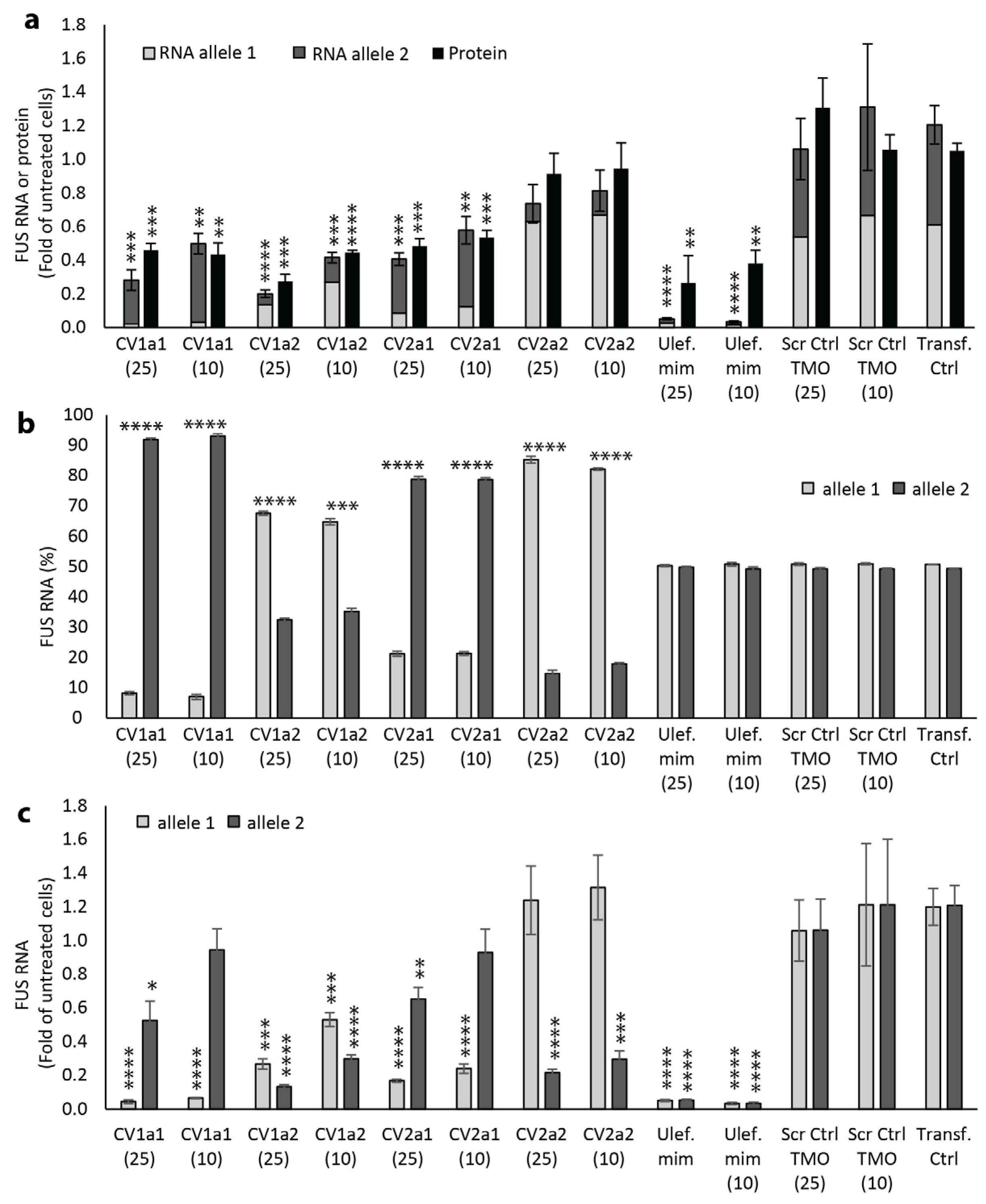
2.4. Effect of Allele-Selective TMOs on FUS Protein Expression
2.5. Effect of Reducing Total Length and Gap Length on Efficacy and Selectivity
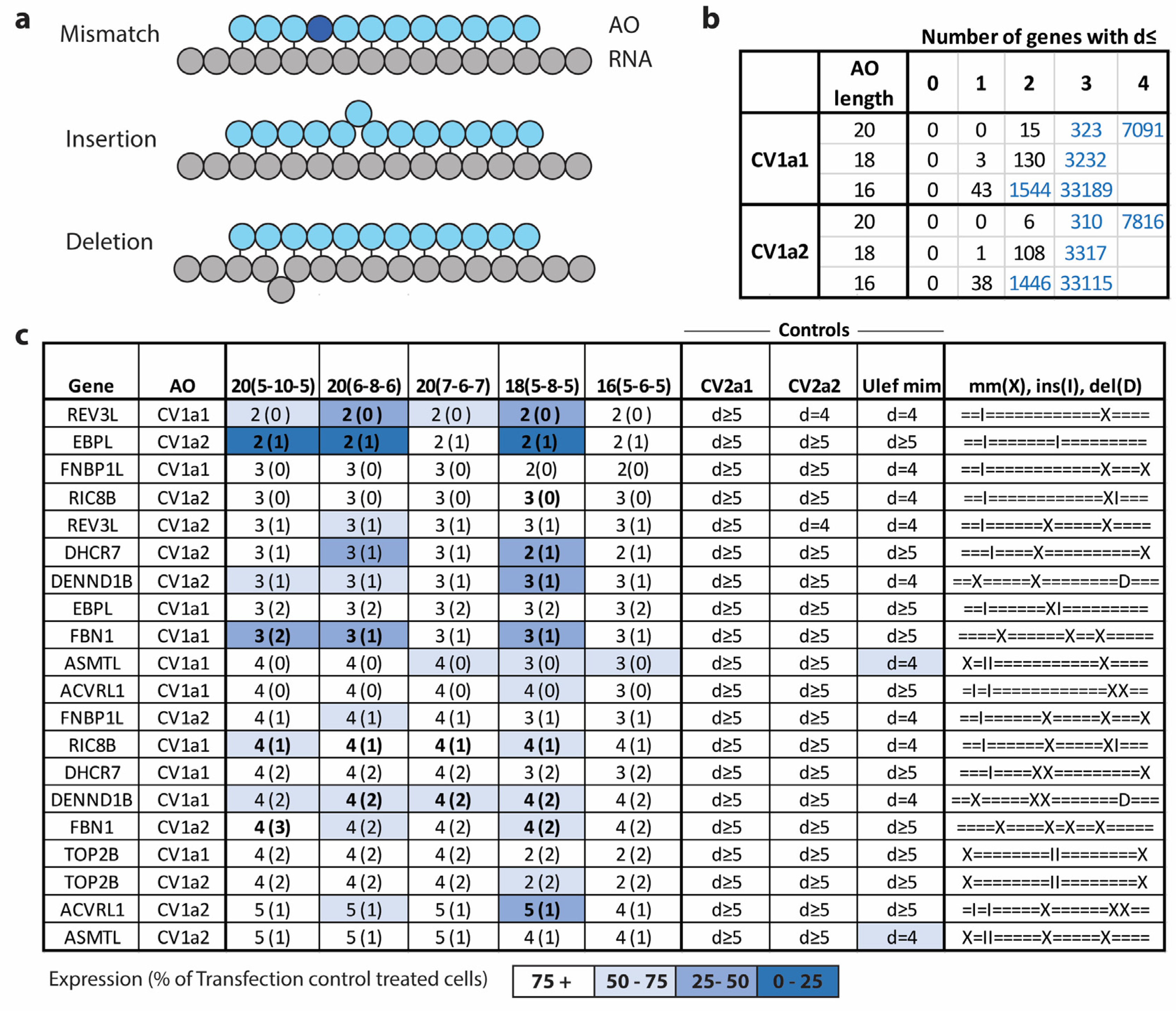
2.6. Hybridization-Dependent Off-Target Gene Knockdown
3. Discussion
4. Materials and Methods
4.1. Antisense Oliginucleotide Synthesis
4.2. Cell Culture and Transfection
4.3. RNA Extraction
4.4. ddPCR Analysis
4.5. Protein Isolation and Western Blot
4.6. Immunocytochemistry
4.7. cDNA Synthesis and qPCR
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rowland, L.P.; Shneider, N.A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Kirby, J.; Al Sultan, A.; Waller, R.; Heath, P. The genetics of amyotrophic lateral sclerosis: Current insights. Degener. Neurol. Neuromuscul. Dis. 2016, 6, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.-Y.; Cui, L.-Y.; Sun, Q.; Li, X.-G.; Liu, M.-S.; Xu, Y.; Zhou, Y.; Yang, X.-Z. De novo FUS gene mutations are associated with juvenile-onset sporadic amyotrophic lateral sclerosis in China. Neurobiol. Aging 2013, 34, 1312.e1–1312.e8. [Google Scholar] [CrossRef]
- Gromicho, M.; Oliveira Santos, M.; Pinto, A.; Pronto-Laborinho, A.; De Carvalho, M. Young-onset rapidly progressive ALS associated with heterozygous FUS mutation. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Hübers, A.; Just, W.; Rosenbohm, A.; Müller, K.; Marroquin, N.; Goebel, I.; Högel, J.; Thiele, H.; Altmüller, J.; Nürnberg, P.; et al. De novo FUS mutations are the most frequent genetic cause in early-onset German ALS patients. Neurobiol. Aging 2015, 36, 3117.e1–3117.e6. [Google Scholar] [CrossRef]
- Hübers, A.; Volk, A.; Just, W.; Rosenbohm, A.; Bierbaumer, N.; Kathrin, M.; Nicolai, M.; Ingrid, G.; Josef, H.; Janine, A.; et al. V42. De novo mutations in the FUS gene are a frequent cause of sporadic ALS in very young patients. Clin. Neurophysiol. 2015, 126, e87. [Google Scholar] [CrossRef]
- Zou, Z.-Y.; Zhou, Z.-R.; Che, C.-H.; Liu, C.-Y.; He, R.-L.; Huang, H.-P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Lattante, S.; Rouleau, G.A.; Kabashi, E. TARDBP and FUS Mutations Associated with Amyotrophic Lateral Sclerosis: Summary and Update. Hum. Mutat. 2013, 34, 812–826. [Google Scholar] [CrossRef]
- Vance, C.; Scotter, E.L.; Nishimura, A.L.; Troakes, C.; Mitchell, J.C.; Kathe, C.; Urwin, H.; Manser, C.; Miller, C.C.; Hortobágyi, T.; et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum. Mol. Genet. 2013, 22, 2676–2688. [Google Scholar] [CrossRef]
- Niu, C.; Zhang, J.; Gao, F.; Yang, L.; Jia, M.; Zhu, H.; Gong, W. FUS-NLS/Transportin 1 complex structure provides insights into the nuclear targeting mechanism of FUS and the implications in ALS. PLoS ONE 2012, 7, e47056. [Google Scholar] [CrossRef]
- Shang, Y.; Huang, E.J. Mechanisms of FUS Mutations in Familial Amyotrophic Lateral Sclerosis. Brain Res. 2016, 1647, 65–78. [Google Scholar] [CrossRef]
- Nomura, T.; Watanabe, S.; Kaneko, K.; Yamanaka, K.; Nukina, N.; Furukawa, Y. Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J. Biol. Chem. 2014, 289, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Van Daele, S.H.; Masrori, P.; Van Damme, P.; Van Den Bosch, L. The sense of antisense therapies in ALS. Trends Mol. Med. 2024, 30, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Hyjek, M.; Figiel, M.; Nowotny, M. RNases H: Structure and mechanism. DNA Repair 2019, 84, 102672. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A.; Subasinghe, C.; Shinozuka, K.; Cohen, J.S. Physicochemical properties of phosphorothioate oligodeoxynucleotides. Nucleic Acids Res. 1988, 16, 3209–3221. [Google Scholar] [CrossRef]
- Crooke, S.T.; Vickers, T.A.; Liang, X.H. Phosphorothioate modified oligonucleotide-protein interactions. Nucleic Acids Res. 2020, 48, 5235–5253. [Google Scholar] [CrossRef] [PubMed]
- Pollak, A.J.; Zhao, L.; Vickers, T.A.; Huggins, I.J.; Liang, X.H.; Crooke, S.T. Insights into innate immune activation via PS-ASO-protein-TLR9 interactions. Nucleic Acids Res. 2022, 50, 8107–8126. [Google Scholar] [CrossRef]
- Collotta, D.; Bertocchi, I.; Chiapello, E.; Collino, M. Antisense oligonucleotides: A novel Frontier in pharmacological strategy. Front. Pharmacol. 2023, 14, 1304342. [Google Scholar] [CrossRef]
- Hill, A.C.; Hall, J. The MOE Modification of RNA: Origins and Widescale Impact on the Oligonucleotide Therapeutics Field. Helv. Chim. Acta 2023, 106, e202200169. [Google Scholar] [CrossRef]
- Summerton, J.; Weller, D. Morpholino antisense oligomers: Design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187. [Google Scholar] [CrossRef]
- Hudziak, R.M.; Barofsky, E.; Barofsky, D.F.; Weller, D.L.; Huang, S.B.; Weller, D.D. Resistance of morpholino phosphorodiamidate oligomers to enzymatic degradation. Antisense Nucleic Acid Drug Dev. 1996, 6, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.; Powers, J.; Duda, P.; Eliopoulos, H. Clinical safety of eteplirsen, a phosphorodiamidate morpholino oligomer (PMO), in Duchenne muscular dystrophy (DMD) patients amenable to skipping exon 51 of the DMD gene. Neuromuscul. Disord. 2016, 26, S153–S154. [Google Scholar] [CrossRef]
- Crooke, S.T.; Liang, X.-H.; Baker, B.F.; Crooke, R.M. Antisense technology: A review. J. Biol. Chem. 2021, 296, 100416. [Google Scholar] [CrossRef] [PubMed]
- Langner, H.K.; Jastrzebska, K.; Caruthers, M.H. Synthesis and Characterization of Thiophosphoramidate Morpholino Oligonucleotides and Chimeras. J. Am. Chem. Soc. 2020, 142, 16240–16253. [Google Scholar] [CrossRef] [PubMed]
- Korobeynikov, V.A.; Lyashchenko, A.K.; Blanco-Redondo, B.; Jafar-Nejad, P.; Shneider, N.A. Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat. Med. 2022, 28, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.; Fairley, S.; Zheng-Bradley, X.; Streeter, I.; Perry, E.; Lowy, E.; Tassé, A.-M.; Flicek, P. The international Genome sample resource (IGSR): A worldwide collection of genome variation incorporating the 1000 Genomes Project data. Nucleic Acids Res. 2016, 45, D854–D859. [Google Scholar] [CrossRef]
- Flynn, L.L.; Li, R.; Pitout, I.L.; Larcher, L.M.; Cooper, J.A.; Aung-Htut, M.T.H.; Hubbard, A.; Griffiths, L.; Bond, C.S.; Wilton, S.D.; et al. Single stranded fully modified-phosphorothioate oligonucleotides rapidly induce structured nuclear inclusions and global alterations to the transcriptome in vitro. Front. Genet. 2022, 13, 791416. [Google Scholar] [CrossRef]
- Qiu, H.; Lee, S.; Shang, Y.; Wang, W.-Y.; Au, K.F.; Kamiya, S.; Barmada, S.J.; Finkbeiner, S.; Lui, H.; Carlton, C.E.; et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Investig. 2014, 124, 981–999. [Google Scholar] [CrossRef]
- Yasuhara, H.; Yoshida, T.; Sasaki, K.; Obika, S.; Inoue, T. Reduction of Off-Target Effects of Gapmer Antisense Oligonucleotides by Oligonucleotide Extension. Mol. Diagn. Ther. 2022, 26, 117–127. [Google Scholar] [CrossRef]
- López-Erauskin, J.; Tadokoro, T.; Baughn, M.W.; Myers, B.; McAlonis-Downes, M.; Chillon-Marinas, C.; Asiaban, J.N.; Artates, J.; Bui, A.T.; Vetto, A.P.; et al. ALS/FTD-Linked Mutation in FUS Suppresses Intra-axonal Protein Synthesis and Drives Disease Without Nuclear Loss-of-Function of FUS. Neuron 2018, 100, 816–830.e7. [Google Scholar] [CrossRef]
- Scekic-Zahirovic, J.; Sendscheid, O.; El Oussini, H.; Jambeau, M.; Sun, Y.; Mersmann, S.; Wagner, M.; Dieterlé, S.; Sinniger, J.; Dirrig-Grosch, S.; et al. Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J. 2016, 35, 1077–1097. [Google Scholar] [CrossRef]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef]
- Ishigaki, S.; Sobue, G. Importance of Functional Loss of FUS in FTLD/ALS. Front. Mol. Biosci. 2018, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, B.; Günther, R.; Japtok, J.; Frech, M.J.; Naumann, M.; Lee, H.O.; Hermann, A. FUS ALS neurons activate major stress pathways and reduce translation as an early protective mechanism against neurodegeneration. Cell Rep. 2023, 42, 112025. [Google Scholar] [CrossRef]
- Ratti, A.; Buratti, E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J. Neurochem. 2016, 138, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.-C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488. [Google Scholar] [CrossRef]
- Colombrita, C.; Onesto, E.; Buratti, E.; de la Grange, P.; Gumina, V.; Baralle, F.E.; Silani, V.; Ratti, A. From transcriptomic to protein level changes in TDP-43 and FUS loss-of-function cell models. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2015, 1849, 1398–1410. [Google Scholar] [CrossRef] [PubMed]
- Reber, S.; Stettler, J.; Filosa, G.; Colombo, M.; Jutzi, D.; Lenzken, S.C.; Schweingruber, C.; Bruggmann, R.; Bachi, A.; Barabino, S.M.; et al. Minor intron splicing is regulated by FUS and affected by ALS-associated FUS mutants. EMBO J. 2016, 35, 1504–1521. [Google Scholar] [CrossRef]
- Ishigaki, S.; Fujioka, Y.; Okada, Y.; Riku, Y.; Udagawa, T.; Honda, D.; Yokoi, S.; Endo, K.; Ikenaka, K.; Takagi, S.; et al. Altered Tau Isoform Ratio Caused by Loss of FUS and SFPQ Function Leads to FTLD-like Phenotypes. Cell Rep. 2017, 18, 1118–1131. [Google Scholar] [CrossRef]
- Orozco, D.; Tahirovic, S.; Rentzsch, K.; Schwenk, B.M.; Haass, C.; Edbauer, D. Loss of fused in sarcoma (FUS) promotes pathological Tau splicing. EMBO Rep. 2012, 13, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Rösler, T.W.; Tayaranian Marvian, A.; Brendel, M.; Nykänen, N.P.; Höllerhage, M.; Schwarz, S.C.; Hopfner, F.; Koeglsperger, T.; Respondek, G.; Schweyer, K.; et al. Four-repeat tauopathies. Prog. Neurobiol. 2019, 180, 101644. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, S.; Riku, Y.; Fujioka, Y.; Endo, K.; Iwade, N.; Kawai, K.; Ishibashi, M.; Yokoi, S.; Katsuno, M.; Watanabe, H.; et al. Aberrant interaction between FUS and SFPQ in neurons in a wide range of FTLD spectrum diseases. Brain 2020, 143, 2398–2405. [Google Scholar] [CrossRef]
- Baechtold, H.; Kuroda, M.; Sok, J.; Ron, D.; Lopez, B.S.; Akhmedov, A.T. Human 75-kDa DNA-pairing Protein Is Identical to the Pro-oncoprotein TLS/FUS and Is Able to Promote D-loop Formation. J. Biol. Chem. 1999, 274, 34337–34342. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, A.S.; Kim, S.H.; Trinh, A.T.; Rodenkirch, L.A.; Tibbetts, R.S. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J. Biol. Chem. 2013, 288, 24731–24741. [Google Scholar] [CrossRef]
- Wang, W.-Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.A.; Huang, E.J.; Tsai, L.-H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383. [Google Scholar] [CrossRef] [PubMed]
- Sukhanova, M.V.; Singatulina, A.S.; Pastré, D.; Lavrik, O.I. Fused in Sarcoma (FUS) in DNA Repair: Tango with Poly(ADP-ribose) Polymerase 1 and Compartmentalisation of Damaged DNA. Int. J. Mol. Sci. 2020, 21, 7020. [Google Scholar] [CrossRef]
- Levone, B.R.; Lenzken, S.C.; Antonaci, M.; Maiser, A.; Rapp, A.; Conte, F.; Reber, S.; Mechtersheimer, J.; Ronchi, A.E.; Mühlemann, O.; et al. FUS-dependent liquid–liquid phase separation is important for DNA repair initiation. J. Cell Biol. 2021, 220, e202008030. [Google Scholar] [CrossRef]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef]
- Sama, R.R.K.; Ward, C.L.; Kaushansky, L.J.; Lemay, N.; Ishigaki, S.; Urano, F.; Bosco, D.A. FUS/TLS assembles into stress granules and is a prosurvival factor during hyperosmolar stress. J. Cell. Physiol. 2013, 228, 2222–2231. [Google Scholar] [CrossRef]
- Lenzi, J.; De Santis, R.; De Turris, V.; Morlando, M.; Laneve, P.; Calvo, A.; Caliendo, V.; Chiò, A.; Rosa, A.; Bozzoni, I. ALS mutant FUS proteins are recruited into stress granules in induced pluripotent stem cell-derived motoneurons. DMM Dis. Models Mech. 2015, 8, 755–766. [Google Scholar] [CrossRef]
- Wolozin, B.; Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.H.; Nakagawa, S.; Hirose, T.; Bond, C.S. Paraspeckles: Where Long Noncoding RNA Meets Phase Separation. Trends Biochem. Sci. 2018, 43, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Hennig, S.; Kong, G.; Mannen, T.; Sadowska, A.; Kobelke, S.; Blythe, A.; Knott, G.J.; Iyer, K.S.; Ho, D.; Newcombe, E.A.; et al. Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J. Cell Biol. 2015, 210, 529–539. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Skelt, L.; Notaro, A.; Highley, J.R.; Fox, A.H.; La Bella, V.; Buchman, V.L.; Shelkovnikova, T.A. ALS-linked FUS mutations confer loss and gain of function in the nucleus by promoting excessive formation of dysfunctional paraspeckles. Acta Neuropathol. Commun. 2019, 7, 7. [Google Scholar] [CrossRef]
- Shelkovnikova, T.A.; Robinson, H.K.; Troakes, C.; Ninkina, N.; Buchman, V.L. Compromised paraspeckle formation as a pathogenic factor in FUSopathies. Hum. Mol. Genet. 2013, 23, 2298–2312. [Google Scholar] [CrossRef] [PubMed]
- Le, B.T.; Paul, S.; Jastrzebska, K.; Langer, H.; Caruthers, M.H.; Veedu, R.N. Thiomorpholino oligonucleotides as a robust class of next generation platforms for alternate mRNA splicing. Proc. Natl. Acad. Sci. USA 2022, 119, e2207956119. [Google Scholar] [CrossRef] [PubMed]
- Monia, B.P.; Johnston, J.F.; Ecker, D.J.; Zounes, M.A.; Lima, W.F.; Freier, S.M. Selective inhibition of mutant Ha-ras mRNA expression by antisense oligonucleotides. J. Biol. Chem. 1992, 267, 19954–19962. [Google Scholar] [CrossRef]
- Rukov, J.L.; Hagedorn, P.H.; Høy, I.B.; Feng, Y.; Lindow, M.; Vinther, J. Dissecting the target specificity of RNase H recruiting oligonucleotides using massively parallel reporter analysis of short RNA motifs. Nucleic Acids Res. 2015, 43, 8476–8487. [Google Scholar] [CrossRef] [PubMed]
- Lima, W.F.; Rose, J.B.; Nichols, J.G.; Wu, H.; Migawa, M.T.; Wyrzykiewicz, T.K.; Vasquez, G.; Swayze, E.E.; Crooke, S.T. The Positional Influence of the Helical Geometry of the Heteroduplex Substrate on Human RNase H1 Catalysis. Mol. Pharmacol. 2007, 71, 73–82. [Google Scholar] [CrossRef]
- Giles, R.V.; Ruddell, C.J.; Spiller, D.G.; Green, J.A.; Tidd, D.M. Single base discrimination for ribonuclease H-dependent antisense effects within intact human leukaemia cells. Nucleic Acids Res. 1995, 23, 954–961. [Google Scholar] [CrossRef]
- Magner, D.; Biala, E.; Lisowiec-Wachnicka, J.; Kierzek, E.; Kierzek, R. A Tandem Oligonucleotide Approach for SNP-Selective RNA Degradation Using Modified Antisense Oligonucleotides. PLoS ONE 2015, 10, e0142139. [Google Scholar] [CrossRef] [PubMed]
- Kiełpiński, Ł.J.; Hagedorn, P.H.; Lindow, M.; Vinther, J. RNase H sequence preferences influence antisense oligonucleotide efficiency. Nucleic Acids Res. 2017, 45, 12932–12944. [Google Scholar] [CrossRef]
- Wu, H.; Lima, W.F.; Zhang, H.; Fan, A.; Sun, H.; Crooke, S.T. Determination of the Role of the Human RNase H1 in the Pharmacology of DNA-like Antisense Drugs. J. Biol. Chem. 2004, 279, 17181–17189. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T. Molecular Mechanisms of Antisense Oligonucleotides. Nucleic Acid Ther. 2017, 27, 70–77. [Google Scholar] [CrossRef]
- Wu, H.; Lima, W.F.; Crooke, S.T. Properties of cloned and expressed human RNase H1. J. Biol. Chem. 1999, 274, 28270–28278. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.; Sciabola, S.; Salatto, C.; Weng, Y.; Moshinsky, D.; Little, J.; Walters, E.; Kreeger, J.; DiMattia, D.; Chen, T.; et al. Chemical Modification Study of Antisense Gapmers. Nucleic Acid Ther. 2012, 22, 344–359. [Google Scholar] [CrossRef]
- Straarup, E.M.; Fisker, N.; Hedtjarn, M.; Lindholm, M.W.; Rosenbohm, C.; Aarup, V.; Hansen, H.F.; Ørum, H.; Hansen, J.B.; Koch, T. Short locked nucleic acid antisense oligonucleotides potently reduce apolipoprotein B mRNA and serum cholesterol in mice and non-human primates. Nucleic Acids Res. 2010, 38, 7100–7111. [Google Scholar] [CrossRef]
- Pedersen, L.; Hagedorn, P.H.; Lindholm, M.W.; Lindow, M. A Kinetic Model Explains Why Shorter and Less Affine Enzyme-recruiting Oligonucleotides Can Be More Potent. Mol. Ther. Nucleic. Acids 2014, 3, e149. [Google Scholar] [CrossRef]
- Herschlag, D. Implications of ribozyme kinetics for targeting the cleavage of specific RNA molecules in vivo: More isn’t always better. Proc. Natl. Acad. Sci. USA 1991, 88, 6921–6925. [Google Scholar] [CrossRef]
- Magner, D.; Biala, E.; Lisowiec-wachnicka, J.; Kierzek, R. Influence of mismatched and bulged nucleotides on SNP-preferential RNase H cleavage of RNA-antisense gapmer heteroduplexes. Sci. Rep. 2017, 7, 12532. [Google Scholar] [CrossRef]
- Xiao, X.; Li, M.; Ye, Z.; He, X.; Wei, J.; Zha, Y. FUS gene mutation in amyotrophic lateral sclerosis: A new case report and systematic review. Amyotroph Lateral Scler Front. Degener 2024, 25, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Populations | CV1 | CV2 | CV1 and/or CV2 | CV1 and CV2 |
|---|---|---|---|---|
| European | 42.9 | 45.9 | 56.5 | 32.3 |
| American | 45.8 | 49.9 | 57.9 | 37.8 |
| Asian | 41.1 | 41.4 | 42.6 | 39.9 |
| African | 5.3 | 34.2 | 38.8 | 0.7 |
| AO Name | Gapmer Configuration | Complementary to | Sequence (5′-3′) |
|---|---|---|---|
| CV1a1 | 20 (5-10-5) | CV1 Allele 1 | TCTGGCCATATCCTGAAGTG |
| CV1a1-1 | 20 (6-8-6) | CV1 Allele 1 | TCTGGCCATATCCTGAAGTG |
| CV1a1-2 | 20 (7-6-7) | CV1 Allele 1 | TCTGGCCATATCCTGAAGTG |
| CV1a1-3 | 18 (5-8-5) | CV1 Allele 1 | CTGGCCATATCCTGAAGT |
| CV1a1-4 | 16 (5-6-5) | CV1 Allele 1 | TGGCCATATCCTGAAG |
| CV1a2 | 20 (5-10-5) | CV1 Allele 2 | TCTGGCCATAGCCTGAAGTG |
| CV1a2-1 | 20 (6-8-6) | CV1 Allele 2 | TCTGGCCATAGCCTGAAGTG |
| CV1a2-2 | 20 (7-6-7) | CV1 Allele 2 | TCTGGCCATAGCCTGAAGTG |
| CV1a2-3 | 18 (5-8-5) | CV1 Allele 2 | CTGGCCATAGCCTGAAGT |
| CV1a2-4 | 16 (5-6-5) | CV1 Allele 2 | TGGCCATAGCCTGAAG |
| CV1a2b | 20 (5-10-5) | CV1 Allele 2 | GCCATAGCCTGAAGTGTCCG |
| CV2a1 | 20 (5-10-5) | CV2 Allele 1 | CATAGCCAGGGTAGGAGGAC |
| CV2a1-1 | 20 (6-8-6) | CV2 Allele 1 | CATAGCCAGGGTAGGAGGAC |
| CV2a1-2 | 20 (7-6-7) | CV2 Allele 1 | CATAGCCAGGGTAGGAGGAC |
| CV2a1-3 | 18 (5-8-5) | CV2 Allele 1 | ATAGCCAGGGTAGGAGGA |
| CV2a1-4 | 16 (5-6-5) | CV2 Allele 1 | TAGCCAGGGTAGGAGG |
| CV2a2 | 20 (5-10-5) | CV2 Allele 2 | CATAGCCAGGATAGGAGGAC |
| CV2a2-1 | 20 (6-8-6) | CV2 Allele 2 | CATAGCCAGGATAGGAGGAC |
| CV2a2-2 | 20 (7-6-7) | CV2 Allele 2 | CATAGCCAGGATAGGAGGAC |
| CV2a2-3 | 18 (5-8-5) | CV2 Allele 2 | ATAGCCAGGATAGGAGGA |
| CV2a2-4 | 16 (5-6-5) | CV2 Allele 2 | TAGCCAGGATAGGAGG |
| Scramble Control | 20 (5-10-5) | None | ACCTTATCCAATAGCGCCTC |
| Selectivity Control | 20 (5-10-5) | Both alleles | CCACTGTAACTCTGCTGTCC |
| Ulefnersen mimic | 20 (5-10-5) | Both alleles | GC*A*A*T*GTCACCTTTCA*T*ACC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mejzini, R.; Caruthers, M.H.; Schafer, B.; Kostov, O.; Sudheendran, K.; Ciba, M.; Danielsen, M.; Wilton, S.; Akkari, P.A.; Flynn, L.L. Allele-Selective Thiomorpholino Antisense Oligonucleotides as a Therapeutic Approach for Fused-in-Sarcoma Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2024, 25, 8495. https://doi.org/10.3390/ijms25158495
Mejzini R, Caruthers MH, Schafer B, Kostov O, Sudheendran K, Ciba M, Danielsen M, Wilton S, Akkari PA, Flynn LL. Allele-Selective Thiomorpholino Antisense Oligonucleotides as a Therapeutic Approach for Fused-in-Sarcoma Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2024; 25(15):8495. https://doi.org/10.3390/ijms25158495
Chicago/Turabian StyleMejzini, Rita, Marvin H. Caruthers, Balazs Schafer, Ondrej Kostov, Kavitha Sudheendran, Marija Ciba, Mathias Danielsen, Steve Wilton, Patrick Anthony Akkari, and Loren L. Flynn. 2024. "Allele-Selective Thiomorpholino Antisense Oligonucleotides as a Therapeutic Approach for Fused-in-Sarcoma Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 25, no. 15: 8495. https://doi.org/10.3390/ijms25158495