

Mutation Spectrum of GAA Gene in Pompe Disease: Current Knowledge and Results of an Italian Study

 ,
,
Abstract
1. Introduction
Sentinel Symptoms of PD, Diagnostic Tests, and Therapeutic Procedures
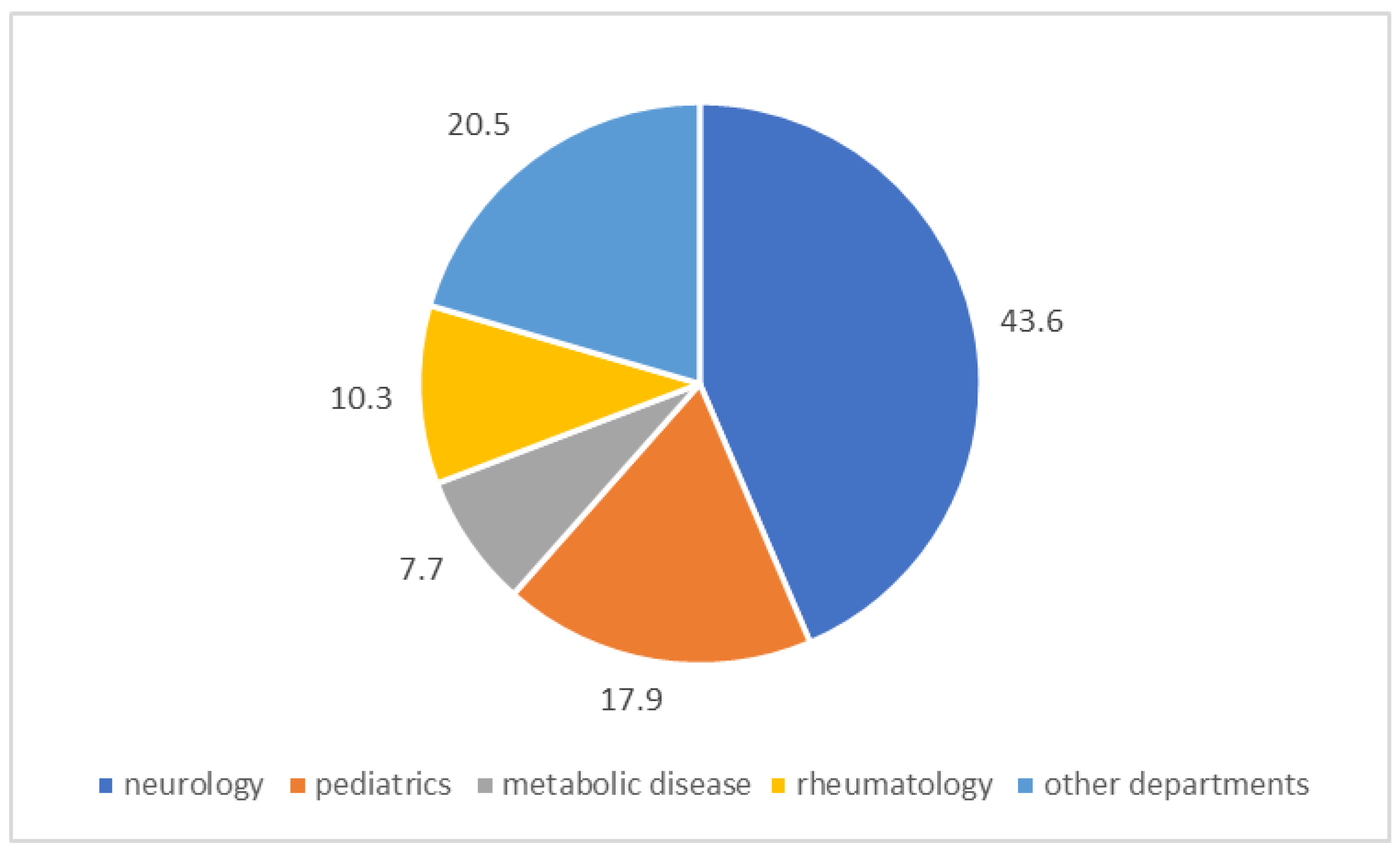
2. Results
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Sample Collection
4.3. Enzymatic Assay
4.4. DNA Extraction
4.5. Genetic Analysis-PCR, Sanger Sequencing, and MPLA
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raben, N.; Plotz, P.; Byrne, B.J. Acid alpha-glucosidase deficiency (glycogenosis type II, Pompe disease). Curr. Mol. Med. 2002, 2, 145–166. [Google Scholar] [CrossRef]
- Herzog, A.; Hartung, R.; Reuser, A.J.; Hermanns, P.; Runz, H.; Karabul, N.; Gökce, S.; Pohlenz, J.; Kampmann, C.; Lampe, C.; et al. A cross-sectional single-centre study on the spectrum of Pompe disease, German patients: Molecular analysis of the GAA gene, manifestation and genotype-phenotype correlations. Orphanet J. Rare Dis. 2012, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- van den Hout, H.M.; Hop, W.; van Diggelen, O.P.; Smeitink, J.A.; Smit, G.P.; Poll-The, B.T.; Bakker, H.D.; Loonen, M.C.; de Klerk, J.B.; Reuser, A.J.; et al. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics 2003, 112, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, R.; Huie, M.L. Frequency of mutations for glycogen storage disease type II in different populations: The delta525T and deltaexon 18 mutations are not generally "common" in white populations. J. Med. Genet. 1999, 36, 85–86. [Google Scholar]
- De Filippi, P.; Saeidi, K.; Ravaglia, S.; Dardis, A.; Angelini, C.; Mongini, T.; Morandi, L.; Moggio, M.; Di Muzio, A.; Filosto, M.; et al. Genotype-phenotype correlation in Pompe disease, a step forward. Orphanet J. Rare. Dis. 2014, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Lee, N.C.; Thurberg, B.L.; Chiang, S.C.; Zhang, X.K.; Keutzer, J.; Huang, A.C.; Wu, M.H.; Huang, P.H.; Tsai, F.J.; et al. Pompe disease in infants: Improving the prognosis by newborn screening and early treatment. Pediatrics 2009, 124, e1116–e1125. [Google Scholar] [CrossRef]
- Er, T.K.; Chen, C.C.; Chien, Y.H.; Liang, W.C.; Kan, T.M.; Jong, Y.J. Development of a feasible assay for the detection of GAA mutations in patients with Pompe disease. Clin. Chim. Acta 2014, 429, 18–25. [Google Scholar] [CrossRef]
- Mechtler, T.P.; Stary, S.; Metz, T.F.; De Jesús, V.R.; Greber-Platzer, S.; Pollak, A.; Herkner, K.R.; Streubel, B.; Kasper, D.C. Neonatal screening for lysosomal storage disorders: Feasibility and incidence from a nationwide study in Austria. Lancet 2012, 379, 335–341. [Google Scholar] [CrossRef]
- Montalvo, A.L.; Bembi, B.; Donnarumma, M.; Filocamo, M.; Parenti, G.; Rossi, M.; Merlini, L.; Buratti, E.; De Filippi, P.; Dardis, A.; et al. Mutation profile of the GAA gene in 40 Italian patients with late onset glycogen storage disease type II. Hum. Mutat. 2006, 27, 999–1006. [Google Scholar] [CrossRef]
- Martiniuk, F.; Chen, A.; Mack, A.; Arvanitopoulos, E.; Chen, Y.; Rom, W.N.; Codd, W.J.; Hanna, B.; Alcabes, P.; Raben, N.; et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am. J. Med. Genet. 1998, 79, 69–72. [Google Scholar] [CrossRef]
- Tang, H.; Feuchtbaum, L.; Sciortino, S.; Matteson, J.; Mathur, D.; Bishop, T.; Olney, R.S. The First Year Experience of Newborn Screening for Pompe Disease in California. Int. J. Neonatal. Screen. 2020, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Sawada, T.; Kido, J.; Nakamura, K. Newborn Screening for Pompe Disease. Int. J. Neonatal. Screen 2020, 6, 31. [Google Scholar] [CrossRef]
- Rairikar, M.V.; Case, L.E.; Bailey, L.A.; Kazi, Z.B.; Desai, A.K.; Berrier, K.L.; Coats, J.; Gandy, R.; Quinones, R.; Kishnani, P.S. Insight into the phenotype of infants with Pompe disease identified by newborn screening with the common c.-32-13T>G "late-onset" GAA variant. Mol. Genet. Metab. 2017, 122, 99–107. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Steiner, R.D.; Bali, D.; Berger, K.; Byrne, B.J.; Case, L.E.; Crowley, J.F.; Downs, S.; Howell, R.R.; Kravitz, R.M.; et al. Pompe disease diagnosis and management guideline. Genet. Med. 2006, 8, 267–288. [Google Scholar] [CrossRef]
- Slonim, A.E.; Bulone, L.; Ritz, S.; Goldberg, T.; Chen, A.; Martiniuk, F. Identification of two subtypes of infantile acid maltase deficiency. J. Pediatr. 2000, 137, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef]
- Taverna, S.; Cammarata, G.; Colomba, P.; Sciarrino, S.; Zizzo, C.; Francofonte, D.; Zora, M.; Scalia, S.; Brando, C.; Curto, A.L.; et al. Pompe disease: Pathogenesis, molecular genetics and diagnosis. Aging 2020, 12, 15856–15874. [Google Scholar] [CrossRef]
- El Marabti, E.; Abdel-Wahab, O. Therapeutic Modulation of RNA Splicing in Malignant and Non-Malignant Disease. Trends Mol. Med. 2021, 27, 643–659. [Google Scholar] [CrossRef]
- Malekkou, A.; Theodosiou, A.; Alexandrou, A.; Papaevripidou, I.; Sismani, C.; Jacobs, E.H.; Ruijter, G.J.G.; Anastasiadou, V.; Ourani, S.; Athanasiou, E.; et al. Variants associated with reduced enzymatic activity but lack of Pompe-related symptoms, incidentally identified by exome sequencing. Mol. Genet. Metab. Rep. 2023, 36, 100997. [Google Scholar] [CrossRef]
- Jastrzębska, A.; Kostera-Pruszczyk, A. Multisystem presentation of Late Onset Pompe Disease: What every consulting neurologist should know. Neurol. Neurochir. Pol. 2023, 57, 143–150. [Google Scholar] [CrossRef]
- Sun, B.; Bird, A.; Young, S.P.; Kishnani, P.S.; Chen, Y.T.; Koeberl, D.D. Enhanced response to enzyme replacement therapy in Pompe disease after the induction of immune tolerance. Am. J. Hum. Genet. 2007, 81, 1042–1049. [Google Scholar] [CrossRef]
- van der Ploeg, A.T.; Kruijshaar, M.E.; Toscano, A.; Laforêt, P.; Angelini, C.; Lachmann, R.H.; Pascual Pascual, S.I.; Roberts, M.; Rösler, K.; Stulnig, T.; et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: A 10-year experience. Eur. J. Neurol. 2017, 24, 768-e31. [Google Scholar] [CrossRef]
- Desai, A.K.; Kazi, Z.B.; Bali, D.S.; Kishnani, P.S. Characterization of immune response in Cross-Reactive Immunological Material (CRIM)-positive infantile Pompe disease patients treated with enzyme replacement therapy. Mol. Genet. Metab. Rep. 2019, 20, 100475. [Google Scholar] [CrossRef]
- Bali, D.S.; Goldstein, J.L.; Banugaria, S.; Dai, J.; Mackey, J.; Rehder, C.; Kishnani, P.S. Predicting cross-reactive immunological material (CRIM) status in Pompe disease using GAA mutations: Lessons learned from 10 years of clinical laboratory testing experience. Am. J. Med. Genet. Part C Semin. Med. Genet. 2012, 160, 40–49. [Google Scholar] [CrossRef]
- Curelaru, S.; Desai, A.K.; Fink, D.; Zehavi, Y.; Kishnani, P.S.; Spiegel, R. A favorable outcome in an infantile-onset Pompe patient with cross reactive immunological material (CRIM) negative disease with high dose enzyme replacement therapy and adjusted immunomodulation. Mol. Genet. Metab. Rep. 2022, 32, 100893. [Google Scholar] [CrossRef]
- Thurberg, B.L.; Lynch Maloney, C.; Vaccaro, C.; Afonso, K.; Tsai, A.C.; Bossen, E.; Kishnani, P.S.; O’Callaghan, M. Characterization of pre- and post-treatment pathology after enzyme replacement therapy for Pompe disease. Lab. Invest. 2006, 86, 1208–1220. [Google Scholar] [CrossRef]
- Chamoles, N.A.; Niizawa, G.; Blanco, M.; Gaggioli, D.; Casentini, C. Glycogen storage disease type II: Enzymatic screening in dried blood spots on filter paper. Clin. Chim. Acta 2004, 347, 97–102. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Beckemeyer, A.A. New therapeutic approaches for Pompe disease: Enzyme replacement therapy and beyond. Pediatr. Endocrinol. Rev. 2014, 12, 114–124. [Google Scholar]
- Huie, M.L.; Chen, A.S.; Brooks, S.S.; Grix, A.; Hirschhorn, R. A de novo 13 nt deletion, a newly identified C647W missense mutation and a deletion of exon 18 in infantile onset glycogen storage disease type II (GSDII). Hum. Mol. Genet. 1994, 3, 1081–1087. [Google Scholar] [CrossRef]
- Salunkhe, M.; Agarwal, A.; Faruq, M.; Srivastava, A.K. Genetic Testing in Neurology: What Every Neurologist Must Know. Ann. Indian Acad. Neurol. 2022, 25, 350–353. [Google Scholar]
- Meena, N.K.; Raben, N. Pompe Disease: New Developments in an Old Lysosomal Storage Disorder. Biomolecules 2020, 10, 1339. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.O. Emerging strategies for the treatment of hereditary metabolic storage disorders. Rejuvenation Res. 2006, 9, 237–244. [Google Scholar] [CrossRef]
- Magadum, A.; Kaur, K.; Zangi, L. mRNA-Based Protein Replacement Therapy for the Heart. Mol. Ther. 2019, 27, 785–793. [Google Scholar] [CrossRef]
- Coutinho, M.F.; Santos, J.I.; Matos, L.; Alves, S. Genetic Substrate Reduction Therapy: A Promising Approach for Lysosomal Storage Disorders. Diseases 2016, 4, 33. [Google Scholar] [CrossRef]
- Cox-Brinkman, J.; van Breemen, M.J.; van Maldegem, B.T.; Bour, L.; Donker, W.E.; Hollak, C.E.; Wijburg, F.A.; Aerts, J.M. Potential efficacy of enzyme replacement and substrate reduction therapy in three siblings with Gaucher disease type III. J. Inherit. Metab. Dis. 2008, 31, 745–752. [Google Scholar] [CrossRef]
- Coutinho, M.F.; Santos, J.I.; Alves, S. Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders. Int. J. Mol. Sci. 2016, 17, 1065. [Google Scholar] [CrossRef]
- Desnick, R.J.; Schuchman, E.H. Enzyme replacement therapy for lysosomal diseases: Lessons from 20 years of experience and remaining challenges. Annu. Rev. Genomics Hum. Genet. 2012, 13, 307–335. [Google Scholar] [CrossRef]
- Wraith, J.E.; Beck, M.; Lane, R.; van der Ploeg, A.; Shapiro, E.; Xue, Y.; Kakkis, E.D.; Guffon, N. Enzyme replacement therapy in patients who have mucopolysaccharidosis I and are younger than 5 years: Results of a multinational study of recombinant human alpha-L-iduronidase (laronidase). Pediatrics 2007, 120, e37–e46. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, L.; Quan, S. Enzyme replacement therapy for infantile-onset Pompe disease. Cochrane Database Syst. Rev. 2017, 11, CD011539. [Google Scholar] [CrossRef]
- Güngör, D.; Kruijshaar, M.E.; Plug, I.; D’Agostino, R.B.; Hagemans, M.L.; van Doorn, P.A.; Reuser, A.J.; van der Ploeg, A.T. Impact of enzyme replacement therapy on survival in adults with Pompe disease: Results from a prospective international observational study. Orphanet J. Rare Dis. 2013, 8, 49. [Google Scholar] [CrossRef]
- Baruteau, J.; Broomfield, A.; Crook, V.; Finnegan, N.; Harvey, K.; Burke, D.; Burch, M.; Shepherd, G.; Vellodi, A. Successful Desensitisation in a Patient with CRIM-Positive Infantile-Onset Pompe Disease. JIMD Rep. 2014, 12, 99–102. [Google Scholar]
- Messinger, Y.H.; Mendelsohn, N.J.; Rhead, W.; Dimmock, D.; Hershkovitz, E.; Champion, M.; Jones, S.A.; Olson, R.; White, A.; Wells, C.; et al. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet. Med. 2012, 14, 135–142. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef]
- Byrne, B.J.; Fuller, D.D.; Smith, B.K.; Clement, N.; Coleman, K.; Cleaver, B.; Vaught, L.; Falk, D.J.; McCall, A.; Corti, M. Pompe disease gene therapy: Neural manifestations require consideration of CNS directed therapy. Ann. Transl. Med. 2019, 7, 290. [Google Scholar] [CrossRef]
- Stevens, D.; Milani-Nejad, S.; Mozaffar, T. Pompe Disease: A Clinical, Diagnostic, and Therapeutic Overview. Curr. Treat. Options Neurol. 2022, 24, 573–588. [Google Scholar] [CrossRef]
- Desai, A.K.; Baloh, C.H.; Sleasman, J.W.; Rosenberg, A.S.; Kishnani, P.S. Benefits of Prophylactic Short-Course Immune Tolerance Induction in Patients With Infantile Pompe Disease: Demonstration of Long-Term Safety and Efficacy in an Expanded Cohort. Front. Immunol. 2020, 11, 1727. [Google Scholar] [CrossRef]
- Banugaria, S.G.; Prater, S.N.; Patel, T.T.; Dearmey, S.M.; Milleson, C.; Sheets, K.B.; Bali, D.S.; Rehder, C.W.; Raiman, J.A.; Wang, R.A.; et al. Algorithm for the early diagnosis and treatment of patients with cross reactive immunologic material-negative classic infantile pompe disease: A step towards improving the efficacy of ERT. PLoS ONE 2013, 8, e67052. [Google Scholar] [CrossRef]
- Xu, S.; Lun, Y.; Frascella, M.; Garcia, A.; Soska, R.; Nair, A.; Ponery, A.S.; Schilling, A.; Feng, J.; Tuske, S.; et al. Improved efficacy of a next-generation ERT in murine Pompe disease. JCI Insight 2019, 4, e125358. [Google Scholar] [CrossRef]
- Toscano, A.; Rodolico, C.; Musumeci, O. Multisystem late onset Pompe disease (LOPD): An update on clinical aspects. Ann. Transl. Med. 2019, 7, 284. [Google Scholar] [CrossRef] [PubMed]
- Ravaglia, S.; Malovini, A.; Cirio, S.; Danesino, C.; De Filippi, P.; Moggio, M.; Mongini, T.; Maggi, L.; Servidei, S.; Vianello, A.; et al. Polymorphism in exercise genes and respiratory function in late-onset Pompe disease. J. Appl. Physiol. 2021, 131, 1762–1771. [Google Scholar] [CrossRef]
- Spada, M.; Porta, F.; Vercelli, L.; Pagliardini, V.; Chiadò-Piat, L.; Boffi, P.; Pagliardini, S.; Remiche, G.; Ronchi, D.; Comi, G.; et al. Screening for later-onset Pompe’s disease in patients with paucisymptomatic hyperCKemia. Mol. Genet. Metab. 2013, 109, 171–173. [Google Scholar] [CrossRef]
- Thirumal Kumar, D.; Umer Niazullah, M.; Tasneem, S.; Judith, E.; Susmita, B.; George Priya Doss, C.; Selvarajan, E.; Zayed, H. A computational method to characterize the missense mutations in the catalytic domain of GAA protein causing Pompe disease. J. Cell Biochem. 2019, 120, 3491–3505. [Google Scholar] [CrossRef] [PubMed]
- Zhong, N.; Martiniuk, F.; Tzall, S.; Hirschhorn, R. Identification of a missense mutation in one allele of a patient with Pompe disease, and use of endonuclease digestion of PCR-amplified RNA to demonstrate lack of mRNA expression from the second allele. Am. J. Hum. Genet. 1991, 49, 635–645. [Google Scholar] [PubMed]
- Peruzzo, P.; Pavan, E.; Dardis, A. Molecular genetics of Pompe disease: A comprehensive overview. Ann. Transl. Med. 2019, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.N.; Huemer, M.; Stulnig, T.M.; Simschitz, P.; Iglseder, S.; Eggers, C.; Moser, H.; Möslinger, D.; Freilinger, M.; Lagler, F.; et al. Pompe disease in Austria: Clinical, genetic and epidemiological aspects. J. Neurol. 2018, 265, 159–164. [Google Scholar] [CrossRef]
- Arslan, A.; Poyrazoğlu, H.G.; Kiraz, A.; Özcan, A.; Işık, H.; Ergul, A.B.; Mungan, N.; Streubel, B.; Ceylaner, S.; Altuner Torun, Y. Combination of two different homozygote mutations in Pompe disease. Pediatr. Int. 2016, 58, 241–243. [Google Scholar] [CrossRef]
- Turaça, L.T.; de Faria, D.O.; Kyosen, S.O.; Teixeira, V.D.; Motta, F.L.; Pessoa, J.G.; Rodrigues E Silva, M.; de Almeida, S.S.; D’Almeida, V.; Munoz Rojas, M.V.; et al. Novel GAA mutations in patients with Pompe disease. Gene 2015, 561, 124–131. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject | Age | Family History | Gender F Female M Male | α-Glucosidase Activity (DBS) | Variant 1 | Variant 2 |
|---|---|---|---|---|---|---|
| #1 | 57 | yes | F | 2.1 | c.-32-13T>G | c.1655 T>C L552P |
| #2 | 54 | yes | M | 0.7 | c.-32-13T>G | c.1655 T>C L552P |
| #3 | 38 | no | M | 0.8 | c.-32-13T>G | c.2431 delC |
| #4 | 45 | no | M | 0.6 | c.-32-13T>G | c.2242dupG |
| #5 | 21 | no | M | 1.6 | c.-32-13T>G | c.1655 T>C L552P |
| #6 | 2 | no | M | 0.6 | c.-32-13T>G | c.1927 G>A G643R |
| #7 | NS | no | M | 0.8 | c.-32-13T>G | c.1465 G>A D489N |
| #8 | 10 | no | M | 1.4 | c.-32-13T>G | c.2238 G>A W746 |
| #9 | 54 | no | M | 1.5 | c.-32-13T>G | c.2560 C>T R854 |
| #10 | 49 | no | F | 1.3 | c.-32-13T>G | c.1210 G>A D404N |
| #11 | 63 | yes | M | 0.9 | c.-32-13T>G | c.526 delG E176R fs45 |
| #12 | NS | no | NS | 0.9 | c.-32-13T>G | c.784 G>A E262K |
| #13 | 82 | yes | M | 1 | c.-32-13T>G | c.784 G>A E262K |
| #14 | 33 | yes | F | 1.3 | c.-32-13T>G | c.525 delT E176R fs45 |
| #15 | 9 | yes | M | 1.5 | c.-32-13T>G | Del ex18 |
| #16 | 35 | no | F | 5.4 | c.670 C>T | c.861C>T |
| #17 | 5 | si | M | 1.1 | c.-32-13T>G homozygous | |
| #18 | 2 month | no | M | 0.4 | c.1465 G>A D489N | c.2238 G>A W746 |
| #19 | 78 | no | M | 1.5 | c.-32-13T>G homozygous | |
| #20 | 24 days | no | M | 0 | c. 1565 C>G P522R homozygous | |
| #21 | 58 | no | M | 0.8 | c.-32-13T>G homozygous | |
| #22 | 59 | yes | M | 0.7 | c.-32-13T>G homozygous | |
| #23 | 52 | yes | M | 1.6 | c.-32-13T>G homozygous | |
| #24 | 3 month | no | M | 0.9 | c.526 delG E176R fs45 | c.2104 C>T R702C |
| #25 | 11 month | no | F | 0.5 | c.1655 T>C L552P | c.2161 ins G E721G fs16 |
| #26 | 1 month | no | F | 0.9 | c.784 G>A E262K | Deletion on exon 9 and 19 |
| #27 | 42 | yes | M | 5 | c.784 G>A E262K | Deletion on exon 9 and 17 |
| #28 | 7 | no | F | 0.4 | c.784 G>A E262K | C.1979 G>A R660H |
| #29 | 5 month | no | M | 0.4 | c.1327-18 A>G | c.1655 T>C L552P |
| #30 | 7 | no | F | 1.1 | c.784 G>A E262K | c.1979 G>A R660H |
| #31 | 12 | no | F | 0.5 | c.2012 T>A M671K homozygous | |
| #32 | 6 mouth | no | M | 0.8 | c.1437+2 T>C | c.1655 T>C L552P |
| #33 | 53 | no | M | 20.3 | c.-32-13T>G | c.1551 +1 G>C |
| #34 | 47 | yes | F | 5.4 | c.-32-13T>G | c.655 G>A G219R |
| #35 | 15 | no | M | 4.2 | c.2332-40 C>G homozygous | |
| #36 | 70 | yes | F | 3.3 | c.-32-13T>G | c.1551+1 G>C etero |
| #37 | 45 | no | M | 0.6 | c.-32-13T>G | c.2242dupG E748G |
| #38 | 24 | yes | M | 3.3 | c.-32-13T>G | Deletion on exon 18 |
| #39 | 54 | yes | F | 0.6 | c.118 C>T R40 | c.2647-7 G>A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moschetti, M.; Lo Curto, A.; Giacomarra, M.; Francofonte, D.; Zizzo, C.; Messina, E.; Duro, G.; Colomba, P. Mutation Spectrum of GAA Gene in Pompe Disease: Current Knowledge and Results of an Italian Study. Int. J. Mol. Sci. 2024, 25, 9139. https://doi.org/10.3390/ijms25179139
Moschetti M, Lo Curto A, Giacomarra M, Francofonte D, Zizzo C, Messina E, Duro G, Colomba P. Mutation Spectrum of GAA Gene in Pompe Disease: Current Knowledge and Results of an Italian Study. International Journal of Molecular Sciences. 2024; 25(17):9139. https://doi.org/10.3390/ijms25179139
Chicago/Turabian StyleMoschetti, Marta, Alessia Lo Curto, Miriam Giacomarra, Daniele Francofonte, Carmela Zizzo, Elisa Messina, Giovanni Duro, and Paolo Colomba. 2024. "Mutation Spectrum of GAA Gene in Pompe Disease: Current Knowledge and Results of an Italian Study" International Journal of Molecular Sciences 25, no. 17: 9139. https://doi.org/10.3390/ijms25179139
APA StyleMoschetti, M., Lo Curto, A., Giacomarra, M., Francofonte, D., Zizzo, C., Messina, E., Duro, G., & Colomba, P. (2024). Mutation Spectrum of GAA Gene in Pompe Disease: Current Knowledge and Results of an Italian Study. International Journal of Molecular Sciences, 25(17), 9139. https://doi.org/10.3390/ijms25179139