
Rabies Virus Regulates Inflammatory Response in BV-2 Cells through Activation of Myd88 and NF-κB Signaling Pathways via TLR7

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. RABV Exhibiting Varying Virulence Levels Provoke an Upregulation in TLR7 in Mouse Brain Tissues
2.2. RABV Induces Chemokine Expression In Vitro in a TLR7-Dependent Manner
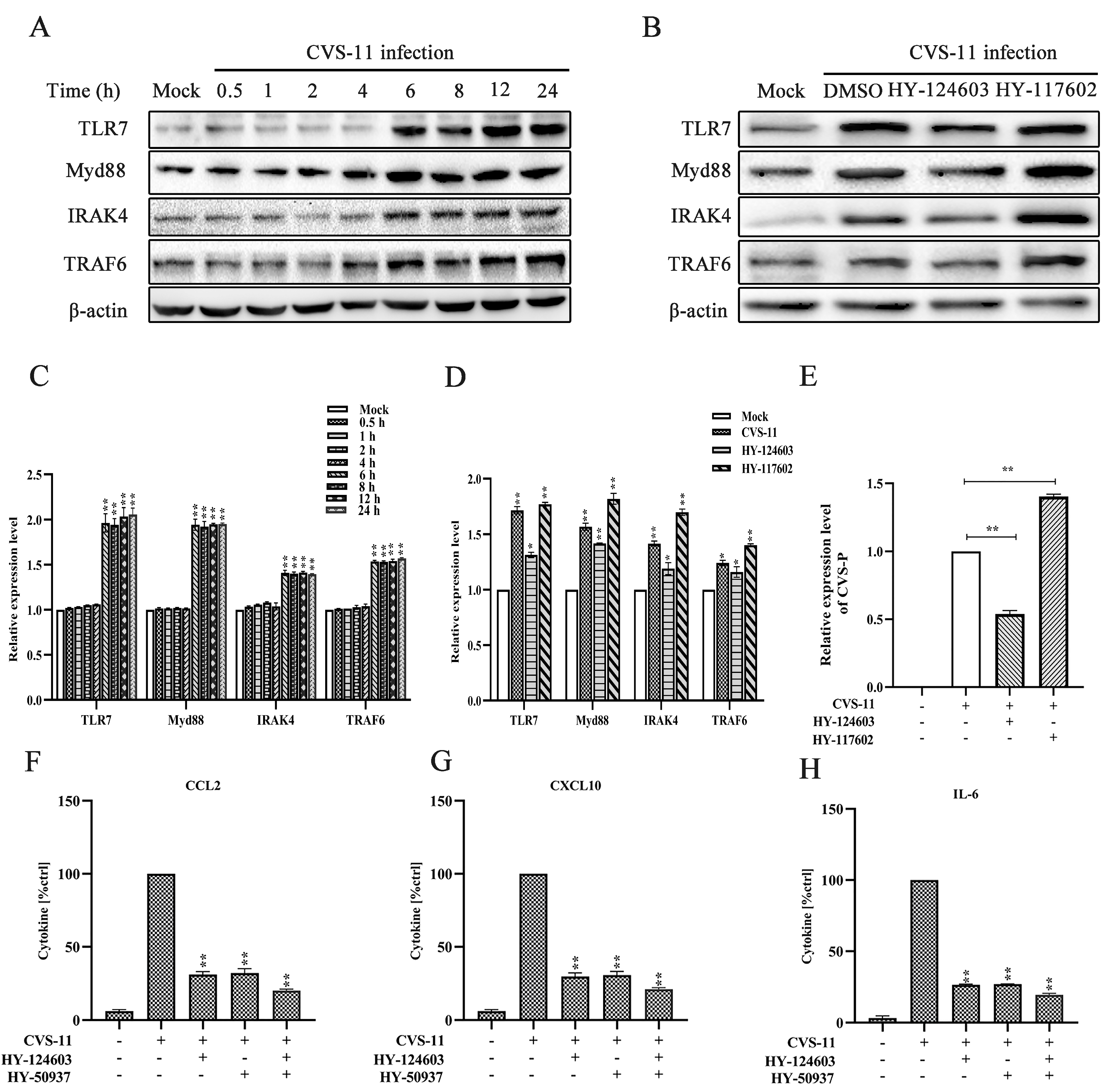
2.3. Activation of Myd88 Pathway Regulates RABV-Induced Cytokine Expression via TLR7 Signal
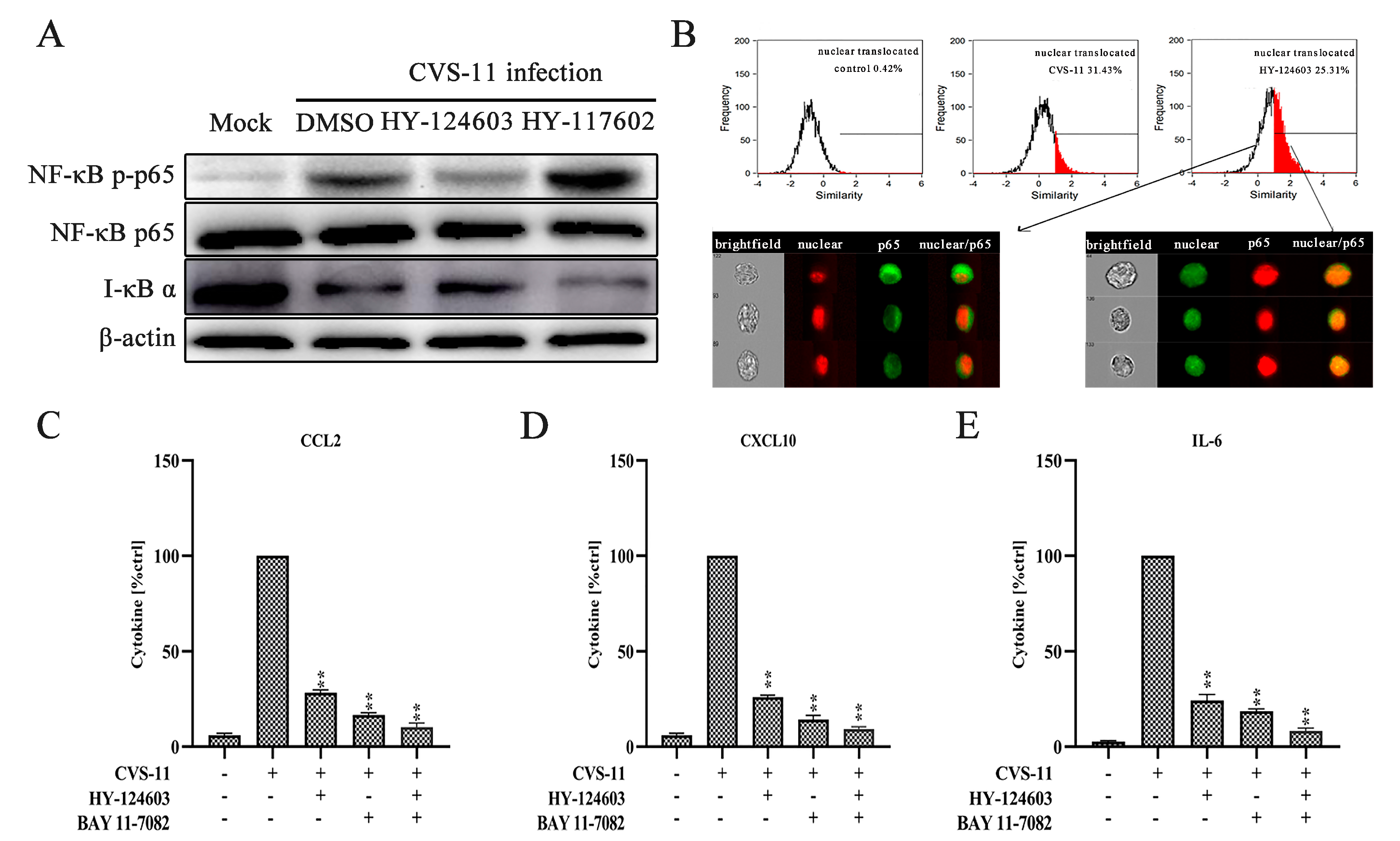
2.4. Activation of NF-κB Pathway Regulates RABV-Induced Cytokine Expression via TLR7 Signal
3. Discussion
4. Materials and Methods
4.1. Mouse Studies
4.2. Cell Culture and Reagents
4.3. Virus Strains
4.4. Viral Titration and Quantitative Real-Time PCR Assay (qRT-PCR)
4.5. Immunohistochemistry
4.6. ELISA
4.7. NF-κB Translocation
4.8. Western Blot
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hampson, K.; Coudeville, L.; Lembo, T.; Sambo, M.; Kieffer, A.; Attlan, M.; Barrat, J.; Blanton, J.D.; Briggs, D.J.; Cleaveland, S.; et al. Estimating the global burden of endemic canine rabies. PLoS Negl. Trop. Dis. 2015, 9, e0003709. [Google Scholar]
- World Health Organization. WHO Expert Consultation on Rabies; Second report; WHO Technical Report Series; World Health Organization: Geneva, Switzerland, 2013; pp. 1–139. [Google Scholar]
- Huang, Y.; Jiao, S.; Tao, X.; Tang, Q.; Jiao, W.; Xiao, J.; Xu, X.; Zhang, Y.; Liang, G. Met-CCL5 represents an immunotherapy strategy to ameliorate rabies virus infection. J. Neuroinflamm. 2014, 11, 146. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Singh, K.P.; Cherian, S.; Saminathan, M.; Kapoor, S.; Manjunatha Reddy, G.B.; Panda, S.; Dhama, K. Rabies–epidemiology, pathogenesis, public health concerns and advances in diagnosis and control: A comprehensive review. Vet. Q. 2017, 37, 212–251. [Google Scholar] [CrossRef] [PubMed]
- Lafon, M. Subversive neuroinvasive strategy of rabies virus. In Emergence and Control of Zoonotic Viral Encephalitides; Archives of Virology. Supplementa; Springer: Wien, Austria, 2004; pp. 149–159. [Google Scholar]
- Kufer, T.A.; Creagh, E.M.; Bryant, C.E. Guardians of the Cell: Effector-Triggered Immunity Steers Mammalian Immune Defense. Trends Immunol. 2019, 40, 939–951. [Google Scholar] [CrossRef]
- Wang, C.; Wang, G.; Zhang, C.; Zhu, P.K.; Dai, H.L.; Yu, N.; He, Z.; Xu, L.; Wang, E. OsCERK1-Mediated Chitin Perception and Immune Signaling Requires Receptor-like Cytoplasmic Kinase 185 to Activate an MAPK Cascade in Rice. Mol. Plant 2017, 10, 619–633. [Google Scholar] [CrossRef]
- Kadota, Y.; Sklenar, J.; Derbyshire, P.; Stransfeld, L.; Asai, S.; Ntoukakis, V.; Jones, J.D.G.; Shirasu, K.; Menke, F.; Jones, A.; et al. Direct regulation of the NADPH oxidase RBOHD by the PRR-associated kinase BIK1 during plant immunity. Mol. Cell 2014, 54, 43–55. [Google Scholar] [CrossRef]
- Goodwin, M.; Lee, E.; Lakshmanan, U.; Shipp, S.; Froessl, L.; Barzaghi, F.; Passerini, L.; Narula, M.; Sheikali, A.; Lee, C.M.; et al. CRISPR-based gene editing enables FOXP3 gene repair in IPEX patient cells. Sci. Adv. 2020, 6, eaaz0571. [Google Scholar] [CrossRef]
- Rodet, F.; Tasiemski, A.; Boidin-Wichlacz, C.; Van Camp, C.; Vuillaume, C.; Slomianny, C.; Salzet, M. Hm-MyD88 and Hm-SARM: Two key regulators of the neuroimmune system and neural repair in the medicinal leech. Sci. Rep. 2015, 5, 9624. [Google Scholar] [CrossRef]
- Kargas, V.; Marzinek, J.K.; Holdbrook, D.A.; Yin, H.; Ford, R.C.; Bond, P.J. A polar SxxS motif drives assembly of the transmembrane domains of Toll-like receptor 4. Biochim. Biophys. Acta Biomembr. 2017, 1859, 2086–2095. [Google Scholar] [CrossRef]
- Su, L.C.; Xu, W.D.; Huang, A.F. IRAK family in inflammatory autoimmune diseases. Autoimmun. Rev. 2020, 19, 102461. [Google Scholar] [CrossRef]
- Leventhal, J.S. Lose appetite, lose control: Integrins and noncanonical autophagy regulate germinal center reactions. J. Clin. Invest. 2018, 128, 3752–3753. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Sanjo, H.; Uematsu, S.; Kaisho, T.; Hoshino, K.; Takeuchi, O.; Kobayashi, M.; Fujita, T.; et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature 2002, 420, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sato, S.; Mori, K.; Hoshino, K.; Takeuchi, O.; Takeda, K.; Akira, S. Cutting edge: A novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J. Immunol. 2002, 169, 6668–6672. [Google Scholar] [CrossRef]
- Honda, K.; Yanai, H.; Mizutani, T.; Negishi, H.; Shimada, N.; Suzuki, N.; Ohba, Y.; Takaoka, A.; Yeh, W.-C.; Taniguchi, T. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 15416–15421. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Sato, S.; Ishii, K.J.; Coban, C.; Hemmi, H.; Yamamoto, M.; Terai, K.; Matsuda, M.; Inoue, J.-I.; Uematsu, S.; et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 2004, 5, 1061–1068. [Google Scholar] [CrossRef]
- Davidson, S.; Kaiko, G.; Loh, Z.; Lalwani, A.; Zhang, V.; Spann, K.; Foo, S.Y.; Hansbro, N.; Uematsu, S.; Akira, S.; et al. Plasmacytoid dendritic cells promote host defense against acute pneumovirus infection via the TLR7-MyD88-dependent signaling pathway. J. Immunol. 2011, 186, 5938–5948. [Google Scholar] [CrossRef]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef]
- Mandl, J.N.; Akondy, R.; Lawson, B.; Kozyr, N.; Staprans, S.I.; Ahmed, R.; Feinberg, M.B. Distinctive TLR7 signaling, type I IFN production, and attenuated innate and adaptive immune responses to yellow fever virus in a primate reservoir host. J. Immunol. 2011, 186, 6406–6416. [Google Scholar] [CrossRef]
- Schlaepfer, E.; Audige, A.; Joller, H.; Speck, R.F. TLR7/8 triggering exerts opposing effects in acute versus latent HIV infection. J. Immunol. 2006, 176, 2888–2895. [Google Scholar] [CrossRef] [PubMed]
- Town, T.; Bai, F.; Wang, T.; Kaplan, A.T.; Qian, F.; Montgomery, R.R.; Anderson, J.F.; Flavell, R.A.; Fikrig, E. Toll-like receptor 7 mitigates lethal West Nile encephalitis via interleukin 23-dependent immune cell infiltration and homing. Immunity 2009, 30, 242–253. [Google Scholar] [CrossRef]
- Miller, R.L.; Meng, T.C.; Tomai, M.A. The antiviral activity of Toll-like receptor 7 and 7/8 agonists. Drug News Perspect. 2008, 21, 69–87. [Google Scholar]
- Liu, R.; Wang, J.; Yang, Y.; Khan, I.; Zhu, N. Rabies virus lipopeptide conjugated to a TLR7 agonist improves the magnitude and quality of the Th1-biased humoral immune response in mice. Virology 2016, 497, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Faber, M.; Dietzschold, B.; Hooper, D.C. The role of toll-like receptors in the induction of immune responses during rabies virus infection. Adv. Virus Res. 2011, 79, 115–126. [Google Scholar] [PubMed]
- Miyamoto, K.; Matsumoto, S. Comparative studies between pathogenesis of street and fixed rabies infection. J. Exp. Med. 1967, 125, 447–456. [Google Scholar] [CrossRef]
- Chai, Q.; He, W.Q.; Zhou, M.; Lu, H.; Fu, Z.F. Enhancement of blood-brain barrier permeability and reduction of tight junction protein expression are modulated by chemokines/cytokines induced by rabies virus infection. J. Virol. 2014, 88, 4698–4710. [Google Scholar] [CrossRef]
- Chai, Q.; She, R.; Huang, Y.; Fu, Z.F. Expression of neuronal CXCL10 induced by rabies virus infection initiates infiltration of inflammatory cells, production of chemokines and cytokines, and enhancement of blood-brain barrier permeability. J. Virol. 2015, 89, 870–876. [Google Scholar] [CrossRef]
- Gnanadurai, C.W.; Fu, Z.F. CXCL10 and blood-brain barrier modulation in rabies virus infection. Oncotarget 2016, 7, 10694. [Google Scholar] [CrossRef]
- Madhu, B.P.; Singh, K.P.; Saminathan, M.; Singh, R.; Tiwari, A.K.; Manjunatha, V.; Harish, C.; Manjunathareddy, G.B. Correlation of inducible nitric oxide synthase (iNOS) inhibition with TNF-α, caspase-1, FasL and TLR-3 in pathogenesis of rabies in mouse model. Virus Genes 2016, 52, 61–70. [Google Scholar] [CrossRef]
- Liu, S.Q.; Gao, X.; Xie, Y.; Wang, Q.; Zhu, W.Y. Rabies viruses of different virulence regulates inflammatory responses both in vivo and in vitro via MAPK and NF-kappaB pathway. Mol. Immunol. 2020, 125, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Li, Y.; Zhou, M.; Lv, L.; Wu, Q.; Chen, C.; Zhang, Y.; Sui, B.; Tu, C.; Cui, M.; et al. Toll-Like Receptor 7 Enhances Rabies Virus-Induced Humoral Immunity by Facilitating the Formation of Germinal Centers. Front. Immunol. 2019, 10, 429. [Google Scholar] [CrossRef]
- Wang, L.; Hu, D.; Xie, B.; Xie, L. Blockade of Myd88 signaling by a novel MyD88 inhibitor prevents colitis-associated colorectal cancer development by impairing myeloid-derived suppressor cells. Invest. New Drugs 2022, 40, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Martina, B.E.E.; Smreczak, M.; Orlowska, A.; Marzec, A.; Trebas, P.; Roose, J.M.; Zmudzinski, J.; Gerhauser, I.; Wohlsein, P.; Baumgärtner, W.; et al. Combination drug treatment prolongs survival of experimentally infected mice with silver-haired bat rabies virus. Vaccine 2019, 37, 4736–4742. [Google Scholar] [CrossRef] [PubMed]
- Manjunatha, V.; Singh, K.P.; Saminathan, M.; Singh, R.; Shivasharanappa, N.; Umeshappa, C.S.; Dhama, K.; Manjunathareddy, G.B. Inhibition of MEK-ERK1/2-MAP kinase signalling pathway reduces rabies virus induced pathologies in mouse model. Microb. Pathog. 2017, 112, 38–49. [Google Scholar] [CrossRef]
- Marosi, A.; Dufkova, L.; Forro, B.; Felde, O.; Erdelyi, K.; Sirmarova, J.; Palus, M.; Hönig, V.; Salát, J.; Tikos, R.; et al. Combination therapy of rabies-infected mice with inhibitors of pro-inflammatory host response, antiviral compounds and human rabies immunoglobulin. Vaccine 2019, 37, 4724–4735. [Google Scholar] [CrossRef]
- Asea, A.; Rehli, M.; Kabingu, E.; Boch, J.A.; Bare, O.; Auron, P.E.; Stevenson, M.A.; Calderwood, S.K. Novel signal transduction pathway utilized by extracellular HSP70: Role of toll-like receptor (TLR) 2 and TLR4. J. Biol. Chem. 2002, 277, 15028–15034. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef]
- Arpaia, N.; Barton, G.M. Toll-like receptors: Key players in antiviral immunity. Curr. Opin. Virol. 2011, 1, 447–454. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Kissner, T.L.; Ruthel, G.; Cisney, E.D.; Ulrich, R.G.; Fernandez, S.; Saikh, K.U. MyD88-dependent pro-inflammatory cytokine response contributes to lethal toxicity of staphylococcal enterotoxin B in mice. Innate Immun. 2011, 17, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.-Y.; Lin, C.-C.; Chen, Y.-M.; Lan, J.-L.; Hung, W.-T.; Chen, H.-H.; Lai, K.-L.; Hsieh, C.-W. Involvement of TLR7 MyD88-dependent signaling pathway in the pathogenesis of adult-onset Still’s disease. Arthritis Res. Ther. 2013, 15, R39. [Google Scholar] [CrossRef]
- Matsushima, H.; Yamada, N.; Matsue, H.; Shimada, S. TLR3-, TLR7-, and TLR9-mediated production of proinflammatory cytokines and chemokines from murine connective tissue type skin-derived mast cells but not from bone marrow-derived mast cells. J. Immunol. 2004, 173, 531–541. [Google Scholar] [CrossRef]
- Ashall, L.; Horton, C.A.; Nelson, D.E.; Paszek, P.; Harper, C.V.; Sillitoe, K.; Ryan, S.; Spiller, D.G.; Unitt, J.F.; Broomhead, D.S.; et al. Pulsatile stimulation determines timing and specificity of NF-kappaB-dependent transcription. Science 2009, 324, 242–246. [Google Scholar] [CrossRef]
- Guo, Z.; Tao, X.; Yin, C.; Han, N.; Yu, J.; Li, H.; Liu, H.; Fang, W.; Adams, J.; Wang, J.; et al. National borders effectively halt the spread of rabies: The current rabies epidemic in China is dislocated from cases in neighboring countries. PLoS Negl. Trop. Dis. 2013, 7, e2039. [Google Scholar] [CrossRef]
- Anindita, P.D.; Sasaki, M.; Okada, K.; Ito, N.; Sugiyama, M.; Saito-Tarashima, N.; Minakawa, N.; Shuto, S.; Otsuguro, S.; Ichikawa, S.; et al. Ribavirin-related compounds exert in vitro inhibitory effects toward rabies virus. Antivir. Res. 2018, 154, 1–9. [Google Scholar] [CrossRef]
- Hooper, D.C.; Morimoto, K.; Bette, M.; Weihe, E.; Koprowski, H.; Dietzschold, B. Collaboration of antibody and inflammation in clearance of rabies virus from the central nervous system. J. Virol. 1998, 72, 3711–3719. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Q.; Xie, Y.; Gao, X.; Wang, Q.; Zhu, W.Y. Inflammatory response and MAPK and NF-kappaB pathway activation induced by natural street rabies virus infection in the brain tissues of dogs and humans. Virol. J. 2020, 17, 157. [Google Scholar] [CrossRef]
- du Prel, J.B.; Hommel, G.; Rohrig, B.; Blettner, M. Confidence interval or p-value?: Part 4 of a series on evaluation of scientific publications. Dtsch. Arztebl. Int. 2009, 106, 335–339. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Y.; Chi, Y.; Tao, X.; Yu, P.; Liu, Q.; Zhang, M.; Yang, N.; Liu, S.; Zhu, W. Rabies Virus Regulates Inflammatory Response in BV-2 Cells through Activation of Myd88 and NF-κB Signaling Pathways via TLR7. Int. J. Mol. Sci. 2024, 25, 9144. https://doi.org/10.3390/ijms25179144
Xie Y, Chi Y, Tao X, Yu P, Liu Q, Zhang M, Yang N, Liu S, Zhu W. Rabies Virus Regulates Inflammatory Response in BV-2 Cells through Activation of Myd88 and NF-κB Signaling Pathways via TLR7. International Journal of Molecular Sciences. 2024; 25(17):9144. https://doi.org/10.3390/ijms25179144
Chicago/Turabian StyleXie, Yuan, Yinglin Chi, Xiaoyan Tao, Pengcheng Yu, Qian Liu, Minghui Zhang, Nuo Yang, Shuqing Liu, and Wuyang Zhu. 2024. "Rabies Virus Regulates Inflammatory Response in BV-2 Cells through Activation of Myd88 and NF-κB Signaling Pathways via TLR7" International Journal of Molecular Sciences 25, no. 17: 9144. https://doi.org/10.3390/ijms25179144