

Reproductive Tract Microbial Transitions from Late Gestation to Early Postpartum Using 16S rRNA Metagenetic Profiling in First-Pregnancy Heifers


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
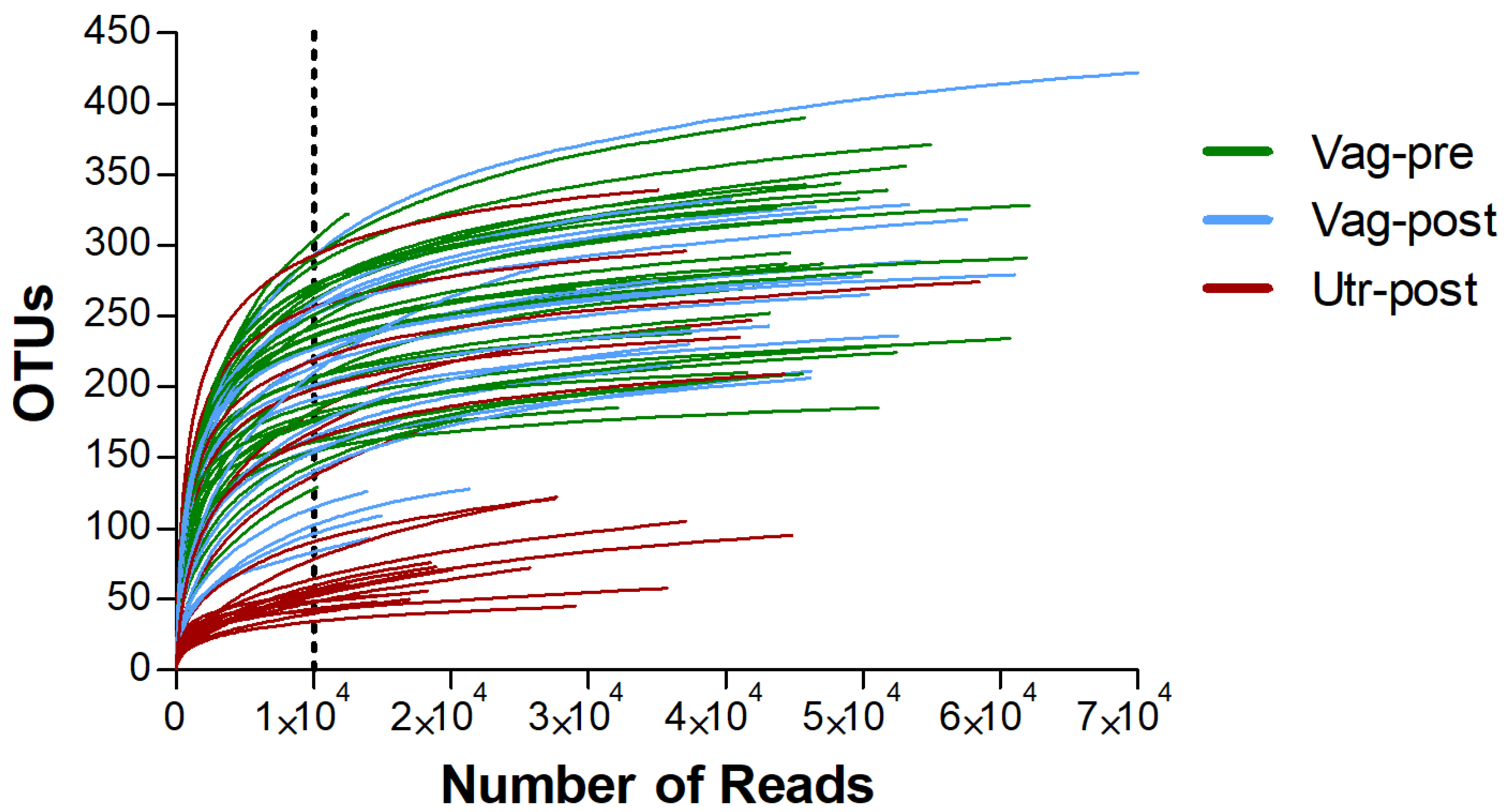
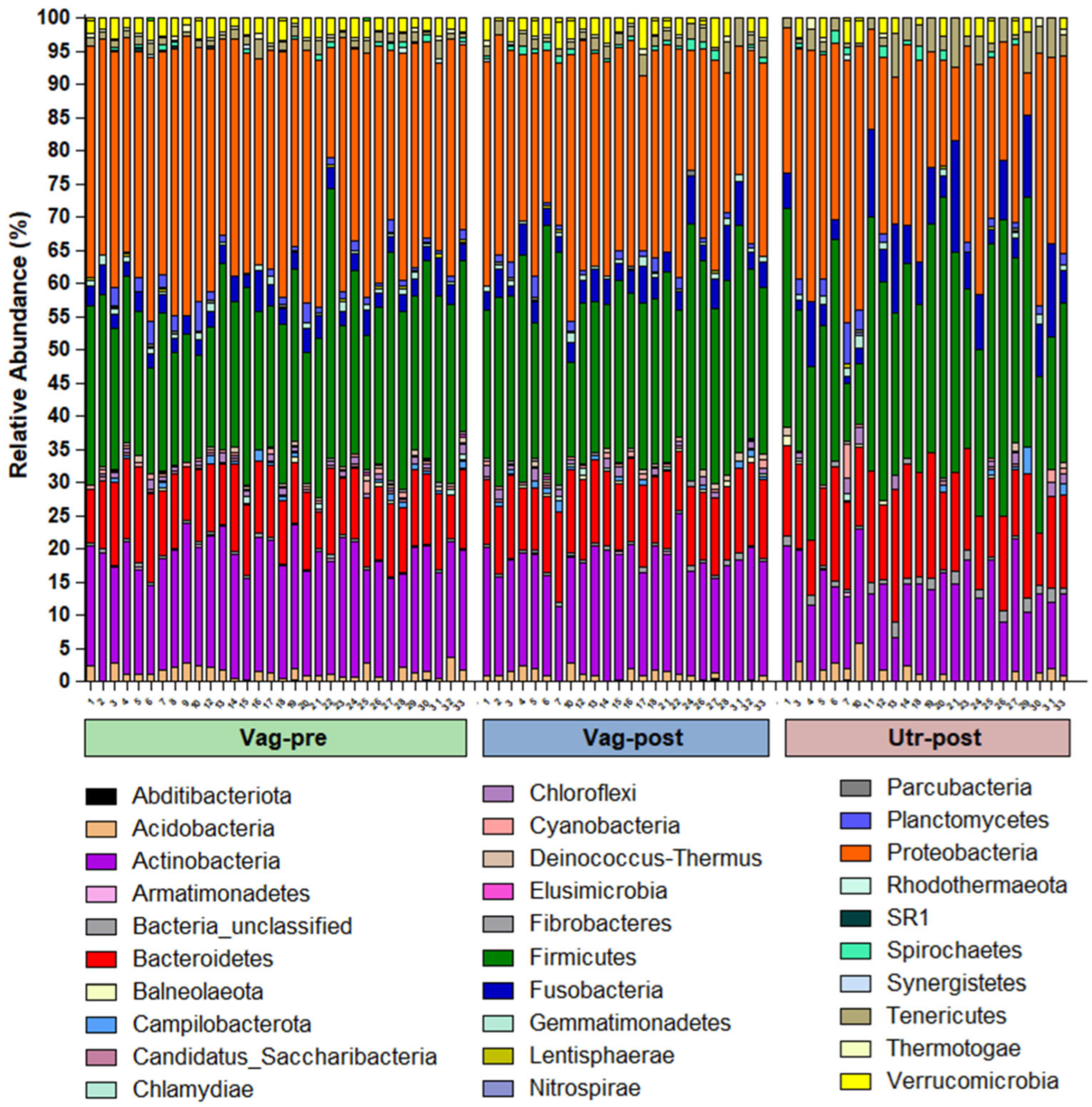
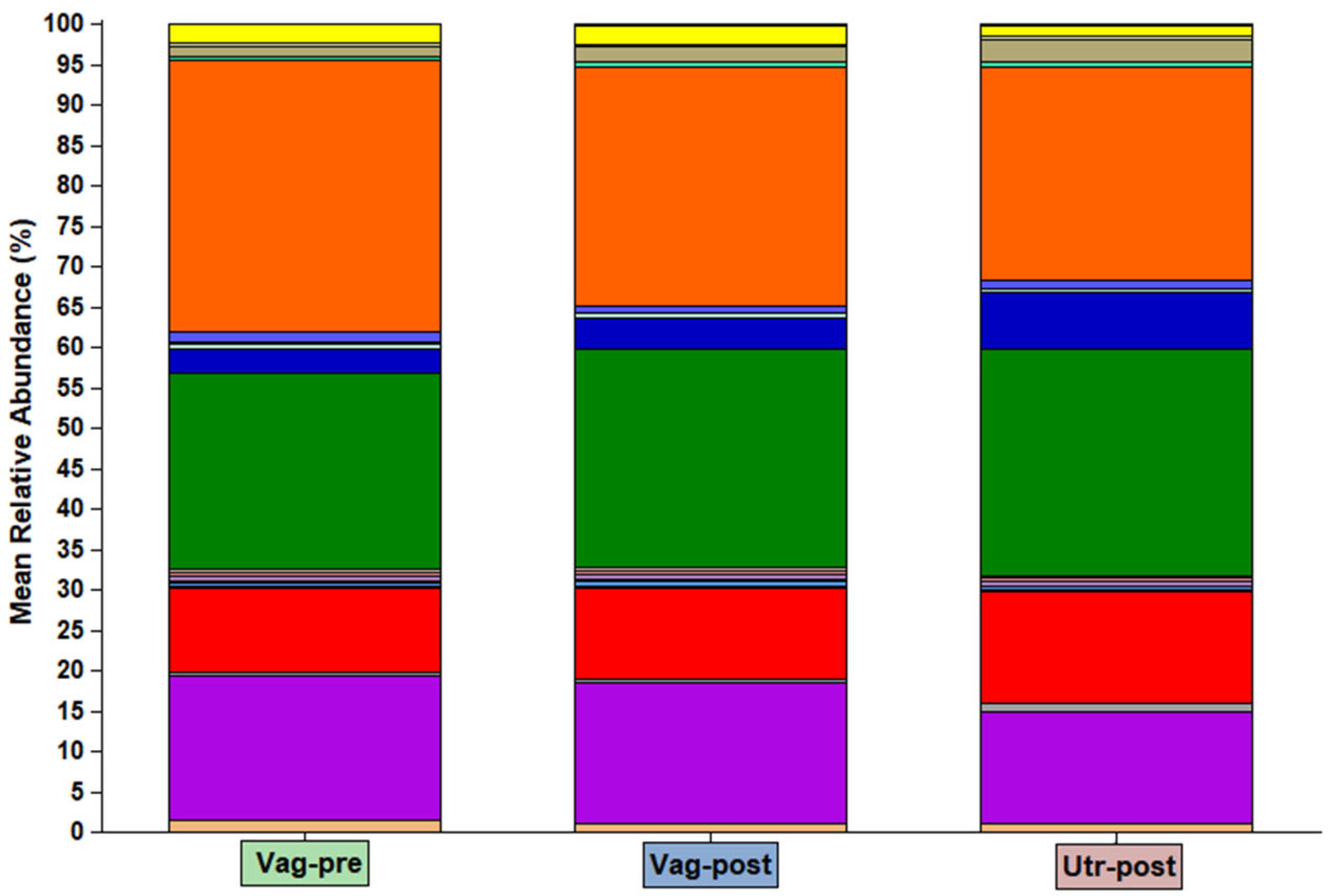
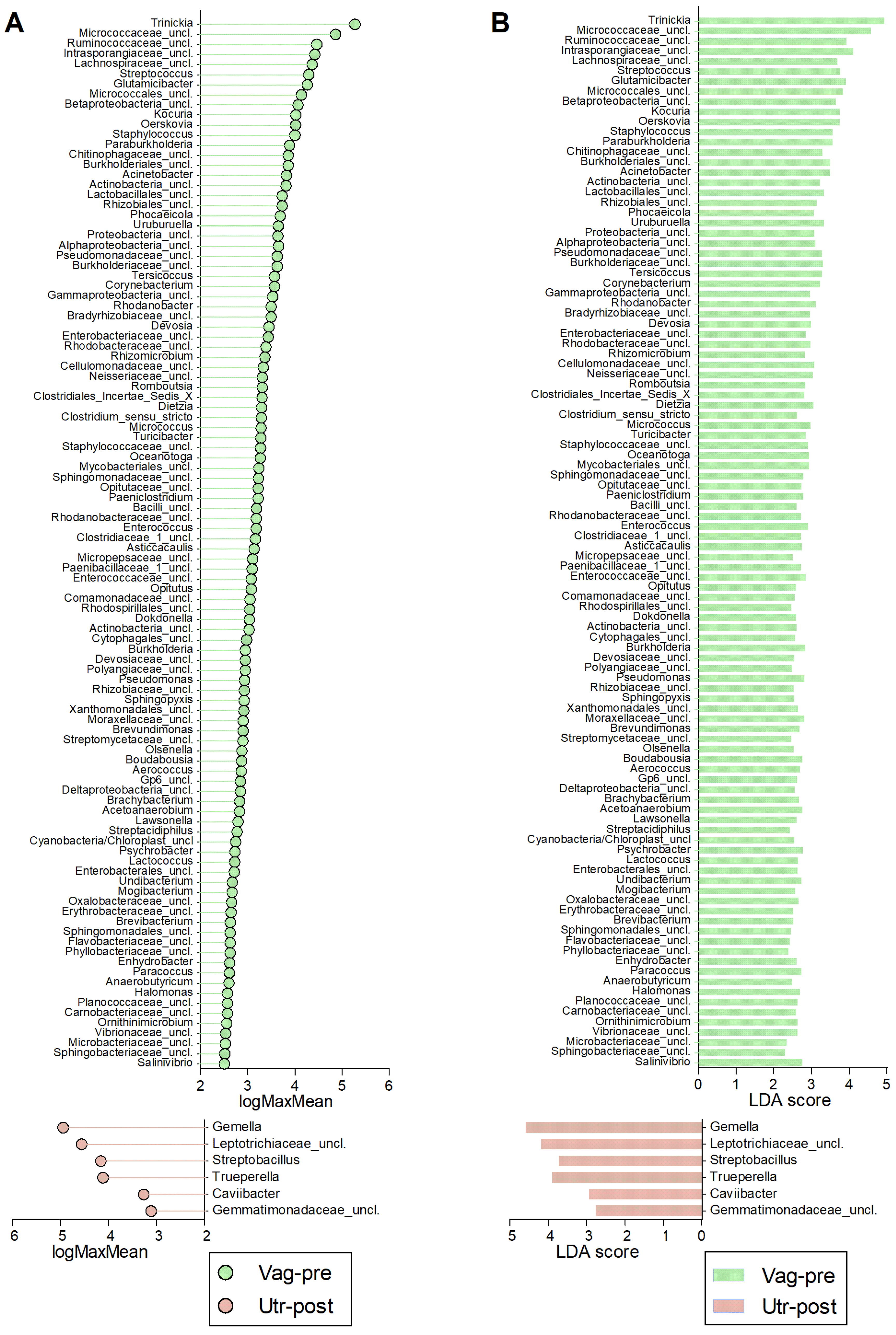
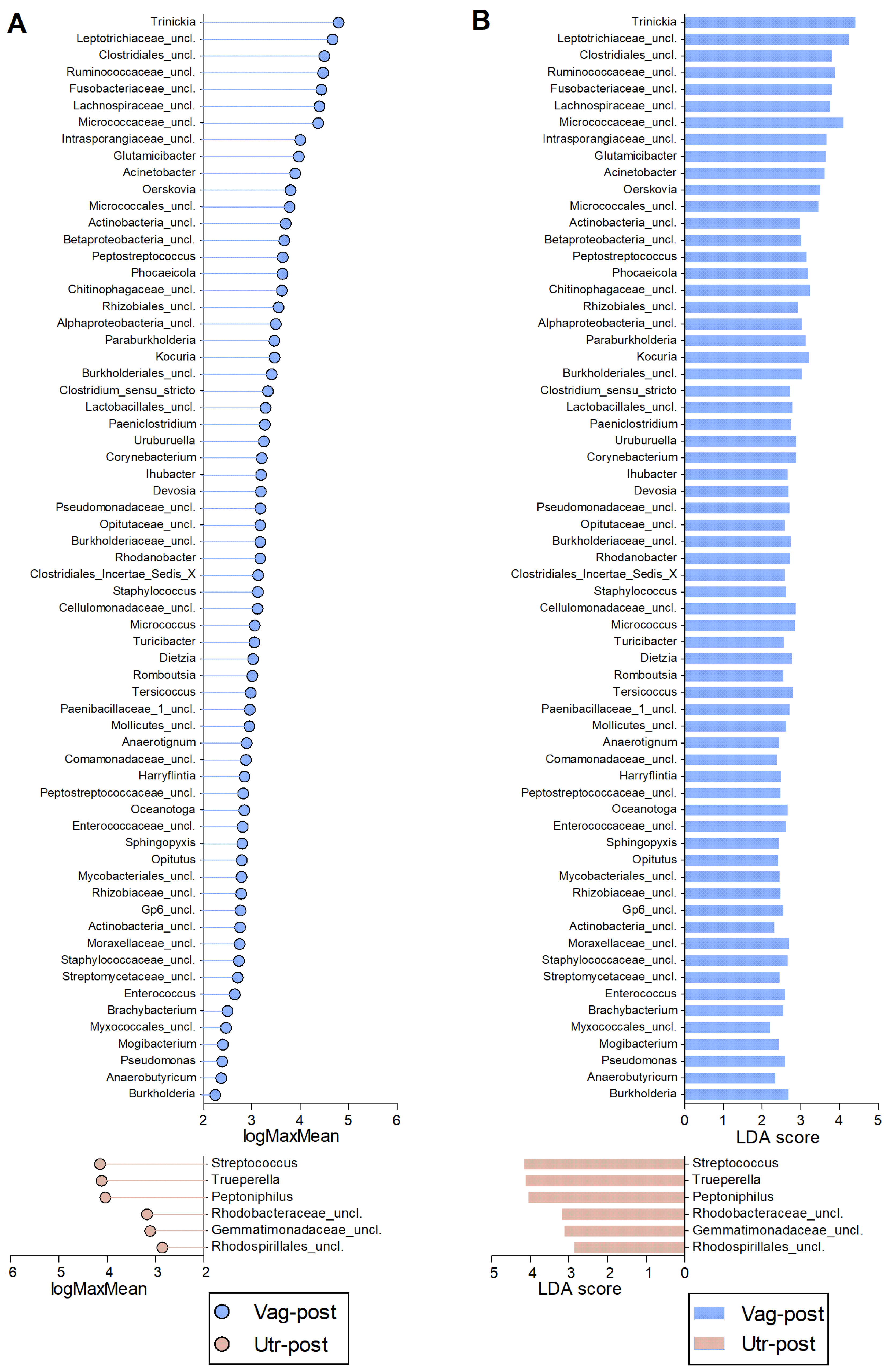
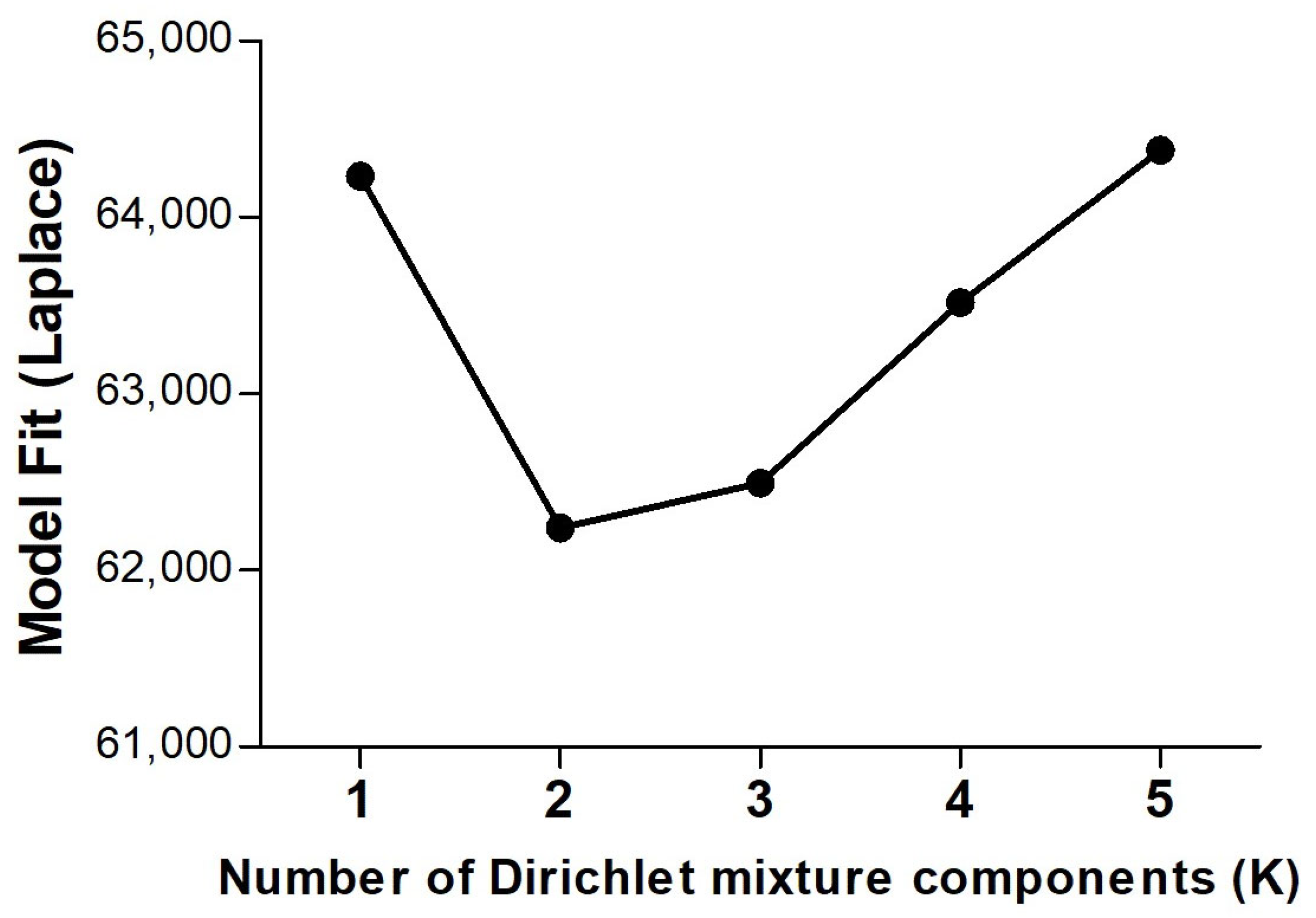
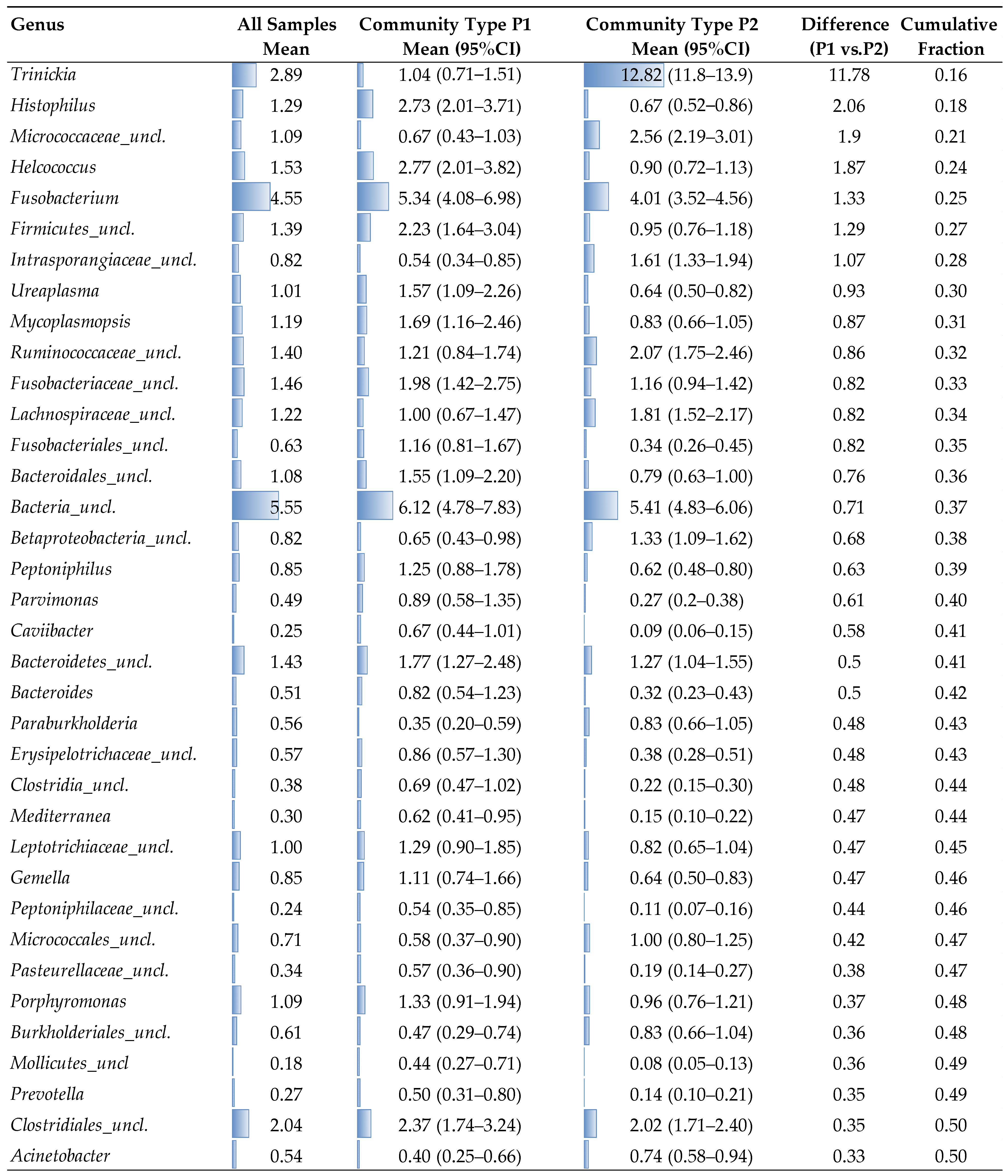
2. Results
3. Discussion
4. Materials and Methods
4.1. Animals and Sampling Procedures
4.2. DNA Extraction
4.3. 16S rRNA Gene Amplicon Libraries Preparation and NGS Sequencing
4.4. Bioinformatic Analysis
4.5. Additional Statistical Analysis & Plotting
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, B.; Tao, Z.; Edupuganti, L.; Serrano, M.G.; Buck, G.A. Roles of the Microbiota of the Female Reproductive Tract in Gynecological and Reproductive Health. Microbiol. Mol. Biol. Rev. 2022, 86, e0018121. [Google Scholar] [CrossRef]
- Ong, C.T.; Turni, C.; Blackall, P.J.; Boe-Hansen, G.; Hayes, B.J.; Tabor, A.E. Interrogating the bovine reproductive tract metagenomes using culture-independent approaches: A systematic review. Anim. Microbiome 2021, 3, 41. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Wang, X.; Wang, J. Research progress on the correlation between gut microbiota and preeclampsia: Microbiome changes, mechanisms and treatments. Front. Cell. Infect. Microbiol. 2023, 13, 1256940. [Google Scholar] [CrossRef]
- Timmons, B.; Akins, M.; Mahendroo, M. Cervical remodeling during pregnancy and parturition. Trends Endocrinol. Metab. 2010, 21, 353–361. [Google Scholar] [CrossRef]
- Yellon, S.M. Immunobiology of Cervix Ripening. Front. Immunol. 2019, 10, 3156. [Google Scholar] [CrossRef]
- Sheldon, I.M.; Williams, E.J.; Miller, A.N.; Nash, D.M.; Herath, S. Uterine diseases in cattle after parturition. Vet. J. 2008, 176, 115–121. [Google Scholar] [CrossRef]
- LeBlanc, S.J. Review: Postpartum reproductive disease and fertility in dairy cows. Animal 2023, 17 (Suppl. S1), 100781. [Google Scholar] [CrossRef] [PubMed]
- Sicsic, R.; Goshen, T.; Dutta, R.; Kedem-Vaanunu, N.; Kaplan-Shabtai, V.; Pasternak, Z.; Gottlieb, Y.; Shpigel, N.Y.; Raz, T. Microbial communities and inflammatory response in the endometrium differ between normal and metritic dairy cows at 5–10 days post-partum. Vet. Res. 2018, 49, 77. [Google Scholar] [CrossRef]
- Pascottini, O.B.; Aurich, C.; England, G.; Grahofer, A. General and comparative aspects of endometritis in domestic species: A review. Reprod. Domest. Anim. 2023, 58 (Suppl. S2), 49–71. [Google Scholar] [CrossRef] [PubMed]
- Pu, S.; Wang, M.; Wang, J.; Zhang, Q.; Ma, X.; Wang, R.; Yu, S.; Wang, L.; Pan, Y. Metagenomic analysis reveals a dynamic microbiome with diversified adaptive functions that respond to ovulation regulation in the mouse endometrium. BMC Genom. 2023, 24, 615. [Google Scholar] [CrossRef]
- Ababneh, M.M.; Degefa, T. Bacteriological findings and hormonal profiles in the postpartum Balady goats. Reprod. Domest. Anim. 2006, 41, 12–16. [Google Scholar] [CrossRef]
- Chauhan, S.P.; Rice, M.M.; Grobman, W.A.; Bailit, J.; Reddy, U.M.; Wapner, R.J.; Varner, M.W.; Thorp, J.M., Jr.; Caritis, S.N.; Prasad, M.; et al. Neonatal and Maternal Composite Adverse Outcomes Among Low-Risk Nulliparous Women Compared with Multiparous Women at 39–41 Weeks of Gestation. Obstet. Gynecol. 2020, 136, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Ngonzi, J.; Bebell, L.M.; Fajardo, Y.; Boatin, A.A.; Siedner, M.J.; Bassett, I.V.; Jacquemyn, Y.; Van Geertruyden, J.P.; Kabakyenga, J.; Wylie, B.J.; et al. Incidence of postpartum infection, outcomes and associated risk factors at Mbarara regional referral hospital in Uganda. BMC Pregnancy Childbirth 2018, 18, 270. [Google Scholar] [CrossRef]
- Stephansson, O.; Sandstrom, A.; Petersson, G.; Wikstrom, A.K.; Cnattingius, S. Prolonged second stage of labour, maternal infectious disease, urinary retention and other complications in the early postpartum period. BJOG 2016, 123, 608–616. [Google Scholar] [CrossRef]
- Williams, E.J.; Fischer, D.P.; Pfeiffer, D.U.; England, G.C.; Noakes, D.E.; Dobson, H.; Sheldon, I.M. Clinical evaluation of postpartum vaginal mucus reflects uterine bacterial infection and the immune response in cattle. Theriogenology 2005, 63, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Moreno, C.; Torres Luque, A.; Oliszewski, R.; Rosa, R.J.; Otero, M.C. Characterization of native Escherichia coli populations from bovine vagina of healthy heifers and cows with postpartum uterine disease. PLoS ONE 2020, 15, e0228294. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, I.M.; Rycroft, A.N.; Dogan, B.; Craven, M.; Bromfield, J.J.; Chandler, A.; Roberts, M.H.; Price, S.B.; Gilbert, R.O.; Simpson, K.W. Specific strains of Escherichia coli are pathogenic for the endometrium of cattle and cause pelvic inflammatory disease in cattle and mice. PLoS ONE 2010, 5, e9192. [Google Scholar] [CrossRef]
- Chen, H.; Fu, K.; Pang, B.; Wang, J.; Li, H.; Jiang, Z.; Feng, Y.; Tian, W.; Cao, R. Determination of uterine bacterial community in postpartum dairy cows with metritis based on 16S rDNA sequencing. Vet. Anim. Sci. 2020, 10, 100102. [Google Scholar] [CrossRef] [PubMed]
- Segre, J.A. What does it take to satisfy Koch’s postulates two centuries later? Microbial genomics and Propionibacteria acnes. J. Investig. Dermatol. 2013, 133, 2141–2142. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef]
- Wasimuddin; Schlaeppi, K.; Ronchi, F.; Leib, S.L.; Erb, M.; Ramette, A. Evaluation of primer pairs for microbiome profiling from soils to humans within the One Health framework. Mol. Ecol. Resour. 2020, 20, 1558–1571. [Google Scholar] [CrossRef] [PubMed]
- Moonsamy, P.V.; Williams, T.; Bonella, P.; Holcomb, C.L.; Hoglund, B.N.; Hillman, G.; Goodridge, D.; Turenchalk, G.S.; Blake, L.A.; Daigle, D.A.; et al. High throughput HLA genotyping using 454 sequencing and the Fluidigm Access Array System for simplified amplicon library preparation. Tissue Antigens 2013, 81, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef]
- Mather, P.M. Computational Methods of Multivariate Analysis in Physical Geography; John Wiley and Sons: London, UK, 1976; p. 532. [Google Scholar]
- Holmes, I.; Harris, K.; Quince, C. Dirichlet multinomial mixtures: Generative models for microbial metagenomics. PLoS ONE 2012, 7, e30126. [Google Scholar] [CrossRef]
- Knott, J.C. A Study of the Gestation Period of Holstein-Friesian Cows1. J. Dairy Sci. 1932, 15, 87–98. [Google Scholar] [CrossRef]
- Eaglen, S.A.; Coffey, M.P.; Woolliams, J.A.; Wall, E. Direct and maternal genetic relationships between calving ease, gestation length, milk production, fertility, type, and lifespan of Holstein-Friesian primiparous cows. J. Dairy Sci. 2013, 96, 4015–4025. [Google Scholar] [CrossRef]
- Taverne, M.A.; Breeveld-Dwarkasing, V.N.; van Dissel-Emiliani, F.M.; Bevers, M.M.; de Jong, R.; van der Weijden, G.C. Between prepartum luteolysis and onset of expulsion. Domest. Anim. Endocrinol. 2002, 23, 329–337. [Google Scholar] [CrossRef]
- Jeon, S.J.; Vieira-Neto, A.; Gobikrushanth, M.; Daetz, R.; Mingoti, R.D.; Parize, A.C.; de Freitas, S.L.; da Costa, A.N.; Bicalho, R.C.; Lima, S.; et al. Uterine Microbiota Progression from Calving until Establishment of Metritis in Dairy Cows. Appl. Environ. Microbiol. 2015, 81, 6324–6332. [Google Scholar] [CrossRef]
- Tasara, T.; Meier, A.B.; Wambui, J.; Whiston, R.; Stevens, M.; Chapwanya, A.; Bleul, U. Interrogating the Diversity of Vaginal, Endometrial, and Fecal Microbiomes in Healthy and Metritis Dairy Cattle. Animals 2023, 13, 1221. [Google Scholar] [CrossRef]
- DiGiulio, D.B.; Callahan, B.J.; McMurdie, P.J.; Costello, E.K.; Lyell, D.J.; Robaczewska, A.; Sun, C.L.; Goltsman, D.S.; Wong, R.J.; Shaw, G.; et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc. Natl. Acad. Sci. USA 2015, 112, 11060–11065. [Google Scholar] [CrossRef]
- Fox, C.; Eichelberger, K. Maternal microbiome and pregnancy outcomes. Fertil. Steril. 2015, 104, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Mallott, E.K.; Borries, C.; Koenig, A.; Amato, K.R.; Lu, A. Reproductive hormones mediate changes in the gut microbiome during pregnancy and lactation in Phayre’s leaf monkeys. Sci. Rep. 2020, 10, 9961. [Google Scholar] [CrossRef] [PubMed]
- Motomura, K.; Miller, D.; Galaz, J.; Liu, T.N.; Romero, R.; Gomez-Lopez, N. The effects of progesterone on immune cellular function at the maternal-fetal interface and in maternal circulation. J. Steroid Biochem. Mol. Biol. 2023, 229, 106254. [Google Scholar] [CrossRef]
- Garcia-Gomez, E.; Vazquez-Martinez, E.R.; Reyes-Mayoral, C.; Cruz-Orozco, O.P.; Camacho-Arroyo, I.; Cerbon, M. Regulation of Inflammation Pathways and Inflammasome by Sex Steroid Hormones in Endometriosis. Front. Endocrinol. 2019, 10, 935. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, G.; Tanyolac Talay, Z.G.; Paerhati, E.; Eren, O.C.; Coskun, N.; Sahin, D.; Alnajjar, I.; Albayrak, O.; Gursoy, A.; Keskin, O.; et al. Dysbiosis in pregnant mice induced by transfer of human vaginal microbiota followed by reversal of pathological changes in the uterus and placenta via progesterone treatment. BMC Pregnancy Childbirth 2024, 24, 427. [Google Scholar] [CrossRef]
- Bradley, F.; Franzen Boger, M.; Kaldhusdal, V.; Ahlberg, A.; Edfeldt, G.; Lajoie, J.; Bergstrom, S.; Omollo, K.; Damdimopoulos, A.; Czarnewski, P.; et al. Multi-omics analysis of the cervical epithelial integrity of women using depot medroxyprogesterone acetate. PLoS Pathog. 2022, 18, e1010494. [Google Scholar] [CrossRef]
- Carosso, A.; Revelli, A.; Gennarelli, G.; Canosa, S.; Cosma, S.; Borella, F.; Tancredi, A.; Paschero, C.; Boatti, L.; Zanotto, E.; et al. Controlled ovarian stimulation and progesterone supplementation affect vaginal and endometrial microbiota in IVF cycles: A pilot study. J. Assist. Reprod. Genet. 2020, 37, 2315–2326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Liu, G.; Wu, Y.; Zhang, T.; Guo, M.; Lei, Y.; Cao, X.; Suo, L.; Brugger, D.; Wang, X.; et al. Gut Microbial Succession Patterns and Metabolic Profiling during Pregnancy and Lactation in a Goat Model. Microbiol. Spectr. 2023, 11, e0295522. [Google Scholar] [CrossRef]
- Kaur, S.; Sarma, S.J.; Marshall, B.L.; Liu, Y.; Kinkade, J.A.; Bellamy, M.M.; Mao, J.; Helferich, W.G.; Schenk, A.K.; Bivens, N.J.; et al. Developmental exposure of California mice to endocrine disrupting chemicals and potential effects on the microbiome-gut-brain axis at adulthood. Sci. Rep. 2020, 10, 10902. [Google Scholar] [CrossRef]
- Wan, L.; Huang, R.J.; Luo, Z.H.; Gong, J.E.; Pan, A.; Manavis, J.; Yan, X.X.; Xiao, B. Reproduction-Associated Hormones and Adult Hippocampal Neurogenesis. Neural Plast. 2021, 2021, 3651735. [Google Scholar] [CrossRef]
- Dockery, P.; Rogers, A.W. The effects of steroids on the fine structure of the endometrium. Baillieres Clin. Obstet. Gynaecol. 1989, 3, 227–248. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, P.V. Uterine involution in Finnish dairy cows. Acta Vet. Scand. 1990, 31, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D. The effects of alignment quality, distance calculation method, sequence filtering, and region on the analysis of 16S rRNA gene-based studies. PLoS Comput. Biol. 2010, 6, e1000844. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Barlund, C.S.; Carruthers, T.D.; Waldner, C.L.; Palmer, C.W. A comparison of diagnostic techniques for postpartum endometritis in dairy cattle. Theriogenology 2008, 69, 714–723. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Jansson, J.K.; Knight, R. The Earth Microbiome project: Successes and aspirations. BMC Biol. 2014, 12, 69. [Google Scholar] [CrossRef]
- Walther-Antonio, M.R.; Jeraldo, P.; Berg Miller, M.E.; Yeoman, C.J.; Nelson, K.E.; Wilson, B.A.; White, B.A.; Chia, N.; Creedon, D.J. Pregnancy’s stronghold on the vaginal microbiome. PLoS ONE 2014, 9, e98514. [Google Scholar] [CrossRef] [PubMed]
- MacIntyre, D.A.; Chandiramani, M.; Lee, Y.S.; Kindinger, L.; Smith, A.; Angelopoulos, N.; Lehne, B.; Arulkumaran, S.; Brown, R.; Teoh, T.G.; et al. The vaginal microbiome during pregnancy and the postpartum period in a European population. Sci. Rep. 2015, 5, 8988. [Google Scholar] [CrossRef]
- Bezirtzoglou, E.; Voidarou, C.; Papadaki, A.; Tsiotsias, A.; Kotsovolou, O.; Konstandi, M. Hormone therapy alters the composition of the vaginal microflora in ovariectomized rats. Microb. Ecol. 2008, 55, 751–759. [Google Scholar] [CrossRef]
- Nikodemova, M.; Holzhausen, E.A.; Deblois, C.L.; Barnet, J.H.; Peppard, P.E.; Suen, G.; Malecki, K.M. The effect of low-abundance OTU filtering methods on the reliability and variability of microbial composition assessed by 16S rRNA amplicon sequencing. Front. Cell Infect. Microbiol. 2023, 13, 1165295. [Google Scholar] [CrossRef]
- Ramette, A. Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 2007, 62, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Oren, A.; Garrity, G.M. Valid publication of the names of forty-two phyla of prokaryotes. Int. J. Syst. Evol. Microbiol. 2021, 71, 005056. [Google Scholar] [CrossRef] [PubMed]
- Estrada-de Los Santos, P.; Palmer, M.; Chavez-Ramirez, B.; Beukes, C.; Steenkamp, E.T.; Briscoe, L.; Khan, N.; Maluk, M.; Lafos, M.; Humm, E.; et al. Whole Genome Analyses Suggests that Burkholderia sensu lato Contains Two Additional Novel Genera (Mycetohabitans gen. nov., and Trinickia gen. nov.): Implications for the Evolution of Diazotrophy and Nodulation in the Burkholderiaceae. Genes 2018, 9, 389. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Zhao, B.N.; Guo, X.Y.; Wu, K.C.; Qiu, L.H. Trinickia mobilis sp. nov. and Trinickia acidisoli sp. nov., isolated from soil. Int. J. Syst. Evol. Microbiol. 2023, 73, 005941. [Google Scholar] [CrossRef]
- Adnane, M.; Chapwanya, A. A Review of the Diversity of the Genital Tract Microbiome and Implications for Fertility of Cattle. Animals 2022, 12, 460. [Google Scholar] [CrossRef]
- Movassagh, M.; Bebell, L.M.; Burgoine, K.; Hehnly, C.; Zhang, L.; Moran, K.; Sheldon, K.; Sinnar, S.A.; Mbabazi-Kabachelor, E.; Kumbakumba, E.; et al. Vaginal microbiome topic modeling of laboring Ugandan women with and without fever. NPJ Biofilms Microbiomes 2021, 7, 75. [Google Scholar] [CrossRef]
- Florova, V.; Romero, R.; Tarca, A.L.; Galaz, J.; Motomura, K.; Ahmad, M.M.; Hsu, C.D.; Hsu, R.; Tong, A.; Ravel, J.; et al. Vaginal host immune-microbiome interactions in a cohort of primarily African-American women who ultimately underwent spontaneous preterm birth or delivered at term. Cytokine 2021, 137, 155316. [Google Scholar] [CrossRef] [PubMed]
- Koziol, J.H.; Sheets, T.; Wickware, C.L.; Johnson, T.A. Composition and diversity of the seminal microbiota in bulls and its association with semen parameters. Theriogenology 2022, 182, 17–25. [Google Scholar] [CrossRef]
- Kumar, B.; Lorusso, E.; Fosso, B.; Pesole, G. A comprehensive overview of microbiome data in the light of machine learning applications: Categorization, accessibility, and future directions. Front. Microbiol. 2024, 15, 1343572. [Google Scholar] [CrossRef]
- Kim, C.; Pongpanich, M.; Porntaveetus, T. Unraveling metagenomics through long-read sequencing: A comprehensive review. J. Transl. Med. 2024, 22, 111. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Handelsman, J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 2005, 71, 1501–1506. [Google Scholar] [CrossRef]
- Massello, F.L.; Donati, E. Effect of heavy metal-induced stress on two extremophilic microbial communities from Caviahue-Copahue, Argentina. Environ. Pollut. 2021, 268, 115709. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L. Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl. Environ. Microbiol. 2011, 77, 3219–3226. [Google Scholar] [CrossRef]
- Huse, S.M.; Welch, D.M.; Morrison, H.G.; Sogin, M.L. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 2010, 12, 1889–1898. [Google Scholar] [CrossRef]
- Nebel, M.; Pfabel, C.; Stock, A.; Dunthorn, M.; Stoeck, T. Delimiting operational taxonomic units for assessing ciliate environmental diversity using small-subunit rRNA gene sequences. Environ. Microbiol. Rep. 2011, 3, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Druker, S.A.; Sicsic, R.; van Straten, M.; Goshen, T.; Kedmi, M.; Raz, T. Cytological endometritis diagnosis in primiparous versus multiparous dairy cows. J. Dairy. Sci. 2022, 105, 665–683. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Taguchi, Y.H.; Oono, Y. Relational patterns of gene expression via non-metric multidimensional scaling analysis. Bioinformatics 2005, 21, 730–740. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Druker, S.; Sicsic, R.; Ravid, S.; Scheinin, S.; Raz, T. Reproductive Tract Microbial Transitions from Late Gestation to Early Postpartum Using 16S rRNA Metagenetic Profiling in First-Pregnancy Heifers. Int. J. Mol. Sci. 2024, 25, 9164. https://doi.org/10.3390/ijms25179164
Druker S, Sicsic R, Ravid S, Scheinin S, Raz T. Reproductive Tract Microbial Transitions from Late Gestation to Early Postpartum Using 16S rRNA Metagenetic Profiling in First-Pregnancy Heifers. International Journal of Molecular Sciences. 2024; 25(17):9164. https://doi.org/10.3390/ijms25179164
Chicago/Turabian StyleDruker, Shaked, Ron Sicsic, Shachar Ravid, Shani Scheinin, and Tal Raz. 2024. "Reproductive Tract Microbial Transitions from Late Gestation to Early Postpartum Using 16S rRNA Metagenetic Profiling in First-Pregnancy Heifers" International Journal of Molecular Sciences 25, no. 17: 9164. https://doi.org/10.3390/ijms25179164
APA StyleDruker, S., Sicsic, R., Ravid, S., Scheinin, S., & Raz, T. (2024). Reproductive Tract Microbial Transitions from Late Gestation to Early Postpartum Using 16S rRNA Metagenetic Profiling in First-Pregnancy Heifers. International Journal of Molecular Sciences, 25(17), 9164. https://doi.org/10.3390/ijms25179164