
Heme Oxygenase-1 and Prostate Cancer: Function, Regulation, and Implication in Cancer Therapy
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
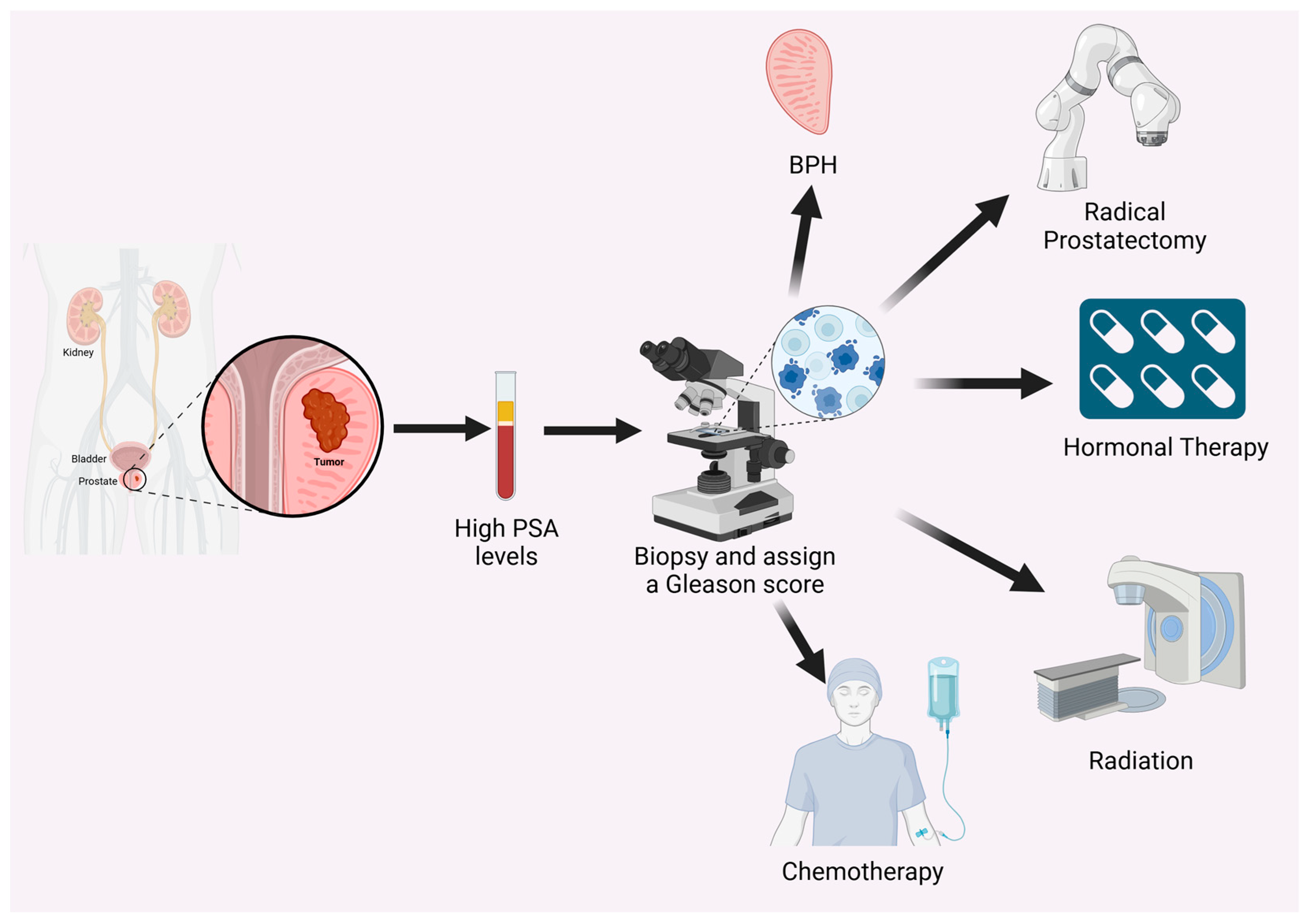
:1. Introduction to PC
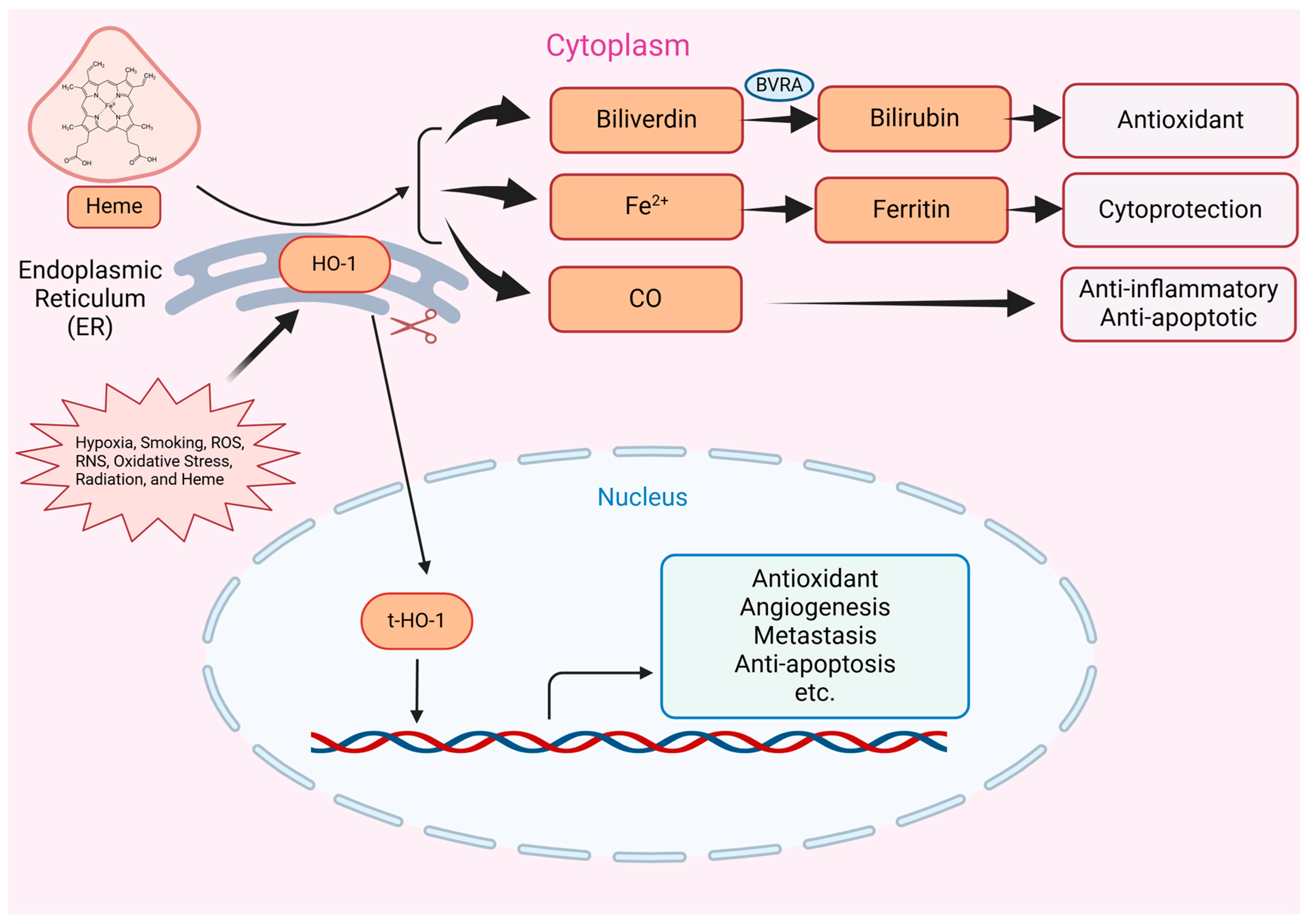
2. Introduction to HO-1
3. Regulation of HO-1 in PC
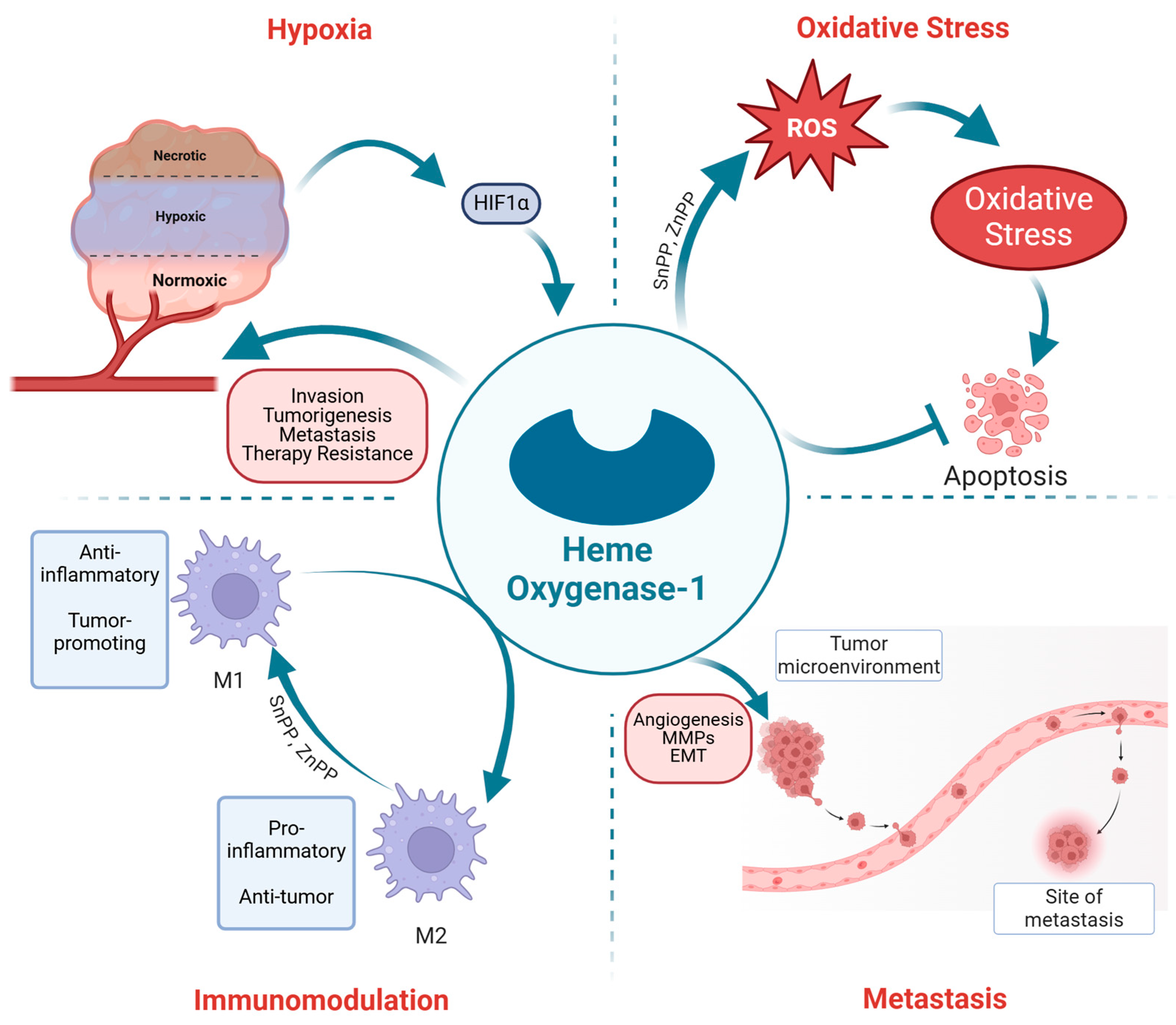
4. Role of HO-1 in PC Progression: Hypoxia
5. The Interplay of HO-1, Oxidative Stress, and PC
6. HO-1 and PC Metastasis
7. HO-1 and Therapy Resistance in PC
8. Immunomodulation by HO-1 in PC
9. Targeting HO-1 as a Therapeutic Strategy
10. Future Perspectives and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef]
- Panigrahi, G.K.; Praharaj, P.P.; Kittaka, H.; Mridha, A.R.; Black, O.M.; Singh, R.; Mercer, R.; van Bokhoven, A.; Torkko, K.C.; Agarwal, C.; et al. Exosome proteomic analyses identify inflammatory phenotype and novel biomarkers in African American prostate cancer patients. Cancer Med. 2019, 8, 1110–1123. [Google Scholar] [CrossRef] [PubMed]
- Shore, N.D.; Morgans, A.K.; El-Haddad, G.; Srinivas, S.; Abramowitz, M. Addressing Challenges and Controversies in the Management of Prostate Cancer with Multidisciplinary Teams. Target. Oncol. 2022, 17, 709–725. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.H. The Prostate Gland: A Review of its Anatomy, Pathology, and Treatment. JAMA 2014, 312, 562. [Google Scholar] [CrossRef]
- Litwin, M.S.; Tan, H.-J. The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef]
- Berenguer, C.V.; Pereira, F.; Câmara, J.S.; Pereira, J.A.M. Underlying Features of Prostate Cancer-Statistics, Risk Factors, and Emerging Methods for Its Diagnosis. Curr. Oncol. 2023, 30, 2300–2321. [Google Scholar] [CrossRef] [PubMed]
- Leitzmann, M.F.; Rohrmann, S. Risk factors for the onset of prostatic cancer: Age, location, and behavioral correlates. Clin. Epidemiol. 2012, 4, 1–11. [Google Scholar] [CrossRef]
- Oskar, B.; Kelly, R.P.; Konstantina, M.; Jonathan, F.; Sean, F.M.; Ola, B.; Freddie, B.; Otis, B.; Amy, N.L.; Lorelei, M.; et al. 2022 Update on Prostate Cancer Epidemiology and Risk Factors—A Systematic Review. Eur. Urol. 2023, 84, 191–206. [Google Scholar] [CrossRef]
- Giorgio, G.; Riccardo, L.; Freddie, B.; Neil, F.; Stephen, J.F.; Adam, K.; Pär, S.; Hendrick, C. Epidemiology and Prevention of Prostate Cancer. Eur. Urol. Oncol. 2021, 4, 877–892. [Google Scholar] [CrossRef]
- Ilic, D.; Djulbegovic, M.; Jung, J.H.; Hwang, E.C.; Zhou, Q.; Cleves, A.; Agoritsas, T.; Dahm, P. Prostate cancer screening with prostate-specific antigen (PSA) test: A systematic review and meta-analysis. BMJ 2018, 362, k3519. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.T. Assay for prostate specific antigen (PSA): Problems and possible solutions. J. Clin. Lab. Anal. 1994, 8, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Fenton, J.J.; Weyrich, M.S.; Durbin, S.; Liu, Y.; Bang, H.; Melnikow, J.U.S. Preventive Services Task Force Evidence Syntheses, formerly Systematic Evidence Reviews. In Prostate-Specific Antigen-Based Screening for Prostate Cancer: A Systematic Evidence Review for the U.S. Preventive Services Task Force; Agency for Healthcare Research and Quality: Rockville, MD, USA, 2018. [Google Scholar]
- Gleason, D.F. Classification of prostatic carcinomas. Cancer Chemother. Rep. 1966, 50, 125–128. [Google Scholar] [PubMed]
- Qiu, J.; Cai, D.; Wang, Z.; Zhou, J.; Gong, Y.; Cai, L.; Gong, K. Prognostic Models for Patients With Gleason Score 9 Prostate Cancer: A Population-Based Study. Front. Oncol. 2021, 11, 633312. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.K.; Gray, K.P.; Nakabayashi, M.; Evan, C.; Kantoff, P.W.; Huang, J.; Galsky, M.D.; Pomerantz, M.; Oh, W.K. Patients with Biopsy Gleason 9 and 10 Prostate Cancer Have Significantly Worse Outcomes Compared to Patients with Gleason 8 Disease. J. Urol. 2015, 194, 91–97. [Google Scholar] [CrossRef]
- Chen, F.Z.; Zhao, X.K. Prostate cancer: Current treatment and prevention strategies. Iran. Red Crescent Med. J. 2013, 15, 279–284. [Google Scholar] [CrossRef]
- Komura, K.; Sweeney, C.J.; Inamoto, T.; Ibuki, N.; Azuma, H.; Kantoff, P.W. Current treatment strategies for advanced prostate cancer. Int. J. Urol. 2018, 25, 220–231. [Google Scholar] [CrossRef]
- Coutinho, I.; Day, T.K.; Tilley, W.D.; Selth, L.A. Androgen receptor signaling in castration-resistant prostate cancer: A lesson in persistence. Endocr. Relat. Cancer 2016, 23, T179–T197. [Google Scholar] [CrossRef]
- Leung, D.K.; Chiu, P.K.; Ng, C.F.; Teoh, J.Y. Novel Strategies for Treating Castration-Resistant Prostate Cancer. Biomedicines 2021, 9, 339. [Google Scholar] [CrossRef]
- Posdzich, P.; Darr, C.; Hilser, T.; Wahl, M.; Herrmann, K.; Hadaschik, B.; Grünwald, V. Metastatic Prostate Cancer-A Review of Current Treatment Options and Promising New Approaches. Cancers 2023, 15, 461. [Google Scholar] [CrossRef]
- Taylor, J.M.; Chen, V.E.; Miller, R.C.; Greenberger, B.A. The Impact of Prostate Cancer Treatment on Quality of Life: A Narrative Review with a Focus on Randomized Data. Res. Rep. Urol. 2020, 12, 533–546. [Google Scholar] [CrossRef]
- Simon, N.I.; Parker, C.; Hope, T.A.; Paller, C.J. Best Approaches and Updates for Prostate Cancer Biochemical Recurrence. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Kapoor, A.; Gu, Y.; Chow, M.J.; Xu, H.; Major, P.; Tang, D. Assessment of biochemical recurrence of prostate cancer (Review). Int. J. Oncol. 2019, 55, 1194–1212. [Google Scholar] [CrossRef]
- Garcia-Marques, F.; Liu, S.; Totten, S.M.; Bermudez, A.; Tanimoto, C.; Hsu, E.C.; Nolley, R.; Hembree, A.; Stoyanova, T.; Brooks, J.D.; et al. Protein signatures to distinguish aggressive from indolent prostate cancer. Prostate 2022, 82, 605–616. [Google Scholar] [CrossRef]
- Seetharaman, A.; Bhattacharya, I.; Chen, L.C.; Kunder, C.A.; Shao, W.; Soerensen, S.J.C.; Wang, J.B.; Teslovich, N.C.; Fan, R.E.; Ghanouni, P.; et al. Automated detection of aggressive and indolent prostate cancer on magnetic resonance imaging. Med. Phys. 2021, 48, 2960–2972. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W. Heme Oxgenase-1, a Cardinal Modulator of Regulated Cell Death and Inflammation. Cells 2021, 10, 515. [Google Scholar] [CrossRef]
- Ryter, S.W. Heme Oxygenase-1: An Anti-Inflammatory Effector in Cardiovascular, Lung, and Related Metabolic Disorders. Antioxidants 2022, 11, 555. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Omata, Y.; Sakamoto, H.; Higashimoto, Y.; Hara, T.; Sagara, Y.; Noguchi, M. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene 2004, 336, 241–250. [Google Scholar] [CrossRef]
- Luu Hoang, K.N.; Anstee, J.E.; Arnold, J.N. The Diverse Roles of Heme Oxygenase-1 in Tumor Progression. Front. Immunol. 2021, 12, 658315. [Google Scholar] [CrossRef]
- Naito, Y.; Takagi, T.; Higashimura, Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch. Biochem. Biophys. 2014, 564, 83–88. [Google Scholar] [CrossRef]
- Halin Bergström, S.; Nilsson, M.; Adamo, H.; Thysell, E.; Jernberg, E.; Stattin, P.; Widmark, A.; Wikström, P.; Bergh, A. Extratumoral Heme Oxygenase-1 (HO-1) Expressing Macrophages Likely Promote Primary and Metastatic Prostate Tumor Growth. PLoS ONE 2016, 11, e0157280. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. The Role of HO-1 and Its Crosstalk with Oxidative Stress in Cancer Cell Survival. Cells 2021, 10, 2401. [Google Scholar] [CrossRef] [PubMed]
- Kiening, M.; Lange, N. A Recap of Heme Metabolism towards Understanding Protoporphyrin IX Selectivity in Cancer Cells. Int. J. Mol. Sci. 2022, 23, 7974. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-J.; Zhang, S. Heme-regulated eIF2α kinase in erythropoiesis and hemoglobinopathies. Blood 2019, 134, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; De La Cruz, L.K.; Yang, X.; Wang, B. Carbon Monoxide Signaling: Examining Its Engagement with Various Molecular Targets in the Context of Binding Affinity, Concentration, and Biologic Response. Pharmacol. Rev. 2022, 74, 823–873. [Google Scholar] [CrossRef]
- Adin, C.A. Bilirubin as a Therapeutic Molecule: Challenges and Opportunities. Antioxidants 2021, 10, 1536. [Google Scholar] [CrossRef]
- Galy, B.; Conrad, M.; Muckenthaler, M. Mechanisms controlling cellular and systemic iron homeostasis. Nat. Rev. Mol. Cell Biol. 2024, 25, 133–155. [Google Scholar] [CrossRef]
- Kim, K.M.; Pae, H.-O.; Zheng, M.; Park, R.; Kim, Y.-M.; Chung, H.-T. Carbon Monoxide Induces Heme Oxygenase-1 via Activation of Protein Kinase R–Like Endoplasmic Reticulum Kinase and Inhibits Endothelial Cell Apoptosis Triggered by Endoplasmic Reticulum Stress. Circ. Res. 2007, 101, 919–927. [Google Scholar] [CrossRef]
- Migita, C.T.; Matera, K.M.; Ikeda-Saito, M.; Olson, J.S.; Fujii, H.; Yoshimura, T.; Zhou, H.; Yoshida, T. The Oxygen and Carbon Monoxide Reactions of Heme Oxygenase. J. Biol. Chem. 1998, 273, 945–949. [Google Scholar] [CrossRef]
- Matera, K.M.; Takahashi, S.; Fujii, H.; Zhou, H.; Ishikawa, K.; Yoshimura, T.; Rousseau, D.L.; Yoshida, T.; Ikeda-Saito, M. Oxygen and one reducing equivalent are both required for the conversion of alpha-hydroxyhemin to verdoheme in heme oxygenase. J. Biol. Chem. 1996, 271, 6618–6624. [Google Scholar] [CrossRef]
- Sugishima, M.; Moffat, K.; Noguchi, M. Discrimination between CO and O2 in heme oxygenase: Comparison of static structures and dynamic conformation changes following CO photolysis. Biochemistry 2012, 51, 8554–8562. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, W. The Nuclear Translocation of Heme Oxygenase-1 in Human Diseases. Front. Cell Dev. Biol. 2022, 10, 890186. [Google Scholar] [CrossRef] [PubMed]
- Chau, L.-Y. Heme oxygenase-1: Emerging target of cancer therapy. J. Biomed. Sci. 2015, 22, 22. [Google Scholar] [CrossRef]
- Lin, Q.; Weis, S.; Yang, G.; Weng, Y.H.; Helston, R.; Rish, K.; Smith, A.; Bordner, J.; Polte, T.; Gaunitz, F.; et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J. Biol. Chem. 2007, 282, 20621–20633. [Google Scholar] [CrossRef] [PubMed]
- Vanella, L.; Barbagallo, I.; Tibullo, D.; Forte, S.; Zappalà, A.; Li Volti, G. The non-canonical functions of the heme oxygenases. Oncotarget 2016, 7, 69075–69086. [Google Scholar] [CrossRef]
- Hsu, F.F.; Chiang, M.T.; Li, F.A.; Yeh, C.T.; Lee, W.H.; Chau, L.Y. Acetylation is essential for nuclear heme oxygenase-1-enhanced tumor growth and invasiveness. Oncogene 2017, 36, 6805–6814. [Google Scholar] [CrossRef] [PubMed]
- Scaffa, A.; Tollefson, G.A.; Yao, H.; Rizal, S.; Wallace, J.; Oulhen, N.; Carr, J.F.; Hegarty, K.; Uzun, A.; Dennery, P.A. Identification of Heme Oxygenase-1 as a Putative DNA-Binding Protein. Antioxidants 2022, 11, 2135. [Google Scholar] [CrossRef]
- Mascaró, M.; Alonso, E.N.; Alonso, E.G.; Lacunza, E.; Curino, A.C.; Facchinetti, M.M. Nuclear Localization of Heme Oxygenase-1 in Pathophysiological Conditions: Does It Explain the Dual Role in Cancer? Antioxidants 2021, 10, 87. [Google Scholar] [CrossRef]
- Canesin, G.; Muralidharan, A.M.; Swanson, K.D.; Wegiel, B. HO-1 and Heme: G-Quadruplex Interaction Choreograph DNA Damage Responses and Cancer Growth. Cells 2021, 10, 1801. [Google Scholar] [CrossRef]
- Miyata, Y.; Kanda, S.; Mitsunari, K.; Asai, A.; Sakai, H. Heme oxygenase-1 expression is associated with tumor aggressiveness and outcomes in patients with bladder cancer: A correlation with smoking intensity. Transl. Res. 2014, 164, 468–476. [Google Scholar] [CrossRef]
- Muliaditan, T.; Opzoomer, J.W.; Caron, J.; Okesola, M.; Kosti, P.; Lall, S.; Van Hemelrijck, M.; Dazzi, F.; Tutt, A.; Grigoriadis, A.; et al. Repurposing Tin Mesoporphyrin as an Immune Checkpoint Inhibitor Shows Therapeutic Efficacy in Preclinical Models of Cancer. Clin. Cancer Res. 2018, 24, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Fang, J.; Liao, L.; Maeda, H.; Su, Q. Upregulation of heme oxygenase-1 in colorectal cancer patients with increased circulation carbon monoxide levels, potentially affects chemotherapeutic sensitivity. BMC Cancer 2014, 14, 436. [Google Scholar] [CrossRef]
- Fest, S.; Soldati, R.; Christiansen, N.M.; Zenclussen, M.L.; Kilz, J.; Berger, E.; Starke, S.; Lode, H.N.; Engel, C.; Zenclussen, A.C.; et al. Targeting of heme oxygenase-1 as a novel immune regulator of neuroblastoma. Int. J. Cancer 2016, 138, 2030–2042. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, B.; Gallo, D.; Csizmadia, E.; Harris, C.; Belcher, J.; Vercellotti, G.M.; Penacho, N.; Seth, P.; Sukhatme, V.; Ahmed, A.; et al. Carbon monoxide expedites metabolic exhaustion to inhibit tumor growth. Cancer Res. 2013, 73, 7009–7021. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.V.; Sapochnik, D.; Garcia Solá, M.; Coso, O. Regulation of the Expression of Heme Oxygenase-1: Signal Transduction, Gene Promoter Activation, and Beyond. Antioxid. Redox Signal. 2020, 32, 1033–1044. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Tong, K.I.; Kobayashi, A.; Katsuoka, F.; Yamamoto, M. Two-site substrate recognition model for the Keap1-Nrf2 system: A hinge and latch mechanism. Biol. Chem. 2006, 387, 1311–1320. [Google Scholar] [CrossRef]
- Tong, K.I.; Padmanabhan, B.; Kobayashi, A.; Shang, C.; Hirotsu, Y.; Yokoyama, S.; Yamamoto, M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol. Cell. Biol. 2007, 27, 7511–7521. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, S.; Jain, A.K.; Bloom, D.A.; Jaiswal, A.K. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J. Biol. Chem. 2005, 280, 16891–16900. [Google Scholar] [CrossRef]
- Reichard, J.F.; Motz, G.T.; Puga, A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res. 2007, 35, 7074–7086. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta. Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.L.; Sun, L.; Wang, Y.X.; Sun, B.H.; Li, Y.F.; Jin, Y.L. Association between HO-1 gene promoter polymorphisms and diseases (Review). Mol. Med. Rep. 2022, 25, 29. [Google Scholar] [CrossRef] [PubMed]
- Global, regional, and national burden of colorectal cancer and its risk factors, 1990-2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Gastroenterol. Hepatol. 2022, 7, 627–647. [CrossRef] [PubMed]
- Pezzuto, A.; Carico, E. Role of HIF-1 in Cancer Progression: Novel Insights. A Review. Curr. Mol. Med. 2018, 18, 343–351. [Google Scholar] [CrossRef]
- Maruyama, A.; Mimura, J.; Itoh, K. Non-coding RNA derived from the region adjacent to the human HO-1 E2 enhancer selectively regulates HO-1 gene induction by modulating Pol II binding. Nucleic Acids Res. 2014, 42, 13599–13614. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.D.; Chen, C.; Nguyen, J.; Thayanithy, V.; Subramanian, S.; Steer, C.J.; Vercellotti, G.M. Regulation of heme oxygenase-1 protein expression by miR-377 in combination with miR-217. J. Biol. Chem. 2011, 286, 3194–3202. [Google Scholar] [CrossRef]
- Skrzypek, K.; Tertil, M.; Golda, S.; Ciesla, M.; Weglarczyk, K.; Collet, G.; Guichard, A.; Kozakowska, M.; Boczkowski, J.; Was, H.; et al. Interplay between heme oxygenase-1 and miR-378 affects non-small cell lung carcinoma growth, vascularization, and metastasis. Antioxid. Redox Signal. 2013, 19, 644–660. [Google Scholar] [CrossRef]
- Gao, N.; Li, Y.; Li, J.; Gao, Z.; Yang, Z.; Li, Y.; Liu, H.; Fan, T. Long Non-Coding RNAs: The Regulatory Mechanisms, Research Strategies, and Future Directions in Cancers. Front. Oncol. 2020, 10, 598817. [Google Scholar] [CrossRef] [PubMed]
- Bolton, E.M.; Tuzova, A.V.; Walsh, A.L.; Lynch, T.; Perry, A.S. Noncoding RNAs in Prostate Cancer: The Long and the Short of It. Clin. Cancer Res. 2014, 20, 35–43. [Google Scholar] [CrossRef]
- Bu, T.; Li, L.; Tian, J. Unlocking the role of non-coding RNAs in prostate cancer progression: Exploring the interplay with the Wnt signaling pathway. Front. Pharmacol. 2023, 14, 1269233. [Google Scholar] [CrossRef]
- Zinatizadeh, M.R.; Schock, B.; Chalbatani, G.M.; Zarandi, P.K.; Jalali, S.A.; Miri, S.R. The Nuclear Factor Kappa B (NF-kB) signaling in cancer development and immune diseases. Genes. Dis. 2021, 8, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Cook, J.L. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am. J. Respir. Cell Mol. Biol. 2007, 36, 166–174. [Google Scholar] [CrossRef]
- Campbell, N.K.; Fitzgerald, H.K.; Dunne, A. Regulation of inflammation by the antioxidant haem oxygenase 1. Nat. Rev. Immunol. 2021, 21, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-M.; Lin, C.-C.; Yang, C.-C.; Cho, R.-L.; Hsiao, L.-D. Mevastatin-Induced AP-1-Dependent HO-1 Expression Suppresses Vascular Cell Adhesion Molecule-1 Expression and Monocyte Adhesion on Human Pulmonary Alveolar Epithelial Cells Challenged with TNF-α. Biomolecules 2020, 10, 381. [Google Scholar] [CrossRef]
- Seo, S.H.; Jeong, G.S. Fisetin inhibits TNF-α-induced inflammatory action and hydrogen peroxide-induced oxidative damage in human keratinocyte HaCaT cells through PI3K/AKT/Nrf-2-mediated heme oxygenase-1 expression. Int. Immunopharmacol. 2015, 29, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.L.; Lin, W.N.; Wang, C.Y.; Yang, C.C.; Hsiao, L.D.; Lin, C.C.; Yang, C.M. Heme oxygenase-1 induction by rosiglitazone via PKCα/AMPKα/p38 MAPKα/SIRT1/PPARγ pathway suppresses lipopolysaccharide-mediated pulmonary inflammation. Biochem. Pharmacol. 2018, 148, 222–237. [Google Scholar] [CrossRef]
- Salloom, R.J.; Ahmad, I.M.; Abdalla, M.Y. Targeting heme degradation pathway augments prostate cancer cell sensitivity to docetaxel-induced apoptosis and attenuates migration. Front. Oncol. 2024, 14, 1431362. [Google Scholar] [CrossRef]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef]
- Mohamed, O.A.A.; Tesen, H.S.; Hany, M.; Sherif, A.; Abdelwahab, M.M.; Elnaggar, M.H. The role of hypoxia on prostate cancer progression and metastasis. Mol. Biol. Rep. 2023, 50, 3873–3884. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Fraga, A.; Ribeiro, R.; Príncipe, P.; Lopes, C.; Medeiros, R. Hypoxia and Prostate Cancer Aggressiveness: A Tale With Many Endings. Clin. Genitourin. Cancer 2015, 13, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.D.; Ross, J.A.; McLaren, D.B.; Parker, C.C.; Habib, F.K.; Riddick, A.C. The relevance of a hypoxic tumour microenvironment in prostate cancer. BJU Int. 2010, 105, 8–13. [Google Scholar] [CrossRef]
- Muñoz-Sánchez, J.; Chánez-Cárdenas, M.E. A review on hemeoxygenase-2: Focus on cellular protection and oxygen response. Oxid. Med. Cell. Longev. 2014, 2014, 604981. [Google Scholar] [CrossRef]
- Neubauer, J.A.; Sunderram, J. Heme oxygenase-1 and chronic hypoxia. Respir. Physiol. Neurobiol. 2012, 184, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.J.; Jiang, B.H.; Chin, B.Y.; Iyer, N.V.; Alam, J.; Semenza, G.L.; Choi, A.M. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J. Biol. Chem. 1997, 272, 5375–5381. [Google Scholar] [CrossRef]
- Kitamuro, T.; Takahashi, K.; Ogawa, K.; Udono-Fujimori, R.; Takeda, K.; Furuyama, K.; Nakayama, M.; Sun, J.; Fujita, H.; Hida, W.; et al. Bach1 functions as a hypoxia-inducible repressor for the heme oxygenase-1 gene in human cells. J. Biol. Chem. 2003, 278, 9125–9133. [Google Scholar] [CrossRef]
- Linnenbaum, M.; Busker, M.; Kraehling, J.R.; Behrends, S. Heme oxygenase isoforms differ in their subcellular trafficking during hypoxia and are differentially modulated by cytochrome P450 reductase. PLoS ONE 2012, 7, e35483. [Google Scholar] [CrossRef]
- Mbenza, N.M.; Nasarudin, N.; Vadakkedath, P.G.; Patel, K.; Ismail, A.Z.; Hanif, M.; Wright, L.J.; Sarojini, V.; Hartinger, C.G.; Leung, I.K.H. Carbon Monoxide is an Inhibitor of HIF Prolyl Hydroxylase Domain 2. ChemBioChem 2021, 22, 2521–2525. [Google Scholar] [CrossRef]
- Choi, Y.K.; Kim, C.K.; Lee, H.; Jeoung, D.; Ha, K.S.; Kwon, Y.G.; Kim, K.W.; Kim, Y.M. Carbon monoxide promotes VEGF expression by increasing HIF-1alpha protein level via two distinct mechanisms, translational activation and stabilization of HIF-1alpha protein. J. Biol. Chem. 2010, 285, 32116–32125. [Google Scholar] [CrossRef] [PubMed]
- Chin, B.Y.; Jiang, G.; Wegiel, B.; Wang, H.J.; Macdonald, T.; Zhang, X.C.; Gallo, D.; Cszimadia, E.; Bach, F.H.; Lee, P.J.; et al. Hypoxia-inducible factor 1alpha stabilization by carbon monoxide results in cytoprotective preconditioning. Proc. Natl. Acad. Sci. USA 2007, 104, 5109–5114. [Google Scholar] [CrossRef] [PubMed]
- Telarovic, I.; Wenger, R.H.; Pruschy, M. Interfering with Tumor Hypoxia for Radiotherapy Optimization. J. Exp. Clin. Cancer Res. 2021, 40, 197. [Google Scholar] [CrossRef] [PubMed]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M.; et al. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: A novel approach to developing treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Liao, C.; Zhang, Q. Hypoxia-Driven Effects in Cancer: Characterization, Mechanisms, and Therapeutic Implications. Cells 2021, 10, 678. [Google Scholar] [CrossRef] [PubMed]
- Yotnda, P.; Wu, D.; Swanson, A.M. Hypoxic tumors and their effect on immune cells and cancer therapy. Methods Mol. Biol. 2010, 651, 1–29. [Google Scholar] [CrossRef]
- Hassan Venkatesh, G.; Abou Khouzam, R.; Shaaban Moustafa Elsayed, W.; Ahmed Zeinelabdin, N.; Terry, S.; Chouaib, S. Tumor hypoxia: An important regulator of tumor progression or a potential modulator of tumor immunogenicity? Oncoimmunology 2021, 10, 1974233. [Google Scholar] [CrossRef]
- Schöning, J.P.; Monteiro, M.; Gu, W. Drug resistance and cancer stem cells: The shared but distinct roles of hypoxia-inducible factors HIF1α and HIF2α. Clin. Exp. Pharmacol. Physiol. 2017, 44, 153–161. [Google Scholar] [CrossRef]
- Moitra, K.; Lou, H.; Dean, M. Multidrug efflux pumps and cancer stem cells: Insights into multidrug resistance and therapeutic development. Clin. Pharmacol. Ther. 2011, 89, 491–502. [Google Scholar] [CrossRef]
- Gupte, A.; Mumper, R.J. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 2009, 35, 32–46. [Google Scholar] [CrossRef]
- Riley, P.A. Free radicals in biology: Oxidative stress and the effects of ionizing radiation. Int. J. Radiat. Biol. 1994, 65, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, A.; Farhan, M.; Nabi, F.; Khan, R.H.; Adil, M.; Ahmad, A. Transcriptional Control of the Oxidative Stress Response and Implications of Using Plant Derived Molecules for Therapeutic Interventions in Cancer. Curr. Med. Chem. 2021, 28, 8480–8495. [Google Scholar] [CrossRef]
- Loreto Palacio, P.; Godoy, J.R.; Aktas, O.; Hanschmann, E.M. Changing Perspectives from Oxidative Stress to Redox Signaling-Extracellular Redox Control in Translational Medicine. Antioxidants 2022, 11, 1181. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E. Oxidative Stress and Cancer. Curr. Pharm. Des. 2018, 24, 4771–4778. [Google Scholar] [CrossRef] [PubMed]
- Oberley, L.W. Anticancer therapy by overexpression of superoxide dismutase. Antioxid. Redox Signal. 2001, 3, 461–472. [Google Scholar] [CrossRef]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef] [PubMed]
- Aebi, H. Catalase in vitro. Methods Enzym. 1984, 105, 121–126. [Google Scholar] [CrossRef]
- Shimizu, T.; Iwanaga, M.; Yasunaga, A.; Urata, Y.; Goto, S.; Shibata, S.; Kondo, T. Protective role of glutathione synthesis on radiation-induced DNA damage in rabbit brain. Cell. Mol. Neurobiol. 1998, 18, 299–310. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef]
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Invest. 1999, 103, 129–135. [Google Scholar] [CrossRef]
- Yachie, A. Heme Oxygenase-1 Deficiency and Oxidative Stress: A Review of 9 Independent Human Cases and Animal Models. Int. J. Mol. Sci. 2021, 22, 1514. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Wu, L.; Yuan, H.; Wang, J. ROS/Autophagy/Nrf2 Pathway Mediated Low-Dose Radiation Induced Radio-Resistance in Human Lung Adenocarcinoma A549 Cell. Int. J. Biol. Sci. 2015, 11, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, S.; Gai, J.; Guan, J.; Li, J.; Li, Y.; Zhao, J.; Zhao, C.; Fu, L.; Li, Q. SIRT5 Promotes Cisplatin Resistance in Ovarian Cancer by Suppressing DNA Damage in a ROS-Dependent Manner via Regulation of the Nrf2/HO-1 Pathway. Front. Oncol. 2019, 9, 754. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Fujino, M.; Takahara, T.; Li, X.K. Protective role of heme oxygenase-1 in fatty liver ischemia-reperfusion injury. Med. Mol. Morphol. 2019, 52, 61–72. [Google Scholar] [CrossRef]
- Abraham, N.G.; Lin, J.H.; Dunn, M.W.; Schwartzman, M.L. Presence of heme oxygenase and NADPH cytochrome P-450 (c) reductase in human corneal epithelium. Investig. Ophthalmol. Vis. Sci. 1987, 28, 1464–1472. [Google Scholar]
- Lee, P.J.; Alam, J.; Wiegand, G.W.; Choi, A.M. Overexpression of heme oxygenase-1 in human pulmonary epithelial cells results in cell growth arrest and increased resistance to hyperoxia. Proc. Natl. Acad. Sci. USA 1996, 93, 10393–10398. [Google Scholar] [CrossRef]
- Stocker, R.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Antioxidant activities of bile pigments: Biliverdin and bilirubin. Methods Enzym. 1990, 186, 301–309. [Google Scholar] [CrossRef]
- Baranano, D.E.; Rao, M.; Ferris, C.D.; Snyder, S.H. Biliverdin reductase: A major physiologic cytoprotectant. Proc. Natl. Acad. Sci. USA 2002, 99, 16093–16098. [Google Scholar] [CrossRef] [PubMed]
- Jansen, T.; Daiber, A. Direct Antioxidant Properties of Bilirubin and Biliverdin. Is there a Role for Biliverdin Reductase? Front. Pharmacol. 2012, 3, 30. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Brouard, S.; Otterbein, L.E.; Anrather, J.; Tobiasch, E.; Bach, F.H.; Choi, A.M.; Soares, M.P. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J. Exp. Med. 2000, 192, 1015–1026. [Google Scholar] [CrossRef]
- Gutteridge, J.M. Iron promoters of the Fenton reaction and lipid peroxidation can be released from haemoglobin by peroxides. FEBS Lett. 1986, 201, 291–295. [Google Scholar] [CrossRef]
- Birrane, G.; Li, H.; Yang, S.; Tachado, S.D.; Seng, S. Cigarette smoke induces nuclear translocation of heme oxygenase 1 (HO-1) in prostate cancer cells: Nuclear HO-1 promotes vascular endothelial growth factor secretion. Int. J. Oncol. 2013, 42, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, M.Y.; Ahmad, I.M.; Switzer, B.; Britigan, B.E. Induction of heme oxygenase-1 contributes to survival of Mycobacterium abscessus in human macrophages-like THP-1 cells. Redox Biol. 2015, 4, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.M.; Dafferner, A.J.; O’Connell, K.A.; Mehla, K.; Britigan, B.E.; Hollingsworth, M.A.; Abdalla, M.Y. Heme Oxygenase-1 Inhibition Potentiates the Effects of Nab-Paclitaxel-Gemcitabine and Modulates the Tumor Microenvironment in Pancreatic Ductal Adenocarcinoma. Cancers 2021, 13, 2264. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D.; Abrahamsson, P.A. Expression of heme oxygenase-1 (HSP32) in human prostate: Normal, hyperplastic, and tumor tissue distribution. Urology 1996, 47, 727–733. [Google Scholar] [CrossRef]
- Blann, A.D.; Balakrishnan, B.; Ryan, P.; Lip, G.Y. Increased levels of plasma haemoxygenase-1 in prostate cancer. Prostate Cancer Prostatic Dis. 2011, 14, 114–117. [Google Scholar] [CrossRef]
- Alaoui-Jamali, M.A.; Bismar, T.A.; Gupta, A.; Szarek, W.A.; Su, J.; Song, W.; Xu, Y.; Xu, B.; Liu, G.; Vlahakis, J.Z.; et al. A Novel Experimental Heme Oxygenase-1–Targeted Therapy for Hormone-Refractory Prostate Cancer. Cancer Res. 2009, 69, 8017–8024. [Google Scholar] [CrossRef]
- Li, Y.; Su, J.; DingZhang, X.; Zhang, J.; Yoshimoto, M.; Liu, S.; Bijian, K.; Gupta, A.; Squire, J.A.; Alaoui Jamali, M.A.; et al. PTEN deletion and heme oxygenase-1 overexpression cooperate in prostate cancer progression and are associated with adverse clinical outcome. J. Pathol. 2011, 224, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Sacca, P.; Meiss, R.; Casas, G.; Mazza, O.; Calvo, J.C.; Navone, N.; Vazquez, E. Nuclear translocation of haeme oxygenase-1 is associated to prostate cancer. Br. J. Cancer 2007, 97, 1683–1689. [Google Scholar] [CrossRef]
- Gueron, G.; De Siervi, A.; Ferrando, M.; Salierno, M.; De Luca, P.; Elguero, B.; Meiss, R.; Navone, N.; Vazquez, E.S. Critical role of endogenous heme oxygenase 1 as a tuner of the invasive potential of prostate cancer cells. Mol. Cancer Res. 2009, 7, 1745–1755. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.R.; Ingersoll, M.A.; Chou, Y.W.; Kosmacek, E.A.; Oberley-Deegan, R.E.; Lin, M.F. Dynamics of antioxidant heme oxygenase-1 and pro-oxidant p66Shc in promoting advanced prostate cancer progression. Free Radic. Biol. Med. 2022, 193, 274–291. [Google Scholar] [CrossRef]
- Nakayama, T.; Kobayashi, T.; Shimpei, O.; Fukuhara, H.; Namikawa, T.; Inoue, K.; Hanazaki, K.; Takahashi, K.; Nakajima, M.; Tanaka, T.; et al. Photoirradiation after aminolevulinic acid treatment suppresses cancer cell proliferation through the HO-1/p21 pathway. Photodiagnosis Photodyn. Ther. 2019, 28, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Podkalicka, P.; Mucha, O.; Józkowicz, A.; Dulak, J.; Łoboda, A. Heme oxygenase inhibition in cancers: Possible tools and targets. Contemp. Oncol. (Pozn.) 2018, 22, 23–32. [Google Scholar] [CrossRef]
- Romeo, G.; Ciaffaglione, V.; Amata, E.; Dichiara, M.; Calabrese, L.; Vanella, L.; Sorrenti, V.; Grosso, S.; D’Amico, A.G.; D’Agata, V.; et al. Combination of Heme Oxygenase-1 Inhibition and Sigma Receptor Modulation for Anticancer Activity. Molecules 2021, 26, 3860. [Google Scholar] [CrossRef] [PubMed]
- Mucha, O.; Podkalicka, P.; Mikulski, M.; Barwacz, S.; Andrysiak, K.; Biela, A.; Mieczkowski, M.; Kachamakova-Trojanowska, N.; Ryszawy, D.; Białas, A.; et al. Development and characterization of a new inhibitor of heme oxygenase activity for cancer treatment. Arch. Biochem. Biophys. 2019, 671, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Baldessari, C.; Pipitone, S.; Molinaro, E.; Cerma, K.; Fanelli, M.; Nasso, C.; Oltrecolli, M.; Pirola, M.; D’Agostino, E.; Pugliese, G.; et al. Bone Metastases and Health in Prostate Cancer: From Pathophysiology to Clinical Implications. Cancers 2023, 15, 1518. [Google Scholar] [CrossRef]
- Zhang, A.C.; Rasul, R.; Golden, A.; Feuerstein, M.A. Incidence and mortality trends of metastatic prostate cancer: Surveillance, Epidemiology, and End Results database analysis. Can. Urol. Assoc. J. 2021, 15, E637–E643. [Google Scholar] [CrossRef] [PubMed]
- Lange, J.M.; Laviana, A.A.; Penson, D.F.; Lin, D.W.; Bill-Axelson, A.; Carlsson, S.V.; Newcomb, L.F.; Trock, B.J.; Carter, H.B.; Carroll, P.R.; et al. Prostate cancer mortality and metastasis under different biopsy frequencies in North American active surveillance cohorts. Cancer 2020, 126, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Suzuki, T.; Adachi, S.; Naganuma, E.; Suzuki, N.; Hosoya, T.; Itoh, K.; Sporn, M.B.; Yamamoto, M. Distinct Regulations of HO-1 Gene Expression for Stress Response and Substrate Induction. Mol. Cell. Biol. 2021, 41, e0023621. [Google Scholar] [CrossRef]
- Ferrando, M.; Wan, X.; Meiss, R.; Yang, J.; De Siervi, A.; Navone, N.; Vazquez, E. Heme oxygenase-1 (HO-1) expression in prostate cancer cells modulates the oxidative response in bone cells. PLoS ONE 2013, 8, e80315. [Google Scholar] [CrossRef] [PubMed]
- Consoli, V.; Sorrenti, V.; Grosso, S.; Vanella, L. Heme Oxygenase-1 Signaling and Redox Homeostasis in Physiopathological Conditions. Biomolecules 2021, 11, 589. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Kim, Y.M. Regulation of Endothelial and Vascular Functions by Carbon Monoxide via Crosstalk With Nitric Oxide. Front. Cardiovasc. Med. 2021, 8, 649630. [Google Scholar] [CrossRef] [PubMed]
- Stefan, W.R.; Augustine, M.K.C. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl. Res. 2016, 167, 7–34. [Google Scholar] [CrossRef]
- Agnieszka, L.; Alicja, J.; Jozef, D. HO-1/CO system in tumor growth, angiogenesis and metabolism—Targeting HO-1 as an anti-tumor therapy. Vasc. Pharmacol. 2015, 74, 11–22. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef]
- Teleanu, R.I.; Chircov, C.; Grumezescu, A.M.; Teleanu, D.M. Tumor Angiogenesis and Anti-Angiogenic Strategies for Cancer Treatment. J. Clin. Med. 2019, 9, 84. [Google Scholar] [CrossRef]
- Tsai, C.F.; Chen, J.H.; Chang, C.N.; Lu, D.Y.; Chang, P.C.; Wang, S.L.; Yeh, W.L. Fisetin inhibits cell migration via inducing HO-1 and reducing MMPs expression in breast cancer cell lines. Food Chem. Toxicol. 2018, 120, 528–535. [Google Scholar] [CrossRef]
- Gong, Y.; Chippada-Venkata, U.D.; Oh, W.K. Roles of matrix metalloproteinases and their natural inhibitors in prostate cancer progression. Cancers 2014, 6, 1298–1327. [Google Scholar] [CrossRef]
- Goncharov, A.P.; Vashakidze, N.; Kharaishvili, G. Epithelial-Mesenchymal Transition: A Fundamental Cellular and Microenvironmental Process in Benign and Malignant Prostate Pathologies. Biomedicines 2024, 12, 418. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhao, J.; Xue, J.; Zhao, X.; Liu, P. Autophagy inhibition promotes epithelial-mesenchymal transition through ROS/HO-1 pathway in ovarian cancer cells. Am. J. Cancer Res. 2016, 6, 2162–2177. [Google Scholar] [PubMed]
- Adamo, H.H.; Strömvall, K.; Nilsson, M.; Halin Bergström, S.; Bergh, A. Adaptive (TINT) Changes in the Tumor Bearing Organ Are Related to Prostate Tumor Size and Aggressiveness. PLoS ONE 2015, 10, e0141601. [Google Scholar] [CrossRef]
- Trujillo, K.A.; Jones, A.C.; Griffith, J.K.; Bisoffi, M. Markers of field cancerization: Proposed clinical applications in prostate biopsies. Prostate Cancer 2012, 2012, 302894. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.; Gong, H.; Luo, S.; Cui, Y. The Role of Exosomes and Their Applications in Cancer. Int. J. Mol. Sci. 2021, 22, 12204. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhao, H.; Chen, Y.; Li, K.; Li, T.; Chen, J.; Zhang, B.; Guo, C.; Qing, L.; Shen, J.; et al. Exosomal long noncoding RNA HOXD-AS1 promotes prostate cancer metastasis via miR-361-5p/FOXM1 axis. Cell Death Dis. 2021, 12, 1129. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.L.; Chen, K.C.; Hsieh, J.T.; Shen, T.L. Exosomes in cancer development and clinical applications. Cancer Sci. 2018, 109, 2364–2374. [Google Scholar] [CrossRef]
- Dai, J.; Su, Y.; Zhong, S.; Cong, L.; Liu, B.; Yang, J.; Tao, Y.; He, Z.; Chen, C.; Jiang, Y. Exosomes: Key players in cancer and potential therapeutic strategy. Signal Transduct. Target. Ther. 2020, 5, 145. [Google Scholar] [CrossRef]
- Ben-Eltriki, M.; Gayle, E.J.; Walker, N.; Deb, S. Pharmacological Significance of Heme Oxygenase 1 in Prostate Cancer. Curr. Issues Mol. Biol. 2023, 45, 4301–4316. [Google Scholar] [CrossRef]
- Wang, H.; Cheng, Q.; Bao, L.; Li, M.; Chang, K.; Yi, X. Cytoprotective Role of Heme Oxygenase-1 in Cancer Chemoresistance: Focus on Antioxidant, Antiapoptotic, and Pro-Autophagy Properties. Antioxidants 2023, 12, 1217. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Abboud, K. Targeting the androgen receptor signaling pathway in advanced prostate cancer. Am. J. Health Syst. Pharm. 2022, 79, 1224–1235. [Google Scholar] [CrossRef]
- Huang, J.; Lin, B.; Li, B. Anti-Androgen Receptor Therapies in Prostate Cancer: A Brief Update and Perspective. Front. Oncol. 2022, 12, 865350. [Google Scholar] [CrossRef] [PubMed]
- Turco, F.; Gillessen, S.; Cathomas, R.; Buttigliero, C.; Vogl, U.M. Treatment Landscape for Patients with Castration-Resistant Prostate Cancer: Patient Selection and Unmet Clinical Needs. Res. Rep. Urol. 2022, 14, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Spratt, D.E.; Shore, N.; Sartor, O.; Rathkopf, D.; Olivier, K. Treating the patient and not just the cancer: Therapeutic burden in prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Mondal, D.; Narwani, D.; Notta, S.; Ghaffar, D.; Mardhekar, N.; Quadri, S.S.A. Oxidative stress and redox signaling in CRPC progression: Therapeutic potential of clinically-tested Nrf2-activators. Cancer Drug Resist. 2021, 4, 96–124. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.-h.; Liu, J.-z.; Wang, B.; Men, Q.-l.; Ju, Y.-q.; Yin, F.-y.; Zheng, C.; Li, W. KLF14 potentiates oxidative adaptation via modulating HO-1 signaling in castrate-resistant prostate cancer. Endocr.-Relat. Cancer 2019, 26, 181–195. [Google Scholar] [CrossRef]
- Bolla, M.; Henry, A.; Mason, M.; Wiegel, T. The role of radiotherapy in localised and locally advanced prostate cancer. Asian J. Urol. 2019, 6, 153–161. [Google Scholar] [CrossRef]
- Lyu, F.; Shang, S.-Y.; Gao, X.-S.; Ma, M.-W.; Xie, M.; Ren, X.-Y.; Liu, M.-Z.; Chen, J.-Y.; Li, S.-S.; Huang, L. Uncovering the Secrets of Prostate Cancer’s Radiotherapy Resistance: Advances in Mechanism Research. Biomedicines 2023, 11, 1628. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Zhang, W.; Tian, Y.; Sethi, G.; Zhang, X.; Qiu, A. Molecular panorama of therapy resistance in prostate cancer: A pre-clinical and bioinformatics analysis for clinical translation. Cancer Metastasis Rev. 2024, 43, 229–260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Qiao, T.; Zha, L. Inhibition of heme oxygenase-1 enhances the radiosensitivity in human nonsmall cell lung cancer a549 cells. Cancer Biother. Radiopharm. 2011, 26, 639–645. [Google Scholar] [CrossRef]
- Nitti, M.; Ortolan, J.; Furfaro, A.L. Role of heme oxygenase-1 in tumor immune escape. Redox Exp. Med. 2023, 2023, e230006. [Google Scholar] [CrossRef]
- Nemeth, Z.; Li, M.; Csizmadia, E.; Döme, B.; Johansson, M.; Liao Persson, J.; Seth, P.; Otterbein, L.; Wegiel, B. Heme oxygenase-1 in macrophages controls prostate cancer progression. Oncotarget 2015, 6, 33675. [Google Scholar] [CrossRef] [PubMed]
- Muliaditan, T.; Caron, J.; Okesola, M.; Opzoomer, J.W.; Kosti, P.; Georgouli, M.; Gordon, P.; Lall, S.; Kuzeva, D.M.; Pedro, L.; et al. Macrophages are exploited from an innate wound healing response to facilitate cancer metastasis. Nat. Commun. 2018, 9, 2951. [Google Scholar] [CrossRef]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Weis, N.; Weigert, A.; von Knethen, A.; Brüne, B. Heme oxygenase-1 contributes to an alternative macrophage activation profile induced by apoptotic cell supernatants. Mol. Biol. Cell 2009, 20, 1280–1288. [Google Scholar] [CrossRef]
- Alaluf, E.; Vokaer, B.; Detavernier, A.; Azouz, A.; Splittgerber, M.; Carrette, A.; Boon, L.; Libert, F.; Soares, M.; Le Moine, A.; et al. Heme oxygenase-1 orchestrates the immunosuppressive program of tumor-associated macrophages. JCI Insight 2020, 5, e133929. [Google Scholar] [CrossRef]
- Arnold, J.N.; Magiera, L.; Kraman, M.; Fearon, D.T. Tumoral immune suppression by macrophages expressing fibroblast activation protein-α and heme oxygenase-1. Cancer Immunol. Res. 2014, 2, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef]
- George, S.D.; Jeffrey, B.; Menachem, G.; David, L.; Nader, G.A. HO-1 overexpression and underexpression: Clinical implications. Arch. Biochem. Biophys. 2019, 673, 108073. [Google Scholar] [CrossRef]
- Alaoui-Jamali, M.; Szarek, W.; Nakatsu, K.; Vlahakis, J.; Gupta, A.; Schipper, H. OB-24, a novel selective and potent HO-1 inhibitor, induces a wide spectrum anti-tumor activity in vitro and in vivo and synergizes with chemotherapy drugs. Mol. Cancer Ther. 2007, 6, C82. [Google Scholar]
- Abdelrahim, M.; Safe, S.; Baker, C.; Abudayyeh, A. RNAi and cancer: Implications and applications. J. RNAi Gene Silenc. 2006, 2, 136–145. [Google Scholar]
- Kim, H.R.; Kim, S.; Kim, E.J.; Park, J.H.; Yang, S.H.; Jeong, E.T.; Park, C.; Youn, M.J.; So, H.S.; Park, R. Suppression of Nrf2-driven heme oxygenase-1 enhances the chemosensitivity of lung cancer A549 cells toward cisplatin. Lung Cancer 2008, 60, 47–56. [Google Scholar] [CrossRef]
- Paez, A.V.; Pallavicini, C.; Schuster, F.; Valacco, M.P.; Giudice, J.; Ortiz, E.G.; Anselmino, N.; Labanca, E.; Binaghi, M.; Salierno, M.; et al. Heme oxygenase-1 in the forefront of a multi-molecular network that governs cell-cell contacts and filopodia-induced zippering in prostate cancer. Cell Death Dis. 2016, 7, e2570. [Google Scholar] [CrossRef]
- Drummond, G.S.; Kappas, A. Chemoprevention of Neonatal Jaundice: Potency of Tin-Protoporphyrin in an Animal Model. Science 1982, 217, 1250–1252. [Google Scholar] [CrossRef]
- Fernández-Fierro, A.; Funes, S.C.; Rios, M.; Covián, C.; González, J.; Kalergis, A.M. Immune Modulation by Inhibitors of the HO System. Int. J. Mol. Sci. 2021, 22, 294. [Google Scholar] [CrossRef]
- Bhutani, V.K.; Poland, R.; Meloy, L.D.; Hegyi, T.; Fanaroff, A.A.; Maisels, M.J. Clinical trial of tin mesoporphyrin to prevent neonatal hyperbilirubinemia. J. Perinatol. 2016, 36, 533–539. [Google Scholar] [CrossRef]
- Costa, D.L.; Namasivayam, S.; Amaral, E.P.; Arora, K.; Chao, A.; Mittereder, L.R.; Maiga, M.; Boshoff, H.I.; Barry, C.E.; Goulding, C.W.; et al. Pharmacological Inhibition of Host Heme Oxygenase-1 Suppresses Mycobacterium tuberculosis Infection <i>In Vivo</i> by a Mechanism Dependent on T Lymphocytes. mBio 2016, 7, 10–1128. [Google Scholar] [CrossRef]
- Andrade, B.B.; Pavan Kumar, N.; Amaral, E.P.; Riteau, N.; Mayer-Barber, K.D.; Tosh, K.W.; Maier, N.; Conceição, E.L.; Kubler, A.; Sridhar, R.; et al. Heme Oxygenase-1 Regulation of Matrix Metalloproteinase-1 Expression Underlies Distinct Disease Profiles in Tuberculosis. J. Immunol. 2015, 195, 2763–2773. [Google Scholar] [CrossRef]
- Fernández-Mendívil, C.; Arreola, M.A.; Hohsfield, L.A.; Green, K.N.; Lopez, M.G. Aging and Progression of Beta-Amyloid Pathology in Alzheimer’s Disease Correlates with Microglial Heme-Oxygenase-1 Overexpression. Antioxidants 2020, 9, 644. [Google Scholar] [CrossRef] [PubMed]
- Stahnke, T.; Richter-Landsberg, C.; Stadelmann, C.; Netzler, A.; Brück, W. Differential upregulation of heme oxygenase-1 (HSP32) in glial cells after oxidative stress and in demyelinating disorders. J. Mol. Neurosci. 2007, 32, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Lilljebjörn, L.V.; Csizmadia, E.; Hedblom, A.; Canesin, G.; Kalbasi, A.; Li, M.; Kramer, F.; Bornfeldt, K.E.; Wegiel, B. A Role of the Heme Degradation Pathway in Shaping Prostate Inflammatory Responses and Lipid Metabolism. Am. J. Pathol. 2020, 190, 830–843. [Google Scholar] [CrossRef]
- Wu, B.; Wu, Y.; Tang, W. Heme Catabolic Pathway in Inflammation and Immune Disorders. Front. Pharmacol. 2019, 10, 825. [Google Scholar] [CrossRef]
- Tsai, J.R.; Wang, H.M.; Liu, P.L.; Chen, Y.H.; Yang, M.C.; Chou, S.H.; Cheng, Y.J.; Yin, W.H.; Hwang, J.J.; Chong, I.W. High expression of heme oxygenase-1 is associated with tumor invasiveness and poor clinical outcome in non-small cell lung cancer patients. Cell. Oncol. (Dordr.) 2012, 35, 461–471. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salloom, R.J.; Ahmad, I.M.; Sahtout, D.Z.; Baine, M.J.; Abdalla, M.Y. Heme Oxygenase-1 and Prostate Cancer: Function, Regulation, and Implication in Cancer Therapy. Int. J. Mol. Sci. 2024, 25, 9195. https://doi.org/10.3390/ijms25179195
Salloom RJ, Ahmad IM, Sahtout DZ, Baine MJ, Abdalla MY. Heme Oxygenase-1 and Prostate Cancer: Function, Regulation, and Implication in Cancer Therapy. International Journal of Molecular Sciences. 2024; 25(17):9195. https://doi.org/10.3390/ijms25179195
Chicago/Turabian StyleSalloom, Ramia J., Iman M. Ahmad, Dania Z. Sahtout, Michael J. Baine, and Maher Y. Abdalla. 2024. "Heme Oxygenase-1 and Prostate Cancer: Function, Regulation, and Implication in Cancer Therapy" International Journal of Molecular Sciences 25, no. 17: 9195. https://doi.org/10.3390/ijms25179195