
The Response of Murine Gut Microbiome in the Presence of Altered rpoS Gene of Klebsiella pneumoniae

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Most Dominant Taxa in Mice in the Presence of Wild-Type and Mutant of K. pneumoniae
2.2. Dominant Microbiota in the Healthy Mice
2.3. Presence of Dominant Microbiota in WT Kp-Treated Mice
2.4. Role of Microbiota in the Mutant Treated Mice
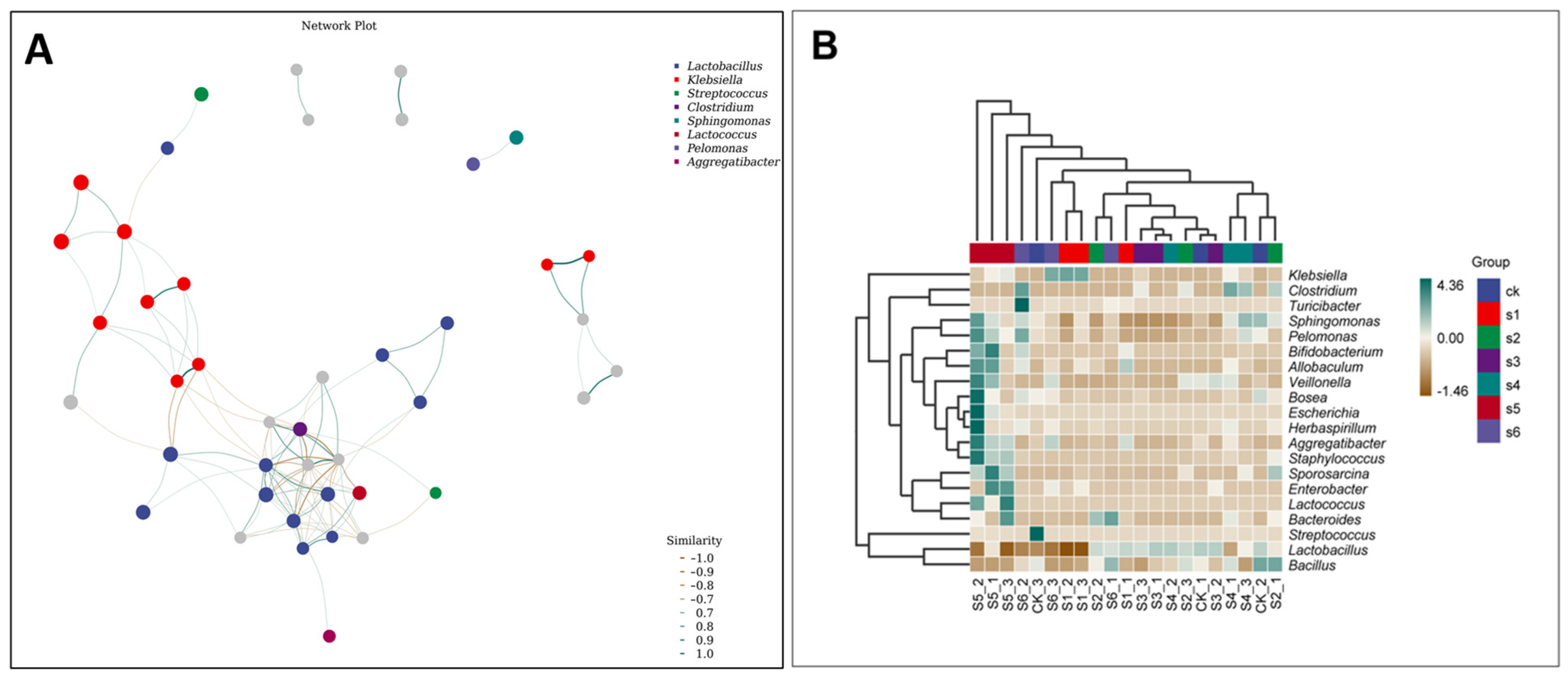
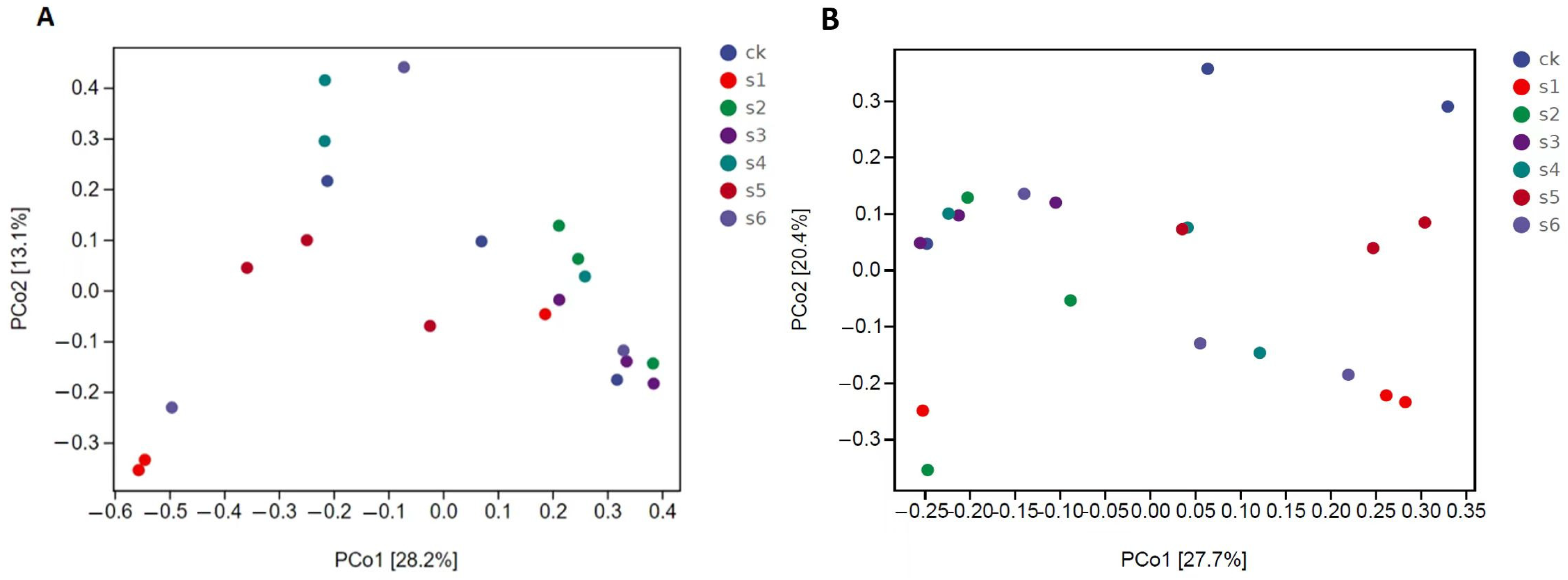
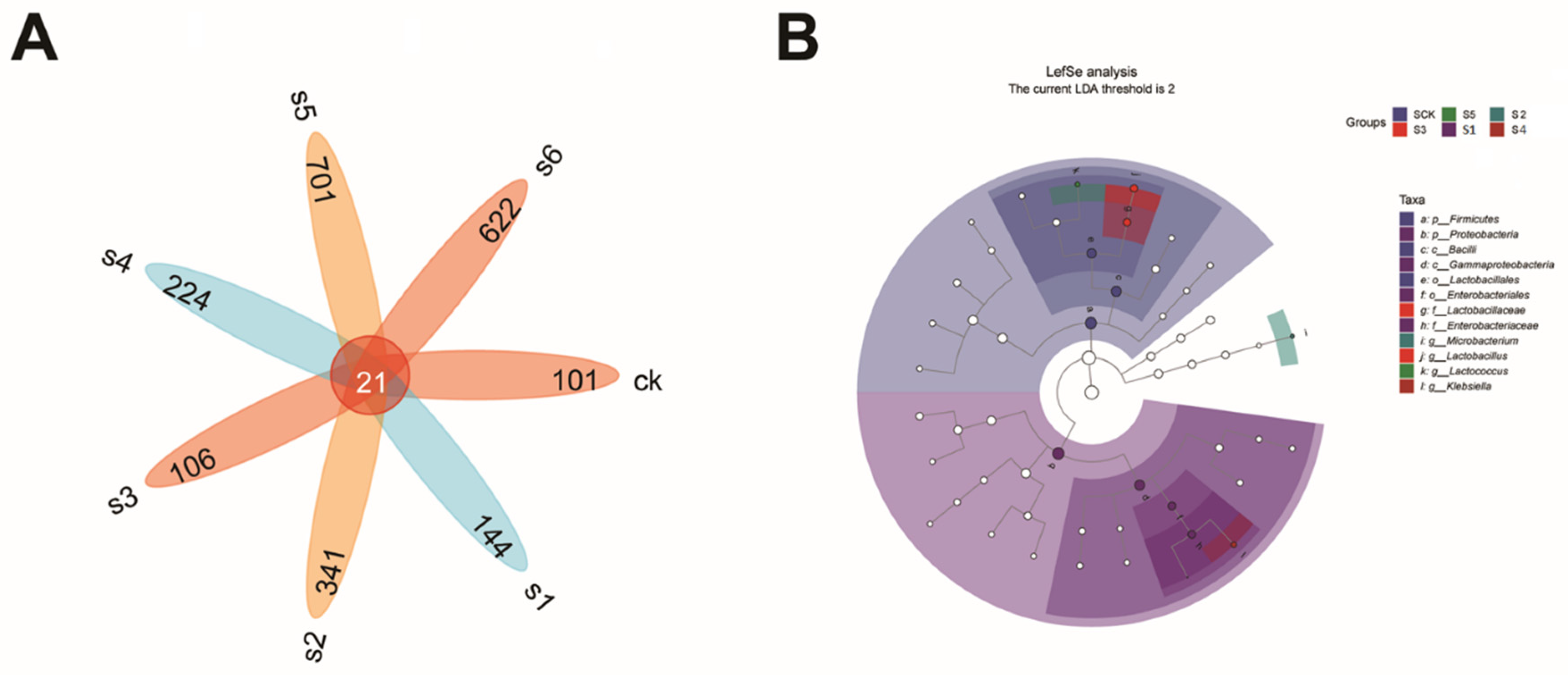
2.5. Microbial Communities’ Comparisons in Healthy, Wild and Mutant Mice
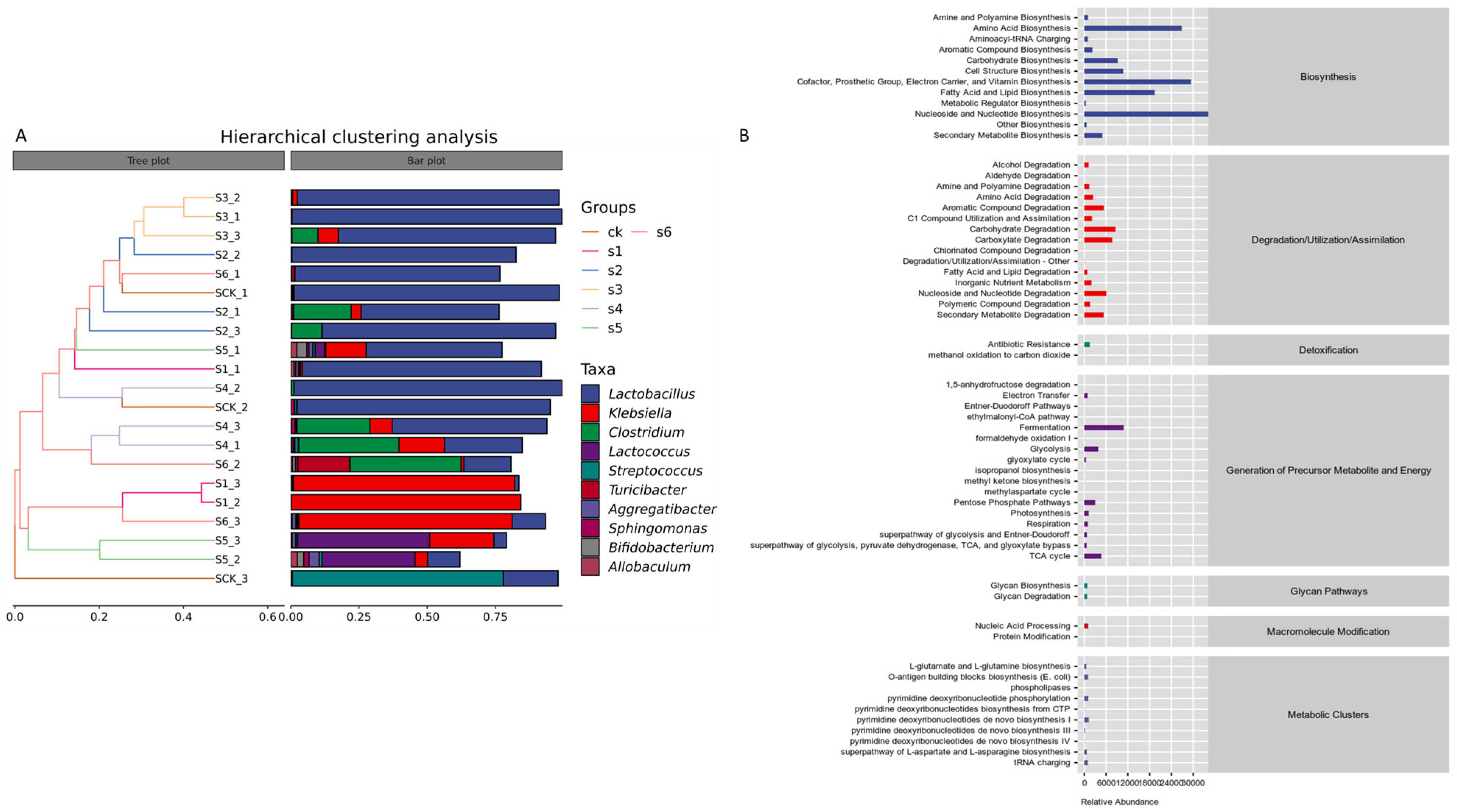
2.6. Functional Predictions of Bacterial Taxa in Healthy, Wild, and Mutant-Treated Mice
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Mice Collection
4.3. Experimental Setup
4.4. Gut-Colonization Model
4.5. Microbiome Analysis in the Presence of Wild-Type and Mutant Strain
4.6. 16S rRNA Gene Amplicon Sequencing
4.7. Sequence Analysis
4.8. Taxonomic Composition Analysis
4.9. Alpha and Beta Diversity Index Analysis
4.10. Analysis of Differentiation among Groupings at Each Taxonomic Level
4.11. Construction of Association Network and Prediction of Microbial Metabolic Functions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ward, N.L.; Pieretti, A.; Dowd, S.E.; Cox, S.B.; Goldstein, A.M. Intestinal aganglionosis is associated with early and sustained disruption of the colonic microbiome. Neurogastroenterol. Motil. 2012, 24, 874-e400. [Google Scholar] [CrossRef]
- Kang, D.-W.; Park, J.G.; Ilhan, Z.E.; Wallstrom, G.; LaBaer, J.; Adams, J.B.; Krajmalnik-Brown, R. Reduced incidence of Prevotella and other fermenters in intestinal microflora of autistic children. PLoS ONE 2013, 8, e68322. [Google Scholar] [CrossRef] [PubMed]
- Pascoe, E.L.; Hauffe, H.C.; Marchesi, J.R.; E Perkins, S. Network analysis of gut microbiota literature: An overview of the research landscape in non-human animal studies. ISME J. 2017, 11, 2644–2651. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, L.; Cai, J.; Zhu, H.; Li, J.; Yu, Y.; Xu, Y.; Shi, G.; Feng, Y. Clinical characteristics of respiratory tract infection caused by Klebsiella pneumoniae in immunocompromised patients: A retrospective cohort study. Front. Cell. Infect. Microbiol. 2023, 13, 1137664. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, S.; Zhan, L.; Jin, Y.; Duan, J.; Hao, Z.; Lv, J.; Qi, X.; Chen, L.; Kreiswirth, B.N.; et al. Microbiological and clinical characteristics of hypermucoviscous Klebsiella pneumoniae isolates associated with invasive infections in China. Front. Cell. Infect. Microbiol. 2017, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-R.; Lee, J.H.; Park, K.S.; Jeon, J.H.; Kim, Y.B.; Cha, C.-J.; Jeong, B.C.; Lee, S.H. Antimicrobial resistance of hypervirulent Klebsiella pneumoniae: Epidemiology, hypervirulence-associated determinants, and resistance mechanisms. Front. Cell. Infect. Microbiol. 2017, 7, 483. [Google Scholar] [CrossRef]
- Alexander, A.D.; Orcutt, R.P.; Henry, J.C.; Baker, J.; Bissahoyo, A.C.; Threadgill, D.W. Quantitative PCR assays for mouse enteric flora reveal strain-dependent differences in composition that are influenced by the microenvironment. Mamm. Genome 2006, 17, 1093–1104. [Google Scholar] [CrossRef]
- Bendtsen, K.M.B.; Krych, L.; Sørensen, D.B.; Pang, W.; Nielsen, D.S.; Josefsen, K.; Hansen, L.H.; Sørensen, S.J.; Hansen, A.K. Gut microbiota composition is correlated to grid floor induced stress and behavior in the BALB/c mouse. PLoS ONE 2012, 7, e46231. [Google Scholar] [CrossRef]
- Dong, T.; Joyce, C.; Schellhorn, H.E. The role of RpoS in bacterial adaptation. In Bacterial Physiology: A Molecular Approach; Springer: Berlin/Heidelberg, Germany, 2008; pp. 313–337. [Google Scholar]
- Hengge-Aronis, R.J.M. Signal transduction and regulatory mechanisms involved in control of the σS (RpoS) subunit of RNA polymerase. Microbiol. Mol. Biol. Rev. 2002, 66, 373–395. [Google Scholar] [CrossRef]
- Fang, F.C.; Libby, S.J.; A Buchmeier, N.; Loewen, P.C.; Switala, J.; Harwood, J.; Guiney, D.G. The alternative sigma factor katF (rpoS) regulates Salmonella virulence. Proc. Natl. Acad. Sci. USA 1992, 89, 11978–11982. [Google Scholar] [CrossRef]
- Yildiz, F.H.; Schoolnik, G.K. Role of rpoS in stress survival and virulence of Vibrio cholerae. J. Bacteriol. 1998, 180, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.-J.; Silo-Suh, L.; Woods, D.E.; Hassett, D.J.; West, S.E.H.; Ohman, D.E. Effect of rpoS mutation on the stress response and expression of virulence factors in Pseudomonas aeruginosa. J. Bacteriol. 1999, 181, 3890–3897. [Google Scholar] [CrossRef]
- Wang, Y.; Kim, K.S. Effect of rpoS mutations on stress-resistance and invasion of brain microvascular endothelial cells in Escherichia coli K1. FEMS Microbiol. Lett. 2000, 182, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Battesti, A.; Majdalani, N.; Gottesman, S. The rpoS-mediated general stress response in Escherichia coli. Annu. Rev. Microbiol. 2011, 65, 189–213. [Google Scholar] [CrossRef]
- Dong, T.; Schellhorn, H.E. Role of RpoS in virulence of pathogens. Infect. Immun. 2010, 78, 887–897. [Google Scholar] [CrossRef]
- Price, S.B.; Cheng, C.-M.; Kaspar, C.W.; Wright, J.C.; DeGraves, F.J.; Penfound, T.A.; Castanie-Cornet, M.-P.; Foster, J.W. Role of rpoS in acid resistance and fecal shedding of Escherichia coli O157:H7. Appl. Environ. Microbiol. 2000, 66, 632–637. [Google Scholar] [CrossRef]
- Hmelo, L.R.; Borlee, B.R.; Almblad, H.; E Love, M.; E Randall, T.; Tseng, B.S.; Lin, C.; Irie, Y.; Storek, K.M.; Yang, J.J.; et al. Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nat. Protoc. 2015, 10, 1820–1841. [Google Scholar] [CrossRef]
- Dong, T.; Coombes, B.K.; Schellhorn, H.E. Role of RpoS in the virulence of Citrobacter rodentium. Infect. Immun. 2009, 77, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Prepr. 2018, 6, e27295v2. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.i.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. ACM SIGMOBILE Mob. Comput. Commun. Rev. 2001, 5, 3–55. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Simpson, E.H. Measurement of diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Good, I.J. The population frequencies of species and the estimation of population parameters. Biometrika 1953, 40, 237–264. [Google Scholar] [CrossRef]
- Pielou, E.C. The measurement of diversity in different types of biological collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Jaccard, P. Nouvelles recherches sur la distribution florale. Bull. Soc. Vaud. Sci. Nat. 1908, 44, 223–270. [Google Scholar]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Ramette, A. Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 2007, 62, 142–160. [Google Scholar] [CrossRef]
- McArdle, B.H.; Anderson, M.J. Fitting multivariate models to community data: A comment on distance-based redundancy analysis. Ecology 2001, 82, 290–297. [Google Scholar] [CrossRef]
- Anderson, M.J.; Ellingsen, K.E.; McArdle, B.H. Multivariate dispersion as a measure of beta diversity. Ecol. Lett. 2006, 9, 683–693. [Google Scholar] [CrossRef]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Warton, D.I.; Wright, S.T.; Wang, Y. Distance-based multivariate analyses confound location and dispersion effects. Methods Ecol. Evol. 2012, 3, 89–101. [Google Scholar] [CrossRef]
- Zaura, E.; Keijser, B.J.; Huse, S.M.; Crielaard, W. Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol. 2009, 9, 259. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.J.B. PICRUSt2: An improved and extensible approach for metagenome inference. bioRxiv 2019. [Google Scholar] [CrossRef]
- Chen, N.; Ling, Z.-X.; Jin, T.-T.; Li, M.; Zhao, S.; Zheng, L.-S.; Xi, X.; Wang, L.-L.; Chen, Y.-Y.; Shen, Y.-L.; et al. Altered profiles of gut microbiota in Klebsiella pneumoniae-induced pyogenic liver abscess. Curr. Microbiol. 2018, 75, 952–959. [Google Scholar] [CrossRef]
- Lynch, S.V.; Pedersen, O. The human intestinal microbiome in health and disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef]
- Roderburg, C.; Luedde, T. The role of the gut microbiome in the development and progression of liver cirrhosis and hepatocellular carcinoma. Gut Microbes 2014, 5, 441–445. [Google Scholar] [CrossRef]
- Sabatino, A.; Regolisti, G.; Cosola, C.; Gesualdo, L.; Fiaccadori, E. Intestinal microbiota in type 2 diabetes and chronic kidney disease. Curr. Diabetes Rep. 2017, 17, 16. [Google Scholar] [CrossRef] [PubMed]
- Claesson, M.J.; van Sinderen, D.; O’Toole, P.W. Lactobacillus phylogenomics–towards a reclassification of the genus. Int. J. Syst. Evol. Microbiol. 2008, 58, 2945–2954. [Google Scholar] [CrossRef]
- Salminen, M.K.; Tynkkynen, S.; Rautelin, H.; Saxelin, M.; Vaara, M.; Ruutu, P.; Sarna, S.; Valtonen, V.; Järvinen, A. Lactobacillus bacteremia during a rapid increase in probiotic use of Lactobacillus rhamnosus GG in Finland. Clin. Infect. Dis. 2002, 35, 1155–1160. [Google Scholar] [CrossRef]
- Sullivan, Å.; Erik Nord, C. Probiotic lactobacilli and bacteraemia in Stockholm. Scand. J. Infect. Dis. 2006, 38, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Bernet-Camard, M.F.; Liévin, V.; Brassart, D.; Neeser, J.R.; Servin, A.L.; Hudault, S. The human Lactobacillus acidophilus strain LA1 secretes a nonbacteriocin antibacterial substance(s) active in vitro and in vivo. Appl. Environ. Microbiol. 1997, 63, 2747–2753. [Google Scholar] [CrossRef] [PubMed]
- Gopal, P.K.; Prasad, J.; Smart, J.; Gill, H.S. In vitro adherence properties of Lactobacillus rhamnosus DR20 and Bifidobacterium lactis DR10 strains and their antagonistic activity against an enterotoxigenic Escherichia coli. Int. J. Food Microbiol. 2001, 67, 207–216. [Google Scholar] [CrossRef]
- Hudault, S.; Liévin, V.; Bernet-Camard, M.F.; Servin, A.L. Antagonistic activity exerted in vitro and in vivo by Lactobacillus casei (strain GG) against Salmonella typhimurium C5 infection. Appl. Environ. Microbiol. 1997, 63, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Reyna-Flores, F.; Barrios-Camacho, H.; Dantán-González, E.; Ramírez-Trujillo, J.A.; Lozano Aguirre Beltrán, L.F.; Rodríguez-Medina, N.; Garza-Ramos, U.; Suárez-Rodríguez, R. Draft genome sequences of endophytic isolates of Klebsiella variicola and Klebsiella pneumoniae obtained from the same sugarcane plant. Genome Announc. 2018, 6, e00147-18. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iqbal, M.Z.; He, P.; He, P.; Wu, Y.; Munir, S.; He, Y. The Response of Murine Gut Microbiome in the Presence of Altered rpoS Gene of Klebsiella pneumoniae. Int. J. Mol. Sci. 2024, 25, 9222. https://doi.org/10.3390/ijms25179222
Iqbal MZ, He P, He P, Wu Y, Munir S, He Y. The Response of Murine Gut Microbiome in the Presence of Altered rpoS Gene of Klebsiella pneumoniae. International Journal of Molecular Sciences. 2024; 25(17):9222. https://doi.org/10.3390/ijms25179222
Chicago/Turabian StyleIqbal, Muhammad Zafar, Pengfei He, Pengbo He, Yixin Wu, Shahzad Munir, and Yueqiu He. 2024. "The Response of Murine Gut Microbiome in the Presence of Altered rpoS Gene of Klebsiella pneumoniae" International Journal of Molecular Sciences 25, no. 17: 9222. https://doi.org/10.3390/ijms25179222