
Plasmodium falciparum Mitochondrial Complex III, the Target of Atovaquone, Is Essential for Progression to the Transmissible Sexual Stages

 ,
,
Abstract
:1. Introduction
2. Results
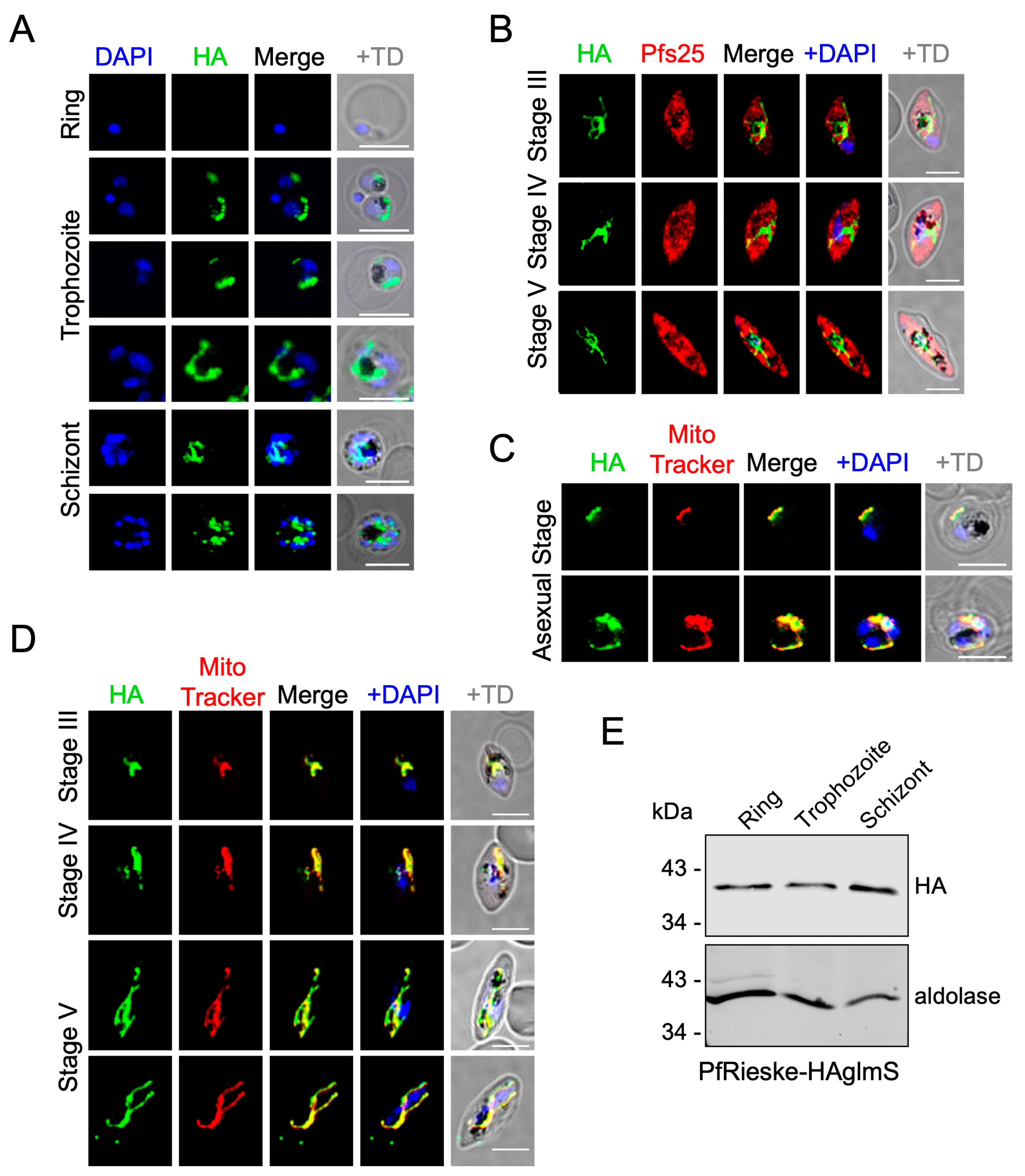
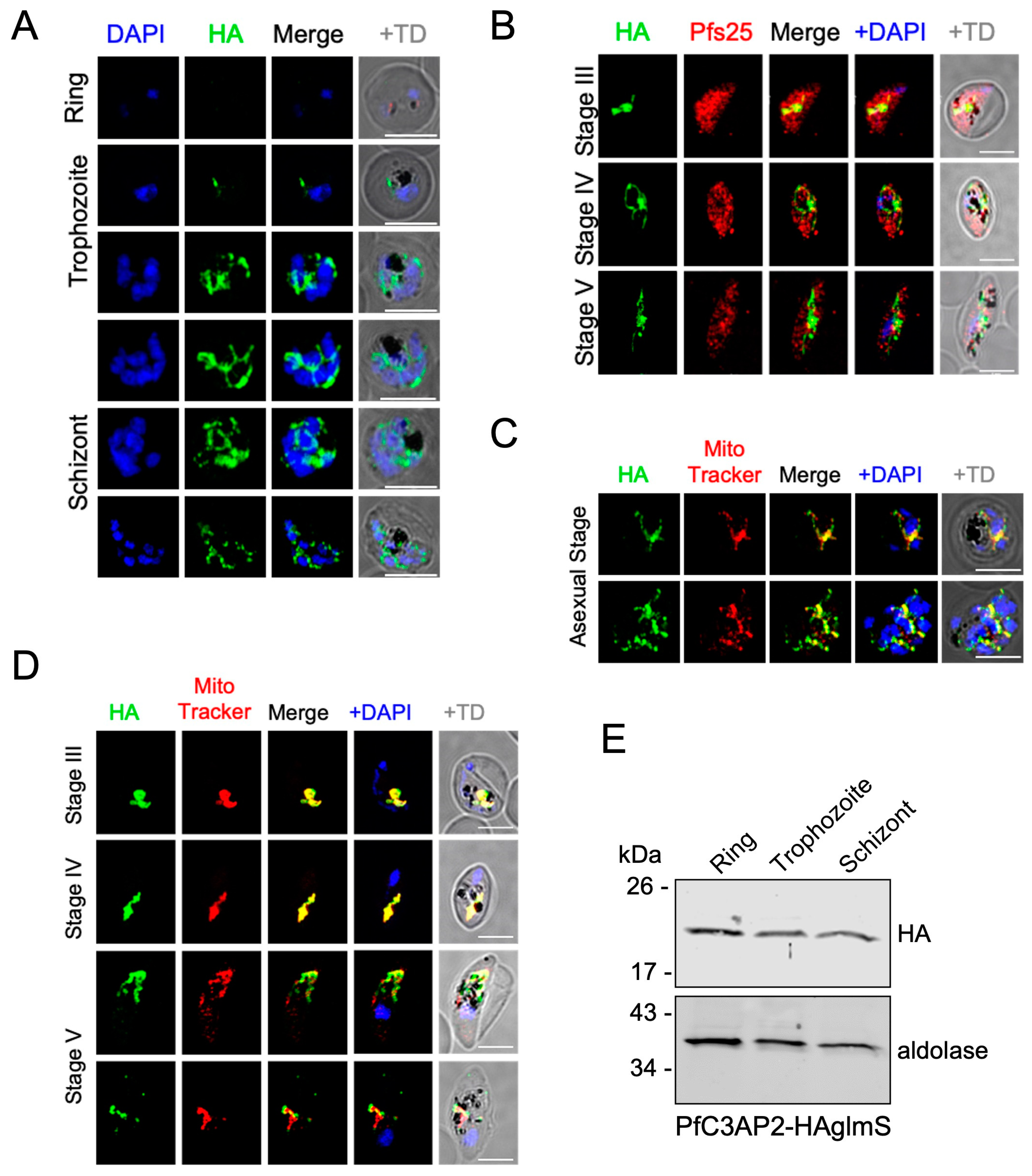
2.1. PfRieske and PfC3AP2 Are Part of Plasmodium falciparum Complex III (PfCIII)
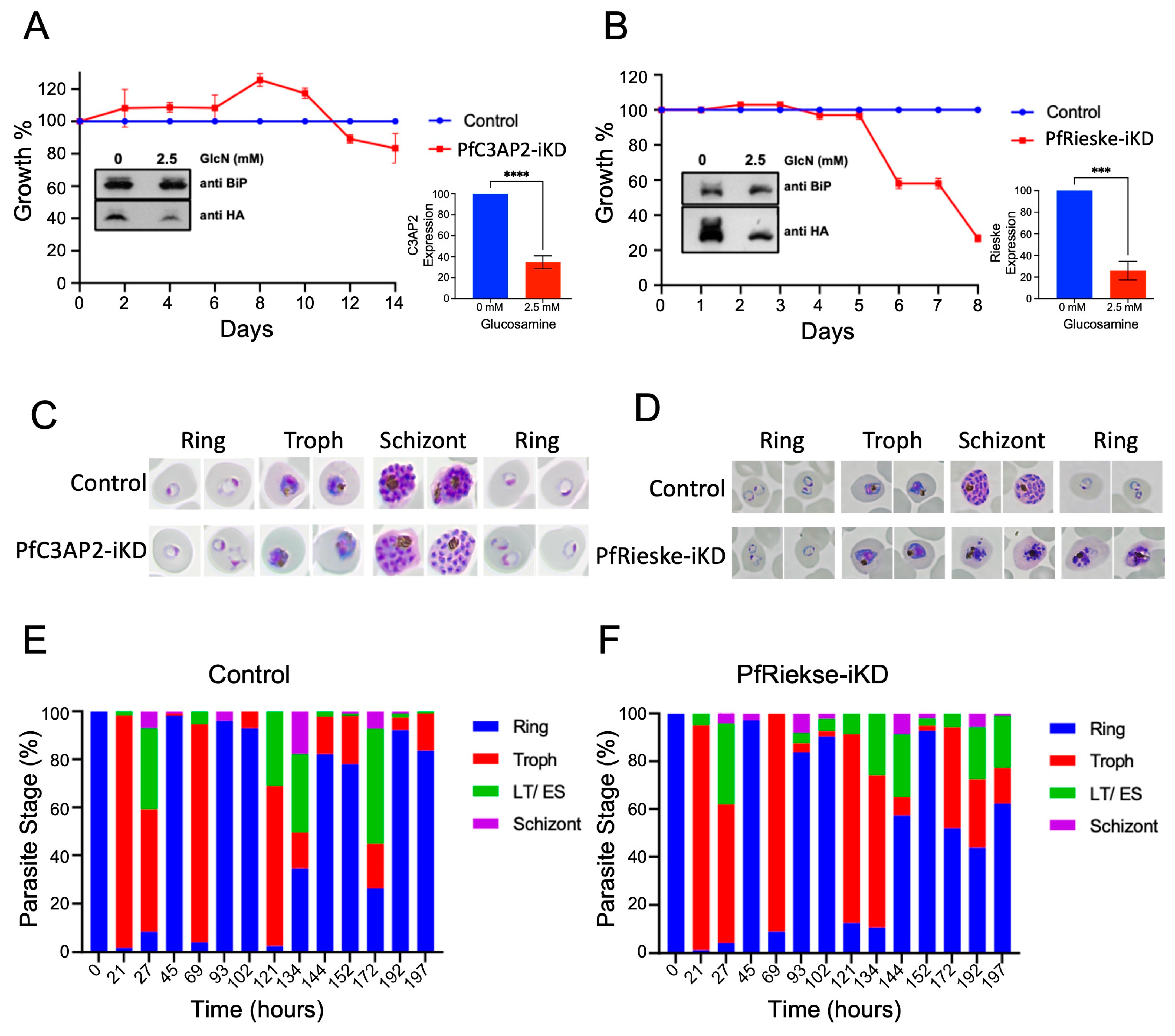
2.2. PfRieske Is Essential for Parasite Growth
2.3. Loss of PfRieske Results in Hypersensitivity to mETC Inhibitors and to Membrane Depolarization by Proguanil
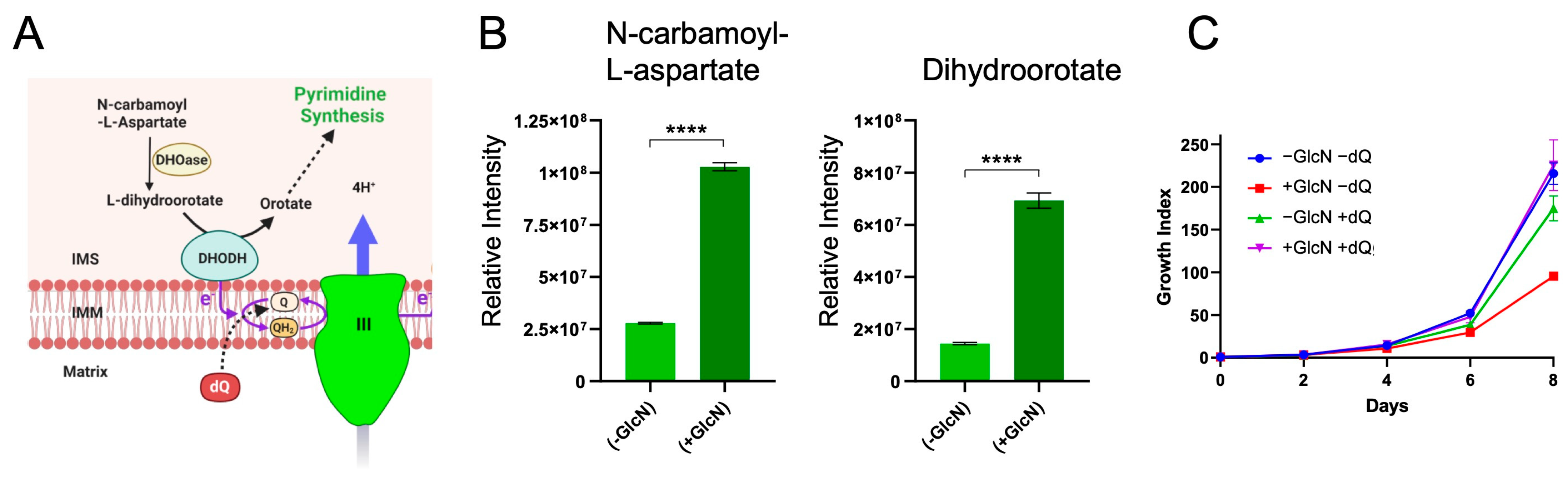
2.4. PfRieske Is Required for the Pyrimidine Synthesis Pathway
2.5. Loss of PfRieske Causes Decrease in Cristae Density and Affects Gametocytes Maturation
3. Discussion
4. Materials and Methods
4.1. Plasmid Construction, Parasite Culture, Transfection, and Transgenic Line Confirmation
4.2. Inducible Knockdown and Parasite Growth Assay
4.3. Immunoprecipitation and Mass Spectrometry
4.4. Denaturing Polyacrylamide Gel Electrophoresis (SDS-PAGE), Blue Native (BN-PAGE), and Western Blotting
4.5. Immuno-Fluorescence Assay (IFA) and Microscopy
4.6. Metabolomic Profiling
4.7. Drug Sensitivity Assay
4.8. Transmission Electron Microscopy
4.9. Gametocyte Commitment and Culturing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. World Malaria Report 2023; World Health Organization: Geneva, Switzerland, 2023. [Google Scholar]
- Bray, R.S.; Garnham, P.C.C. The life-cycle of primate Malaria parasites. Br. Med. Bull. 1982, 38, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Delves, M.; Plouffe, D.; Scheurer, C.; Meister, S.; Wittlin, S.; Winzeler, E.A.; Sinden, R.E.; Leroy, D. The activities of current antimalarial drugs on the life cycle stages of plasmodium: A comparative study with human and rodent parasites. PLoS Med. 2012, 9, e1001169. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Mharakurwa, S.; Ndiaye, D.; Rathod, P.K.; Rosenthal, P.J. Antimalarial Drug Resistance: Literature Review and Activities and Findings of the ICEMR Network. Am. J. Trop. Med. Hyg. 2015, 93, 57–68. [Google Scholar] [CrossRef]
- Siqueira-Neto, J.L.; Wicht, K.J.; Chibale, K.; Burrows, J.N.; Fidock, D.A.; Winzeler, E.A. Antimalarial drug discovery: Progress and approaches. Nat. Rev. Drug Discov. 2023, 22, 807–826. [Google Scholar] [CrossRef] [PubMed]
- Painter, H.J.; Morrisey, J.M.; Mather, M.W.; Vaidya, A.B. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 2007, 446, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Sturm, A.; Mollard, V.; Cozijnsen, A.; Goodman, C.D.; McFadden, G.I. Mitochondrial ATP synthase is dispensable in blood-stage Plasmodium berghei rodent malaria but essential in the mosquito phase. Proc. Natl. Acad. Sci. USA 2015, 112, 10216–10223. [Google Scholar] [CrossRef]
- Fry, M.; Pudney, M. Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4′-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80). Biochem. Pharmacol. 1992, 43, 1545–1553. [Google Scholar] [CrossRef]
- Davies, C.S.; Pudney, M.; Matthews, P.J.; Sinden, R.E. The causal prophylactic activity of the novel hydroxynaphthoquinone 566C80 against Plasmodium berghei infections in rats. Acta Leiden. 1989, 58, 115–128. [Google Scholar]
- Srivastava, I.K.; Rottenberg, H.; Vaidya, A.B. Atovaquone, a broad spectrum antiparasitic drug, collapses mitochondrial membrane potential in a malarial parasite. J. Biol. Chem. 1997, 272, 3961–3966. [Google Scholar] [CrossRef]
- Winter, R.W.; Kelly, J.X.; Smilkstein, M.J.; Dodean, R.; Hinrichs, D.; Riscoe, M.K. Antimalarial quinolones: Synthesis, potency, and mechanistic studies. Exp. Parasitol. 2007, 118, 487–497. [Google Scholar] [CrossRef]
- Nilsen, A.; LaCrue, A.N.; White, K.L.; Forquer, I.P.; Cross, R.M.; Marfurt, J.; Mather, M.W.; Delves, M.J.; Shackleford, D.M.; Saenz, F.E.; et al. Quinolone-3-diarylethers: A new class of antimalarial drug. Sci. Transl. Med. 2013, 5, 177ra37. [Google Scholar] [CrossRef] [PubMed]
- Winter, R.; Kelly, J.X.; Smilkstein, M.J.; Hinrichs, D.; Koop, D.R.; Riscoe, M.K. Optimization of endochin-like quinolones for antimalarial activity. Exp. Parasitol. 2010, 127, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Zong, S.; Wu, M.; Gu, J.; Yang, M. Architecture of Human Mitochondrial Respiratory Megacomplex I2III2IV2. Cell 2017, 170, 1247–1257.e12. [Google Scholar] [CrossRef]
- Evers, F.; Cabrera-Orefice, A.; Elurbe, D.M.; Lindert, M.K.-T.; Boltryk, S.D.; Voss, T.S.; Huynen, M.A.; Brandt, U.; Kooij, T.W.A. Composition and stage dynamics of mitochondrial complexes in Plasmodium falciparum. Nat. Commun. 2021, 12, 3820. [Google Scholar] [CrossRef] [PubMed]
- Espino-Sanchez, T.J.; Wienkers, H.; Marvin, R.G.; Nalder, S.-A.; Garcfa-Guerrero, A.E.; VanNatta, P.E.; Jami-Alahmadi, Y.; Blackwell, A.M.; Whitby, F.G.; Wohlschlegel, J.A.; et al. Direct tests of cytochrome c and c(1) functions in the electron transport chain of malaria para-sites. Proc. Natl. Acad. Sci. USA 2023, 120, e2301047120. [Google Scholar] [CrossRef]
- Maclean, A.E.; Bridges, H.R.; Silva, M.F.; Ding, S.; Ovciarikova, J.; Hirst, J.; Sheiner, L. Complexome profile of Toxoplasma gondii mitochondria identifies divergent subunits of respiratory chain complexes including new subunits of cytochrome bc1 complex. PLoS Pathog. 2021, 17, e1009301. [Google Scholar] [CrossRef]
- Sheokand, P.K.; Narwal, M.; Thakur, V.; Mohmmed, A. GlmS mediated knock-down of a phospholipase expedite alternate pathway to generate phosphocho-line required for phosphatidylcholine synthesis in Plasmodium falciparum. Biochem. J. 2021, 478, 3429–3444. [Google Scholar] [CrossRef]
- van Dooren, G.G.; Marti, M.; Tonkin, C.J.; Stimmler, L.M.; Cowman, A.F.; McFadden, G.I. Development of the endoplasmic reticulum, mitochondrion and apicoplast during the asexual life cycle of Plasmodium falciparum. Mol. Microbiol. 2005, 57, 405–419. [Google Scholar] [CrossRef]
- Okamoto, N.; Spurck, T.P.; Goodman, C.D.; McFadden, G.I. Apicoplast and mitochondrion in gametocytogenesis of Plasmodium falciparum. Eukaryot. Cell 2009, 8, 128–132. [Google Scholar] [CrossRef]
- Goodman, C.D.; Siregar, J.E.; Mollard, V.; Vega-Rodríguez, J.; Syafruddin, D.; Matsuoka, H.; Matsuzaki, M.; Toyama, T.; Sturm, A.; Cozijnsen, A.; et al. Parasites resistant to the antimalarial atovaquone fail to transmit by mosquitoes. Science 2016, 352, 349–353. [Google Scholar] [CrossRef]
- Komatsuya, K.; Sakura, T.; Shiomi, K.; Ōmura, S.; Hikosaka, K.; Nozaki, T.; Kita, K.; Inaoka, D.K. Siccanin Is a Dual-Target Inhibitor of Plasmodium falciparum Mitochondrial Complex II and Complex III. Pharmaceuticals 2022, 15, 903. [Google Scholar] [CrossRef]
- Xiang, H.; McSurdy-Freed, J.; Moorthy, G.S.; Hugger, E.; Bambal, R.; Han, C.; Ferrer, S.; Gargallo, D.; Davis, C.B. Preclinical drug metabolism and pharmacokinetic evaluation of GW844520, A novel anti-malarial mitochondrial electron transport inhibitor. J. Pharm. Sci. 2006, 95, 2657–2672. [Google Scholar] [CrossRef] [PubMed]
- Yeates, C.L.; Batchelor, J.F.; Capon, E.C.; Cheesman, N.J.; Fry, M.; Hudson, A.T.; Pudney, M.; Trimming, H.; Woolven, J.; Bueno, J.M.; et al. Synthesis and structure–activity relationships of 4-pyridones as potential antimalarials. J. Med. Chem. 2008, 51, 2845–2852. [Google Scholar] [CrossRef]
- Winter, R.W.; Kelly, J.X.; Smilkstein, M.J.; Dodean, R.; Bagby, G.C.; Rathbun, R.K.; Levin, J.I.; Hinrichs, D.; Riscoe, M.K. Evaluation and lead optimization of anti-malarial acridones. Exp. Parasitol. 2006, 114, 47–56. [Google Scholar] [CrossRef]
- Coteron, J.M.; Marco, M.; Esquivias, J.; Deng, X.; White, K.L.; White, J.; Koltun, M.; El Mazouni, F.; Kokkonda, S.; Katneni, K.; et al. Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J. Med. Chem. 2011, 54, 5540–5561. [Google Scholar] [CrossRef] [PubMed]
- Pudney, M.; Gutteridge, W.; Zeman, A.; Dickins, M.; Woolley, J.L. Atovaquone and proguanil hydrochloride: A review of nonclinical studies. J. Travel Med. 1999, 6, S8–S12. [Google Scholar] [CrossRef]
- Ling, L.; Mulaka, M.; Munro, J.; Dass, S.; Mather, M.W.; Riscoe, M.K.; Llinás, M.; Zhou, J.; Ke, H. Genetic ablation of the mitoribosome in the malaria parasite Plasmodium falciparum sensitizes it to antimalarials that target mitochondrial functions. J. Biol. Chem. 2020, 295, 7235–7248. [Google Scholar] [CrossRef]
- Ke, H.; Morrisey, J.M.; Ganesan, S.M.; Painter, H.J.; Mather, M.W.; Vaidya, A.B. Variation among Plasmodium falciparum strains in their reliance on mitochondrial electron transport chain func-tion. Eukaryot. Cell 2011, 10, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Falekun, S.; Sepulveda, J.; Jami-Alahmadi, Y.; Park, H.; A Wohlschlegel, J.; A Sigala, P.; Angeles, L.; States, U. Divergent acyl carrier protein decouples mitochondrial Fe-S cluster biogenesis from fatty acid synthesis in malaria parasites. eLife 2021, 10, 71636. [Google Scholar] [CrossRef]
- MacRae, J.I.; Dixon, M.W.; Dearnley, M.K.; Chua, H.H.; Chambers, J.M.; Kenny, S.; Bottova, I.; Tilley, L.; McConville, M.J. Mitochondrial metabolism of sexual and asexual blood stages of the malaria parasite Plasmodium falciparum. BMC Biol. 2013, 11, 67. [Google Scholar] [CrossRef]
- Srivastava, A.; Philip, N.; Hughes, K.R.; Georgiou, K.; MacRae, J.I.; Barrett, M.P.; Creek, D.J.; McConville, M.J.; Waters, A.P. Stage-Specific Changes in Plasmodium Metabolism Required for Differentiation and Adaptation to Different Host and Vector Environments. PLoS Pathog. 2016, 12, e1006094. [Google Scholar] [CrossRef]
- Paton, D.G.; Childs, L.M.; Itoe, M.A.; Holmdahl, I.E.; Buckee, C.O.; Catteruccia, F. Exposing Anopheles mosquitoes to antimalarials blocks Plasmodium parasite transmission. Nature 2019, 567, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Paton, D.G.; Probst, A.S.; Ma, E.; Adams, K.L.; Shaw, W.R.; Singh, N.; Bopp, S.; Volkman, S.K.; Hien, D.F.S.; Paré, P.S.L.; et al. Using an antimalarial in mosquitoes overcomes Anopheles and Plasmodium resistance to malaria control strategies. PLoS Pathog. 2022, 18, e1010609. [Google Scholar] [CrossRef]
- Saliba, K.S. and M. Jacobs-Lorena, Production of Plasmodium falciparum gametocytes in vitro. Methods Mol. Biol. 2013, 923, 17–25. [Google Scholar] [PubMed]
- Krungkrai, J.; Prapunwattana, P.; Krungkrai, S. Ultrastructure and function of mitochondria in gametocytic stage of Plasmodium falciparum. Parasite 2000, 7, 19–26. [Google Scholar] [CrossRef]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef]
- Hayward, J.A.; Makota, F.V.; Cihalova, D.; Leonard, R.A.; Rajendran, E.; Zwahlen, S.M.; Shuttleworth, L.; Wiedemann, U.; Spry, C.; Saliba, K.J.; et al. A screen of drug-like molecules identifies chemically diverse electron transport chain inhibitors in apicomplexan parasites. PLoS Pathog. 2023, 19, e1011517. [Google Scholar] [CrossRef]
- Seidi, A.; Muellner-Wong, L.S.; Rajendran, E.; Tjhin, E.T.; Dagley, L.F.; Aw, V.Y.; Faou, P.; Webb, A.I.; Tonkin, C.J.; Van Dooren, G.G. Elucidating the mitochondrial proteome of Toxoplasma gondii reveals the presence of a divergent cytochrome c oxidase. eLife 2018, 7, e38131. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.F.; Douglas, K.; Sandalli, S.; Maclean, A.E.; Sheiner, L. Functional and biochemical characterization of the Toxoplasma gondii succinate dehydrogenase complex. PLoS Pathog. 2023, 19, e1011867. [Google Scholar] [CrossRef]
- Vercellino, I.; Sazanov, L.A. Structure and assembly of the mammalian mitochondrial supercomplex CIII2CIV. Nature 2021, 598, 364–367. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, C.; Otto, T.D.; Oberstaller, J.; Liao, X.; Adapa, S.R.; Udenze, K.; Bronner, I.F.; Casandra, D.; Mayho, M.; et al. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation muta-genesis. Science 2018, 360, eaap7847. [Google Scholar] [CrossRef] [PubMed]
- Dass, S.; Mather, M.W.; Morrisey, J.M.; Ling, L.; Vaidya, A.B.; Ke, H. Transcriptional changes in Plasmodium falciparum upon conditional knock down of mitochondrial ribosomal proteins RSM22 and L23. PLoS ONE 2022, 17, e0274993. [Google Scholar] [CrossRef] [PubMed]
- Skinner-Adams, T.S.; Fisher, G.M.; Riches, A.G.; Hutt, O.E.; Jarvis, K.E.; Wilson, T.; von Itzstein, M.; Chopra, P.; Antonova-Koch, Y.; Meister, S.; et al. Cyclization-blocked proguanil as a strategy to improve the antimalarial activity of atovaquone. Commun. Biol. 2019, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hino, A.; Hirai, M.; Tanaka, T.Q.; Watanabe, Y.-I.; Matsuoka, H.; Kita, K. Critical roles of the mitochondrial complex II in oocyst formation of rodent malaria parasite Plasmodium berghei. J. Biochem. 2012, 152, 259–268. [Google Scholar] [CrossRef]
- Boysen, K.E.; Matuschewski, K. Arrested oocyst maturation in plasmodium parasites lacking type II NADH:ubiquinone dehydrogenase. J. Biol. Chem. 2011, 286, 32661–32671. [Google Scholar] [CrossRef]
- Mühleip, A.; Flygaard, R.K.; Baradaran, R.; Haapanen, O.; Gruhl, T.; Tobiasson, V.; Maréchal, A.; Sharma, V.; Amunts, A. Structural basis of mitochondrial membrane bending by the I-II-III(2)-IV(2) supercomplex. Nature 2023, 615, 934–938. [Google Scholar] [CrossRef]
- Mallo, N.; Ovciarikova, J.; Martins-Duarte, E.S.; Baehr, S.C.; Biddau, M.; Wilde, M.L.; Uboldi, A.D.; Lemgruber, L.; Tonkin, C.J.; Wideman, J.G.; et al. Depletion of a Toxoplasma porin leads to defects in mitochondrial morphology and contacts with the endoplasmic reticulum. J. Cell Sci. 2021, 134, jcs255299. [Google Scholar] [CrossRef]
- Ovciarikova, J.; Lemgruber, L.; Stilger, K.L.; Sullivan, W.J.; Sheiner, L. Mitochondrial behaviour throughout the lytic cycle of Toxoplasma gondii. Sci. Rep. 2017, 7, srep42746. [Google Scholar] [CrossRef]
- Souza, R.O.O.; Jacobs, K.N.; Back, P.S.; Bradley, P.J.; Arrizabalaga, G. IMC10 and LMF1 mediate mitochondrial morphology through mitochondrion–pellicle contact sites in Toxoplasma gondii. J. Cell Sci. 2022, 135, 260083. [Google Scholar] [CrossRef]
- Ovciarikova, J.; Souza, R.O.O.; Arrizabalaga, G.; Sheiner, L. Protein control of membrane and organelle dynamics: Insights from the divergent eukaryote Toxoplasma gondii. Curr. Opin. Cell Biol. 2022, 76, 102085. [Google Scholar] [CrossRef]
- Connelly, S.V.; Manzella-Lapeira, J.; Levine, Z.C.; Brzostowski, J.; Krymskaya, L.; Rahman, R.S.; Ellis, A.C.; Amin, S.N.; Sá, J.M.; Wellems, T.E. Restructured Mitochondrial-Nuclear Interaction in Plasmodium falciparum Dormancy and Persister Survival after Artemisinin Exposure. mBio 2021, 12, e0075321. [Google Scholar] [CrossRef] [PubMed]
- Crabb, B.S.; Rug, M.; Gilberger, T.-W.; Thompson, J.K.; Triglia, T.; Maier, A.G.; Cowman, A.F. Transfection of the human malaria parasite Plasmodium falciparum. Methods Mol. Biol. 2004, 270, 263–276. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Expected | Detected (Epx1) | Detected (Exp 2) | Accession |
|---|---|---|---|
| Cytb | Yes | Yes | PF3D7_MITO2300 |
| Cytc1 | Yes | Yes | PF3D7_1462700 |
| Rieske | Yes | Yes | PF3D7_1439400 |
| QCR6 | No | No | PF3D7_1426900 |
| QCR7 | Yes | Yes | PF3D7_1012300 |
| QCR8 | No | No | PF3D7_0306000 |
| QCR9 | No | Yes | PF3D7_0622600 |
| MPPalpha | Yes | Yes | PF3D7_0523100 |
| MPPbeta | Yes | Yes | PF3D7_0933600 |
| C3AP1 | No | Yes | PF3D7_0722700 |
| C3AP2 | Yes | Yes | PF3D7_1326000 |
| C3AP3 | Yes | Yes | PF3D7_0817800 |
| Name | Sequence |
|---|---|
| 2448 PfC3AP2-FWD | GCAGATCTCCTTCAGATAGTAGGAAATTTG |
| 2449 PfC3AP2-REV | GCCTGCAGC TGCGTGTTCAACTGA |
| 2535 PfRieske-FWD-B | GCGGATCCCCTGTATTTCCAAGAAAACC |
| 2451 PfRieske-REV | GCCTGCAGCTCCAATTTTTATCGTATTTTC |
| 2718 PfC3AP2-Int1 | CAAAAGAATTCAGGGTAAATTTATACCTTG |
| 2719 PfRieske-Int1 | GTATTCCTCCTGCATCTGAAGATC |
| 2797 HAglmS-REV-1 | TGATCTGCACACTCAGCCGGGA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheokand, P.K.; Pradhan, S.; Maclean, A.E.; Mühleip, A.; Sheiner, L. Plasmodium falciparum Mitochondrial Complex III, the Target of Atovaquone, Is Essential for Progression to the Transmissible Sexual Stages. Int. J. Mol. Sci. 2024, 25, 9239. https://doi.org/10.3390/ijms25179239
Sheokand PK, Pradhan S, Maclean AE, Mühleip A, Sheiner L. Plasmodium falciparum Mitochondrial Complex III, the Target of Atovaquone, Is Essential for Progression to the Transmissible Sexual Stages. International Journal of Molecular Sciences. 2024; 25(17):9239. https://doi.org/10.3390/ijms25179239
Chicago/Turabian StyleSheokand, Pradeep Kumar, Sabyasachi Pradhan, Andrew E. Maclean, Alexander Mühleip, and Lilach Sheiner. 2024. "Plasmodium falciparum Mitochondrial Complex III, the Target of Atovaquone, Is Essential for Progression to the Transmissible Sexual Stages" International Journal of Molecular Sciences 25, no. 17: 9239. https://doi.org/10.3390/ijms25179239